

Abstract
In the past few years there has been a veritable explosion in the field of reporter gene imaging, with the aim of determining the location, duration and extent of gene expression within living subjects. An important application of this approach is the molecular imaging of interacting protein partners, which could pave the way to functional proteomics in living animals and might provide a tool for the whole-body evaluation of new pharmaceuticals targeted to modulate protein–protein interactions. Three general methods are currently available for imaging protein–protein interactions in living subjects using reporter genes: a modified mammalian two-hybrid system, a bioluminescence resonance energy transfer (BRET) system, and split reporter protein complementation and reconstitution strategies. In the future, these innovative approaches are likely to enhance our appreciation of entire biological pathway systems and their pharmacological regulation.
Introduction
A frequent theme pervading biological investigations is that the great majority of proteins generally operate as components of complexes that contain other macromolecules to carry out specific biological functions, and that networks of interactions (interactomes) connect multiple, different cellular processes [1]. Indeed, it is primarily as components of complexes that proteins perform cellular functions. We now fully appreciate that the cell is not a simple aqueous solution, but instead a dense gel of interacting proteins forming the basis of phenomena such as DNA replication and transcription, enzymatic-driven metabolic pathways, signaling pathways, and cell-cycle control [2,3]. Other interacting protein complexes work as components of cellular machines such as ribosomes, which read genetic information and synthesize proteins. Many human diseases can be traced to aberrant protein–protein interactions, resulting either from the loss of an essential interaction or through the formation of an abnormal protein complex at an inappropriate time or location through interactions with endogenous proteins, proteins from pathogens or both [4].
Current research aims to isolate and structurally characterize all the proteins that exist in the cell. Importantly, protein–protein interactions are now considered to be so vital to cellular function that one of the first experiments performed on a protein may be a search for its interaction partners [2]. As of April 2004 there were about 12 000 known structures, from a variety of organisms, of assemblies involving two or more protein chains [5]. Just how many complexes exist in a particular proteome is not easy to deduce, because of the different component types (e.g. proteins, nucleic acids, nucleotides and metal ions) and the varying life spans of the protein complexes (e.g. transient protein–protein interactions, such as those involved in signaling, and stable interactions, such as in the ribosome). Until recently, the most comprehensive information about protein–protein interactions was available for the yeast proteome, consisting of approximately 6200 proteins [5,6]. In yeast, there are about nine protein partners per protein, although not necessarily all direct or interacting at the same time. The human proteome could have an order of magnitude more complexes than the yeast cell [5,7].
Several technologies, grouped together under the term `proteomics' (a term introduced in 1995 by Wasinger et al. [8]), have emerged with the common objective of studying protein function at the scale of an entire pathway, a whole cell or even a whole organism. Proteomic analyses encompass three key aspects: the large-scale study of protein–protein interactions or complexes to establish comprehensive protein interactomes; the global examination of protein expression profiles; and, more recently, the analysis of protein post-translational modifications [9]. Many cell-based experimental techniques (reviewed elsewhere [3]) have been developed and employed using intact cells and cell extracts to study protein–protein interactions and to facilitate these proteomic endeavors. Each of these analytical systems has its own merits and drawbacks, as reviewed previously [10].
The overall modification of existing in vitro and cell-culture-based experimental assays to study protein–protein interactions in living small animal models of disease is dependent on the challenging task of adapting them so that signals can be noninvasively detected from the exterior of living subjects upon the cellular or sub-cellular interaction of two proteins of interest. Only over the past 4–5 years has it been possible to develop such methods, as a result of the increased availability of non-invasive small animal imaging technologies and the rapidly expanding field of molecular imaging, allowing signal detection from deep tissues within a living subject. We previously reviewed in detail the many advantages afforded by molecular imaging in living subjects (such as assessment of whole-body phenomena, repeatability, functionality, and quantification) [11]. One subset of molecular imaging techniques comprises reporter gene expression imaging. This represents an `indirect' imaging method involving multiple components and entails the use of a pre-targeting molecule (an imaging reporter gene) that is subsequently activated upon occurrence of a specific molecular event. Following this event, a molecular probe (a substrate or a ligand) specific for the activated pre-targeting molecule (an enzyme or receptor) is used to image its activation [12]. An important feature of reporter gene imaging techniques is their particular versatility, which allows them to be adapted for imaging diverse protein–protein interactions in intact living subjects, as outlined below (also recently reviewed by our group [10] and others [13–17]).
The ability to noninvasively image protein–protein interactions has important implications for a wide variety of biological research endeavors, including drug discovery and molecular medicine. In particular, the visual representation, characterization, quantification, and timing of these biological processes in living subjects could create unprecedented opportunities to complement available in vitro or cell culture methodologies. In turn, such studies could accelerate the evaluation in living subjects of novel drugs that promote or inhibit active homodimeric or heterodimeric protein assembly, and could be employed to characterize known protein–protein interactions more fully (e.g. the reasons for, and the factors that drive their association) in the context of whole-body physiologically authentic environments [18].
Modified mammalian two-hybrid system
It has been estimated that more than 50% of all protein interactions described in the literature have been detected using the yeast two-hybrid system [19,20]. In a two-hybrid assay, two proteins are expressed in yeast: one fused to a DNA-binding domain (BD) and the other fused to a transcription activation domain (AD). If the two proteins interact, they activate transcription of one or more reporter genes that are distal to binding sites for the BD. Studied first in yeast, this classical two-hybrid system was later adapted for mammalian cells, using different expression plasmids but similar assay principles. Our group has adapted this system further (Figure 1) using pBIND-Id (which contains the yeast GAL4 DNA-binding domain fused to the Id protein [inhibitor of differentiation/DNA binding]) and pACT-MyoD (which contains the herpes simplex virus VP16 activation domain fused to a segment of the murine MyoD protein) as the two-hybrid proteins [21•]. Id and MyoD are members of the helix–loop–helix family of nuclear proteins, and are known to strongly interact in vivo during myogenic differentiation [22]. To inducibly modulate the expression of these two hybrid proteins, the CMV (cytomegalovirus) promoters of pBIND-Id and pACT-MyoD were replaced with tumor necrosis factor α (TNFα) inducible nuclear factor κB (NFκB) response elements. TNFα is a pleio-tropic cytokine secreted by lipopolysaccharide-stimulated macrophages that induces a variety of cell-specific events, including NFκB activation, and causes tumor necrosis in vivo when injected in tumor-bearing mice [23]. Finally, we used pG5-luc (which contains five GAL4-binding sites upstream of a minimal TATA box, followed by the firefly luciferase [Fluc] gene) as a two-hybrid reporter. In cells treated with TNFα, activated endogenous NFκB moves to the nucleus and activates transcription of the two-hybrid proteins: they in turn interact, and activate transcription of the Fluc reporter. We studied this system in detail in cell culture and in 293T cells implanted in mice using cooled CCD (charge-coupled device) camera bioluminescence imaging, while using TNFα to modulate the system [21•].
Figure 1.
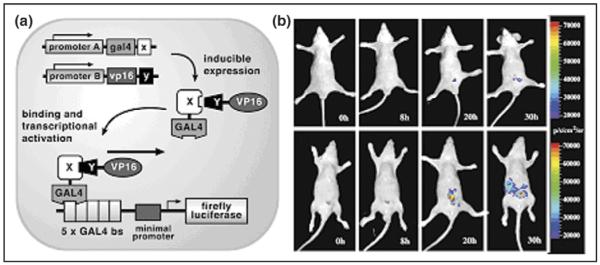
Imaging protein–protein interactions in living mice with a modified yeast two-hybrid strategy. (a) Schematic diagram of the system for imaging the interaction of proteins X and Y. The first step involves the vectors pA-gal4-x and pB-vp16-y, which are used to drive transcription of gal4-x and vp16-y through use of the promoters A and B. In the second step, the two fusion proteins GAL4-X and VP16-Y interact because of the specificity of protein X for protein Y. Subsequently, the GAL4-X-Y-VP16 binds to GAL4-binding sites (five GAL4-binding sites [bs] are available) on a reporter template. This leads to VP16-mediated transactivation of firefly luciferase (Fluc) reporter gene expression under the control of GAL4 response elements in a minimal promoter. Transcription of the Fluc reporter gene leads to Fluc protein which, in turn, leads to a detectable visible light signal in the presence of the appropriate substrate (d-luciferin), ATP, Mg2+ and oxygen. The NFκB promoter was used for either pA or pB and TNFα-mediated induction. (b) In vivo optical CCD imaging of mice carrying transiently transfected 293T cells for induction of the yeast two-hybrid system. All images shown are the visible light image superimposed on the optical CCD bioluminescence image with a scale in photons/sec/cm2/steridian (sr). Mice in top row were imaged after injection of d-luciferin but with no TNFα-mediated induction. Mice in bottom row were imaged after injection of d-luciferin after TNFα-mediated induction, showing a marked gain in signal from the peritoneum over 30 h. (Figure reproduced from [21•] with permission.)
Separately, but following a similar design, Luker et al. [24] developed a tetracycline (or doxycycline)-inducible, bidirectional vector carrying in one direction the tumor suppressor p53 gene fused with Gal4, and in the other direction, the T antigen (TAg) of SV40 fused with VP16. Expression of the p53 and TAg hybrid proteins is induced by doxycycline, resulting in their interaction and formation of a VP16–Gal4 transactivator complex. This complex subsequently binds to the Gal4-binding sequences in the promoter of a HSV1-sr39tk-GFP (green fluorescent protein) reporter fusion protein. Tumor xenografts of HeLa cells stably expressing both the reporter plasmid and the bidirectional two-hybrid expression plasmid were implanted in living mice, and doxycycline-induced protein–protein interactions were imaged by microPET at different time points. Micro positron emission tomography (microPET) imaging was performed using a fluorine-18 positron labeled analog of penciclovir, which is trapped in cells expressing the reporter fusion (through the action of the sr39TK protein). The fusion protein showed increasing expression, as detected by fluorescence microscopy (for the GFP component) and by microPET (for the TK component), when induced with increasing doses of doxycycline both in cells and in tumor xenografts of living mice. A subsequent study defined quantitative (relative differences in amounts of interacting proteins and expression of the reporter gene) and kinetic (time interval between induction of interacting proteins and detection of reporter activity in vivo) parameters pertaining to this system [25].
Bioluminescence resonance energy transfer
Bioluminescence resonance energy transfer (BRET) technology involves the non-radioactive transfer of energy between donor and acceptor molecules by the Förster mechanism [26] (Figure 2). The energy transfer primarily depends on two factors: an overlap between the emission and excitation spectra of the donor and acceptor molecules, respectively, and the close proximity of the donor and the acceptor entities (<100Å). As fluorescence resonance energy transfer (FRET)- and BRET-based technologies assume increasingly prominent roles for studying protein–protein interactions, manufacturers are continually developing new instrumentation for measuring FRET and/or BRET ratios, which are, in general, low-intensity signals. BRET measurements are usually obtained with a microplate reader equipped with specific filter sets for the detection of the donor and acceptor emission peaks. In a study from our laboratory [27•], we used the BRET2 system (Biosignal Packard), which involves Renilla luciferase (Rluc) as a bioluminescent donor and mutant GFP2 as a fluorescent acceptor, adapted for expression in mammalian cells. The resonance energy transfer from the reaction of the reconstructed Rluc protein with its substrate deep blue coelenterazine (DBC) excited the GFP2 protein when the two fused proteins — either Id and MyoD or FKBP12 (FK506-binding protein) and FRB (the FKBP rapamycin-binding domain of FRAP [i.e. mammalian target of rapamycin]) in the presence of a small-molecule mediator (rapamycin) — interact. We demonstrated the ability to detect signal from protein–protein interactions in cultured cells, as well as from the surface and deeper tissues of living animals with implanted cells overexpressing the fusion constructs (Figure 2). This approach, currently under continuing validation, could have important implications for the study of protein–protein interactions in cells maintained in their natural environment, particularly if it can be effectively applied for the evaluation of new pharmaceuticals.
Figure 2.

Imaging protein–protein interactions in living mice using BRET. (a) Schematic showing small-molecule-mediated protein–protein interactions leading to bioluminescence resonance energy transfer (BRET). FKBP12 is fused to the N terminus of Rluc donor protein, and FRB is fused to the C terminus of the GFP2 acceptor protein. When the genes encoding both of these fusion proteins are expressed inside cells and rapamycin is present to mediate the FRB–FKBP12 interaction, then resonance energy transfer occurs. This BRET signal can be detected using the Deep Blue Coelenterazine (DBC) substrate for Rluc. (b,c) Detection of the in vivo BRET2 signal from specific protein–protein interactions. Dorsal view of a nude mouse implanted subcutaneously with 5 × 106 293T cells either transiently transfected with pBRET2 (L) or with pFKBP12-hRluc (LL) alone or co-transfected with pFKBP12-hRluc and pGFP2-FRB (LR) in (b) the presence or (c) absence of rapamycin. Mice that received the small-molecule mediator drug, rapamycin (5 mg/kg) were injected intraperitoneally immediately after cell implantation. The scan was performed 7 h after drug administration. Mice were scanned for 5 min integration time using either GFP2 or DBC filters in succession by injecting with 25 μg DBC intravenously. (Figure reproduced from [27•] with permission.)
Split reporter strategies
In certain circumstances, functional proteins can be assembled from one or more polypeptide fragments and this property has been exploited in strategies to measure real-time protein–protein interactions. Indeed, synthetically separated fragments of some enzymes can reconstitute functionally active proteins, particularly if the interaction is assisted by fusion of the enzyme fragments to strongly interacting moieties. Thus, in the `split protein' strategy, a single reporter protein/enzyme is cleaved into N-terminal and C-terminal segments; each segment is fused to one of two interacting proteins (X and Y). Physical interaction between X and Y reconstitutes the functional reporter protein, leading to signal generation that can be measured. This split protein strategy can work either through protein-fragment complementation assays (PCA) or through intein-mediated reconstitution assays. In the former, non-covalent assembly of the reporter protein occurs; in the latter, reconstitution of the full reporter protein occurs through covalent bonding. To date, several reporter proteins (e.g. β-lactamase, β-galactosidase, ubiquitin, dihydrofolate reductase [DHFR], Fluc, Rluc and GFP) have been adapted for split protein strategies by finding various split sites for each reporter protein [28,29•,30,31]. If a full-length reporter can be imaged in living subjects, and this reporter can be appropriately split, then the split reporter assay could potentially be used to noninvasively image protein–protein interactions. The appropriate split point should lead to two fragments that do not have significant affinity for each other, yet when brought together (through interaction of the two proteins being studied) lead to a detectable signal.
The principle of the PCA strategy for detecting protein–protein interactions was first demonstrated by Pelletier et al. [32] using the enzyme DHFR, following inspiration from a 1994 paper by Johnsson and Varshavsky describing what they called the `ubiquitin split protein sensor' [33]. In all PCAs, splitting a specific reporter protein into two distinct fragments abolishes its function. Bringing the two fragments back together in a controlled manner then restores functional activity [34] (Figure 3). Selected fragments of many proteins can associate to produce functional bimolecular complexes [35]; the PCA system can therefore be generalized for several enzymes for the detection of protein–protein interactions. Many examples have been reported, including DHFR, glycinamide ribo-nucleotide transformoylase, aminoglycoside and hygromycin B phosphotransferases (reviewed by Michnick et al. [34]), Escherichia coli TEM-1 β-lactamase [30,36], GFP and its variants [35], and the molecular imaging reporters Fluc [29•] and Rluc [31].
Figure 3.
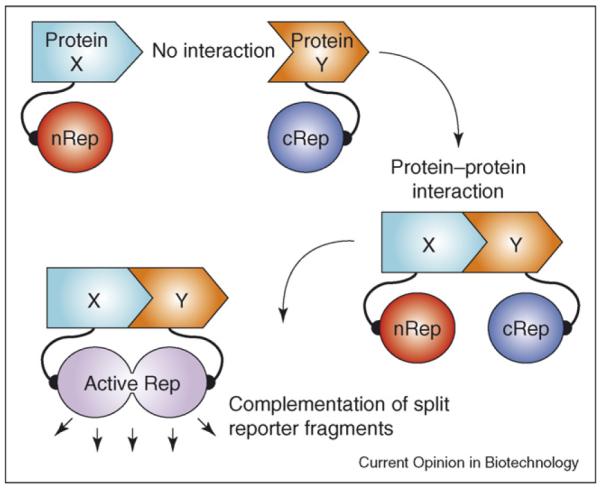
Schematic diagram of the split reporter based complementation strategy used to optically image protein–protein interactions in living mice. The N-terminal half of the reporter protein or enzyme (nRep) is attached to protein X through a short linker peptide, and the C-terminal half of cRep is connected to protein Y through another linker peptide. Interaction of proteins X and Y recovers the reporter protein/enzyme activity through protein complementation.
We demonstrated for the first time the feasibility of imaging protein–protein interactions using split reporters in small living animals. We studied a PCA based on split Fluc — cleaved into two fragments nFluc (residues 1–437) and cFluc (residues 438–550) — using the interaction of Id and MyoD as test proteins [29•] (Figure 3). Separately, Luker et al. [37] described a systematic truncation library yielding alternative complementary N- and C-terminal fragments of Fluc (nFluc residues 2–416 and cFluc residues 398–550). These fragments were used to monitor the rapamycin-mediated interaction of rapamycin-binding proteins FRB and FKBP12 [37]. Likewise, we used the Fluc fragments previously tested with Id and MyoD to study rapamycin-mediated interactions, but found the complementation to be too weak for optical imaging in living animals using the CCD camera (R Paulmurugan, unpublished).
The enzyme Rluc (or the synthetically mutated humanized version, hRluc) is a 36 kDa monomeric bioluminescence imaging reporter protein, being the smallest optical reporter protein identified to date for studying protein–protein interactions in a PCA strategy [31,38]. This PCA strategy, using the N- and C-terminal halves of split Rluc, was found to function in both cell culture and living animals and has been demonstrated with several different protein partners. We used fragments generated by splitting between residues 229 and 230 to study rapamycin-induced interaction of the human proteins FRB and FKBP12 [39•]. Moreover, protein interaction between insulin receptor substrate 1 (IRS-1) and the SH2 domain of phosphotidyl inositol 3-kinase (PI3K) in the insulin signaling pathway was located in living mammalian cells using Rluc split between residues 91 and 92 [31]. One limitation associated with the use of Rluc is its relatively rapid reaction kinetics, requiring early time-point measurements [40]. Nevertheless, this split reporter system appears highly suitable for studying protein–protein interactions in cells and in living animals, owing to its optical bioluminescence and the fact that its signal can be amplified through an enzymatic process. Further, the complementation strategies based on Rluc fragments, having a smaller fragment size than Fluc, cause less hindrance with interacting protein partners and work more efficiently with different imaging assays, including protein–protein interactions, small-molecule-induced protein–protein interactions, small-molecule-mediated inhibition of protein–protein interactions, and protein homodimerization [18,29•,39•]. More recently, we developed a novel fusion protein approach for studying the rapamycin-mediated interaction of fused FRB and FKBP12 with either split hRluc or split enhanced GFP, to achieve a system with greater sensitivity for detecting lower levels of drug-mediated protein–protein interactions in vivo [41].
Ozawa et al. [42] initially demonstrated that Fluc can be split between amino acid positions 437 and 438 and used with inteins (DnaE) in a reconstitution strategy to detect the insulin-induced interaction of phosphorylated IRS-1 and its target N-terminal SH2 domain of PI3K in a cell culture assay. We subsequently reasoned that it might be possible to split Fluc and use split reporter complementation without inteins. We therefore studied PCA and the intein-mediated reconstitution of Fluc fragments and found that a complementation strategy was as sensitive as the intein-mediated reconstitution strategy under the conditions tested [29•]. More recently, Kim et al. [43] developed a genetically encoded bioluminescent indicator for monitoring and imaging the nuclear trafficking of target proteins in vitro and in vivo. The assay is based on the reconstitution of split fragments of Rluc by protein splicing with a DnaE intein. A target cytosolic protein fused to the N-terminal half of Rluc is expressed in mammalian cells. If the protein translocates into the nucleus, the Rluc moiety meets the C-terminal half of Rluc and full-length Rluc is reconstituted by protein splicing. The authors demonstrated quantitative cell-based in vitro sensing and in vivo imaging of ligand-induced translocation of the androgen receptor, which allowed high-throughput screening of exogenous and endogenous agonists and antagonists. Similar assays were used to image glucocorticoid receptor translocation into the nucleus [44].
Conclusions
The three general methods currently available for imaging protein–protein interactions in living subjects have been reviewed. The modified mammalian two-hybrid system is derived from the yeast two-hybrid system. Advantages of this system are that it is fairly simple, rapid and the strategies developed should be generally applicable for many protein partners, as long as the interaction complex can lead to transactivation of the reporters of choice. The main limitation is that it can only be used to detect interacting proteins in the nucleus. Several areas of investigation are required to further refine the use of the modified mammalian two-hybrid system for non-invasive imaging of protein–protein interactions. As well as further quantitative and kinetic evaluations (e.g. characterizing the ability to follow interactions over time based on the half-life of the reporter protein(s) that are transactivated), studies are needed to optimize the choice of transactivator as well as the choice of promoters and levels of fusion proteins. Mathematical models to correlate and study these effects are currently under development.
Unlike FRET, BRET does not require excitation of the donor with an external light source and offers several advantages in this respect. BRET assays show no photobleaching or photoisomerization of the donor protein, no photodamage to cells, and no light scattering or auto-fluorescence from cells or microplates (when used in vitro), which can be caused by incident excitation light. In addition, one main advantage of BRET over FRET is the lack of emission arising from direct excitation of the acceptor. This reduction in background should permit the detection of interacting proteins at much lower concentrations than is possible for FRET. The use of BRET technology for imaging protein–protein interactions in living subjects is currently being validated, with a view to effective applications in the evaluation of new pharmaceuticals.
Split protein strategies are based on absolute stereospecific and regiospecific requirements for complex formation amongst interacting sequences. Although no head-to-head comparison is available, this strategy would therefore appear more specific than the modified yeast two-hybrid system, which suffers from many false-positive outcomes, at least in its standard (non-imaging) laboratory use. Moreover, split protein strategies can be used to image interacting proteins anywhere in the cell. Unlike fluorescence-microscopy-based techniques, studies of the kinetics of protein–protein interactions, including analysis of complementation reversibility, are not possible at present, although this will be an area for further investigation. These future experiments will also require assessment in several cell lines, as well as with a greater variety of protein partners of different sizes and interaction affinities (weak transient to strong obligate), to establish the general applicability of this technique.
The high sensitivity of the assays described for detecting, locating, and quantifying protein–protein interactions, combined with the advantages of doing so in a living subject environment, should make them of potential value in many areas of biological investigation and future clinical applications. Ultimately, we foresee innovative molecular imaging tools, such as the ones presented, enhancing our appreciation of entire biological pathway systems and their pharmacological regulation. Such approaches will accelerate the achievement of a `systems biology' understanding of biological complexity.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Cusick ME, Klitgord N, Vidal M, Hill DE. Interactome: gateway into systems biology. Hum Mol Genet. 2005;14:R171–R181. doi: 10.1093/hmg/ddi335. [DOI] [PubMed] [Google Scholar]
- 2.Mayer BJ. Protein-protein interactions in signalling cascades. Methods Mol Biol. 2006;332:79–99. doi: 10.1385/1-59745-048-0:79. [DOI] [PubMed] [Google Scholar]
- 3.Droit A, Poirier GG, Hunter JM. Experimental and bioinformatic approaches for interrogating protein-protein interactions to determine protein function. J Mol Endocrinol. 2005;34:263–280. doi: 10.1677/jme.1.01693. [DOI] [PubMed] [Google Scholar]
- 4.Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Opin Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Russell RB, Alber F, Aloy P, Davis FP, Korkin D, Pichaud M, Topf M, Sali A. A structural perspective on protein-protein interactions. Curr Opin Struct Biol. 2004;14:313–324. doi: 10.1016/j.sbi.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 6.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 7.Abbott A. Proteomics: the society of proteins. Nature. 2002;417:894–896. doi: 10.1038/417894a. [DOI] [PubMed] [Google Scholar]
- 8.Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, Wilkins MR, Duncan MW, Harris R, Williams KL, Humphrey-Smith I. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis. 1995;16:1090–1094. doi: 10.1002/elps.11501601185. [DOI] [PubMed] [Google Scholar]
- 9.Colland F, Daviet L. Integrating a functional proteomic approach into the target discovery process. Biochimie. 2004;86:625–632. doi: 10.1016/j.biochi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 10.Paulmurugan R, Ray P, De A, Chan CT, Gambhir SS. Imaging protein-protein interactions in living subjects. Trends Anal Chem. 2005;24:446–458. [Google Scholar]
- 11.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17:545–580. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 12.Gambhir SS, Barrio JR, Herschman HR, Phelps ME. Assays for noninvasive imaging of reporter gene expression. Nucl Med Biol. 1999;26:481–490. doi: 10.1016/s0969-8051(99)00021-9. [DOI] [PubMed] [Google Scholar]
- 13.Haberkorn U, Altmann A. Noninvasive imaging of protein-protein interactions in living organisms. Trends Biotechnol. 2003;21:241–243. doi: 10.1016/S0167-7799(03)00116-1. [DOI] [PubMed] [Google Scholar]
- 14.Umezawa Y. Seeing what was unseen: new analytical methods for molecular imaging. Chem Rec. 2003;3:22–28. doi: 10.1002/tcr.10049. [DOI] [PubMed] [Google Scholar]
- 15.Luker GD, Sharma V, Piwnica-Worms D. Visualizing protein-protein interactions in living animals. Methods. 2003;29:110–122. doi: 10.1016/s1046-2023(02)00285-2. [DOI] [PubMed] [Google Scholar]
- 16.Luker KE, Piwnica-Worms D. Optimizing luciferase protein fragment complementation for bioluminescent imaging of protein-protein interactions in live cells and animals. Methods Enzymol. 2004;385:349–360. doi: 10.1016/S0076-6879(04)85019-5. [DOI] [PubMed] [Google Scholar]
- 17.Ozawa T. Designing split reporter proteins for analytical tools. Anal Chim Acta. 2006;556:58–68. doi: 10.1016/j.aca.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Massoud TF, Paulmurugan R, Gambhir SS. Molecular imaging of homodimeric protein-protein interactions in living subjects. FASEB J. 2004;18:1105–1107. doi: 10.1096/fj.03-1128fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 20.Fields S, Sternglanz R. The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 1994;10:286–292. doi: 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- 21•.Ray P, Pimenta H, Paulmurugan R, Berger F, Phelps ME, Iyer M, Gambhir SS. Noninvasive quantitative imaging of protein-protein interactions in living subjects. Proc Natl Acad Sci USA. 2002;99:3105–3110. doi: 10.1073/pnas.052710999. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate for the first time the use of a modified mammalian two-hybrid system to image protein–protein interactions in living mice.
- 22.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 23.Boland MP, O'Neill LA. Ceramide activates NFκB by inducing the processing of p105. J Biol Chem. 1998;273:15494–15500. doi: 10.1074/jbc.273.25.15494. [DOI] [PubMed] [Google Scholar]
- 24.Luker GD, Sharma V, Pica CM, Dahlheimer JL, Li W, Ochesky J, Ryan CE, Piwnica-Worms H, Piwnica-Worms D. Noninvasive imaging of protein-protein interactions in living animals. Proc Natl Acad Sci USA. 2002;99:6961–6966. doi: 10.1073/pnas.092022399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luker GD, Sharma V, Pica CM, Prior JL, Li W, Piwnica-Worms D. Molecular imaging of protein-protein interactions: controlled expression of p53 and large T-antigen fusion proteins in vivo. Cancer Res. 2003;63:1780–1788. [PubMed] [Google Scholar]
- 26.Förster T. Intermolecular energy transference and fluorescence. Ann Phys. 1948;2:54–75. [Google Scholar]
- 27•.De A, Gambhir SS. Noninvasive imaging of protein-protein interactions from live cells and living subjects using bioluminescence resonance energy transfer. FASEB J. 2005;19:2017–2019. doi: 10.1096/fj.05-4628fje. [DOI] [PubMed] [Google Scholar]; The authors demonstrate for the first time the use of BRET to image protein–protein interactions in living mice.
- 28.Wehrman T, Kleaveland B, Her JH, Balint RF, Blau HM. Protein-protein interactions monitored in mammalian cells via complementation of beta-lactamase enzyme fragments. Proc Natl Acad Sci USA. 2002;99:3469–3474. doi: 10.1073/pnas.062043699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Paulmurugan R, Umezawa Y, Gambhir SS. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci USA. 2002;99:15608–15613. doi: 10.1073/pnas.242594299. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate for the first time the use of split firefly luciferase to image protein–protein interactions in living mice.
- 30.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 31.Kaihara A, Kawai Y, Sato M, Ozawa T, Umezawa Y. Locating a protein-protein interaction in living cells via split Renilla luciferase complementation. Anal Chem. 2003;75:4176–4181. doi: 10.1021/ac0300800. [DOI] [PubMed] [Google Scholar]
- 32.Pelletier JN, Campbell-Valois F, Michnick SW. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc Natl Acad Sci USA. 1998;95:12141–12146. doi: 10.1073/pnas.95.21.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnsson N, Varshavsky A. Split ubiquitin as a sensor of protein interactions in vivo. Proc Natl Acad Sci USA. 1994;91:10340–10344. doi: 10.1073/pnas.91.22.10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Balisle A, Pelletier JN. Detection of protein-protein interactions by protein fragment complementation strategies. Methods Enzymol. 2000;328:208–230. doi: 10.1016/s0076-6879(00)28399-7. [DOI] [PubMed] [Google Scholar]
- 35.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spotts JM, Dolmetsch RE, Greenberg ME. Time-lapse imaging of a dynamic phosphorylation-dependent protein-protein interaction in mammalian cells. Proc Natl Acad Sci USA. 2002;99:15142–15147. doi: 10.1073/pnas.232565699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal Chem. 2003;75:1584–1589. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Paulmurugan R, Massoud TF, Huang J, Gambhir SS. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004;64:2113–2119. doi: 10.1158/0008-5472.can-03-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate for the first time the use of split Renilla luciferase to image the modulatory effect of a drug on a protein–protein interaction in living mice.
- 40.Bhaumik S, Gambhir SS. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc Natl Acad Sci USA. 2002;99:377–382. doi: 10.1073/pnas.012611099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulmurugan R, Gambhir SS. Novel fusion protein approach for efficient high-throughput screening of small molecule-mediating protein-protein interactions in cells and living animals. Cancer Res. 2005;65:7413–7420. doi: 10.1158/0008-5472.CAN-05-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal Chem. 2001;73:2516–2521. doi: 10.1021/ac0013296. [DOI] [PubMed] [Google Scholar]
- 43.Kim SB, Ozawa T, Watanabe S, Umezawa Y. High-throughput sensing and noninvasive imaging of protein nuclear transport by using reconstitution of split Renilla luciferase. Proc Natl Acad Sci USA. 2004;101:11542–11547. doi: 10.1073/pnas.0401722101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SB, Ozawa T, Umezawa Y. Genetically encoded stress indicator for noninvasively imaging endogenous corticosterone in living mice. Anal Chem. 2005;77:6588–6593. doi: 10.1021/ac0510078. [DOI] [PubMed] [Google Scholar]