

Abstract
Previous studies have indicated that electrical stimulation of the cerebellar fastigial nucleus in rats may reduce brain infarct size, increase the expression of Ku70 in cerebral ischemia/reperfusion area, and decrease the number of apoptotic neurons. However, the anti-apoptotic mechanism of Ku70 remains unclear. In this study, fastigial nucleus stimulation was given to rats 24, 48, and 72 hours before cerebral ischemia/reperfusion injury. Results from the electrical stimulation group revealed that rats exhibited a reduction in brain infarct size, a significant increase in the expression of Ku70 in cerebral ischemia/reperfusion regions, and a decreased number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells. Double immunofluorescence staining revealed no co-localization of Ku70 with TUNEL-positive cells. However, Ku70 partly co-localized with Bax protein in the cytoplasm of rats with cerebral ischemia/reperfusion injury. These findings suggest an involvement of Ku70 with Bax in the cytoplasm of rats exposed to electrical stimulation of the cerebellar fastigial nucleus, and may thus provide an understanding into the anti-apoptotic activity of Ku70 in cerebral ischemia/reperfusion injury.
Keywords: nerve regeneration, brain injury, apoptosis, Ku70, Bax, electrical stimulation, fastigial nucleus, cerebral ischemia/reperfusion injury, DNA repair, NSFC grant, neural regeneration
Introduction
Electrical stimulation of the cerebellar fastigial nucleus confers remarkable protection from brain injury[1]. Stimulation of the fastigial nucleus for 1 hour can salvage brain injury caused by brain ischemia[2], and this protective effect can be maintained until the tenth day when the middle cerebral artery is occluded immediately after the stimulation[3]. The protection is maximal (~50% reduction), when the middle cerebral artery is occluded 3 days after the stimulation, and gradually disappears when lesions occur 3–4 weeks following stimulation. Fastigial nucleus-stimulated brain protection does not elicit changes in cerebral blood flow and the utilization of cerebral glucose[4]. These data suggest the existence of an intrinsic neuroprotective circuitry in the brain. Excitation of intrinsic neurons of the fastigial nucleus, rather than fibers of passage, is essential for central neurogenic neuroprotection[4]. In recent decades, the mechanism of central neurogenic neuroprotection has been the subject of intensive investigation, and still remains unknown. Although the molecular factors linking fastigial nucleus stimulation need to be identified, its protective effect is mediated, at least in part, by the suppression of apoptosis[5].
Apoptosis is involved in ischemia, trauma, and neurodegenerative diseases[6], thus playing a key role in brain injury. Over the last decade, many neurons in ischemic areas have been demonstrated to undergo necrotic and apoptotic cell death after a stroke event[7]. Two general signaling cascades, the mitochondrial apoptosis pathway and the cell death receptor-mediated pathway, have been identified for the activation of apoptosis in cerebral ischemia[7]. The Bcl-2 family of proteins has been found to participate in these complex apoptotic signaling pathways. Members of the Bcl-2 family are pro-apoptotic or anti-apoptotic and have been studied extensively for the past two decades[8]. Mitochondria are often involved in the initiation of apoptosis[9,10]. Once receiving an apoptotic signal, the permeability of the outer mitochondrial membrane increases, resulting in a reduction of the mitochondrial membrane potential followed by the translocation of the pro-apoptotic factor, cytochrome c, from the mitochondrial intermembrane space to the cytosol, thereby activating caspases and causing cell death[11]. Bax is a pro-apoptotic member of the Bcl-2 family, playing a key role in the induction of apoptosis[12]. The structure of Bax consists mainly of αα-helical segments and three Bcl-2 homology domains (BH1-BH3), which are the signature features of the Bcl-2 family of proteins[13]. In healthy cells, Bax is localized in the cytosol as a monomer. Stress signals induce the cleavage of the BH3-only proteins triggering the insertion/oligomerization of Bax into the mitochondrial outer membrane, and resulting in pore formation and decrease in the mitochondrial membrane potential[14]. This effect in turn leads to the release of apoptogenic proteins, such as cytochrome c, from mitochondria to the cytosol. Ischemia-induced activation of Bax has been the subject of intensive investigation, and Bax-dependent initiation and activation of subsequent apoptotic pathways has been shown to play critical roles in ischemic brain injury[15,16]. Studies in models of global cerebral ischemia indicate that the inhibition of Bax channels can prevent the decrease in mitochondrial membrane potential and the release of cytochrome c from mitochondria, and protect neurons against apoptosis[17]. However, the mechanism of inhibiting Bax activation remains unclear.
The DNA repair proteins, Ku70 and Ku80, are DNA-binding regulatory proteins of the DNA-dependent protein kinase, and contribute to the repair of DNA double-strand breaks[18]. Several studies have shown that Ku70 may play a role in DNA repair mechanisms or inhibit apoptosis after ischemia/reperfusion injury[19,20]. Cells from Ku70-deficient mice show increased sensitivity to pro-apoptotic agents[21]. Previous findings have also revealed that Ku70 protein is located in the cytoplasm of cultured primate cells[22] and predominantly in the cytosol of adult rat cerebellum and hippocampus[23]. Cytoplasmic Ku70 is involved in the control of cell apoptosis, specifically Bax-mediated apoptosis[24,25]. Pro-apoptotic stimuli initiate Ku70 to directly interact with Bax through its C terminus, thereby sequestering and releasing Bax in the cytosol of mitochondria[26]. Therefore, Ku70 is an important regulator of Bax activity and has a physiological role in the regulation of Bax-mediated apoptosis.
In our previous study, electrical stimulation of the fastigial nucleus in rats increased the expression of Ku70 in ischemia/reperfusion lesions and reduced the number of apoptotic cells[27]. However, the mechanism by which Ku70 inhibits apoptosis during this process remains unclear. Recently, in vitro studies have confirmed that Ku70 inhibits Bax-mediated apoptosis by preventing the translocation of Bax from the cytoplasm to the mitochondrial membrane[26]. Moreover, a Bax-inhibiting peptide designed from the Bax-binding domain of Ku70 has been developed and was shown to suppress Bax-mediated apoptosis[28,29,30,31]. These results suggest that the protective effect of Ku70 during ischemia/reperfusion injury following electrical stimulation of the fastigial nucleus is also associated with the Bax-mediated mitochondrial apoptotic pathway. In the present study, we investigated Bax as a possible molecular mechanism underlying the up-regulation and apoptotic effect of Ku70 after electrical stimulation of the fastigial nucleus in cerebral ischemia/reperfusion lesions.
Results
Quantitative analysis of experimental animals
A total of 78 Wistar rats were randomly divided into two groups, the model group or electric stimulation group. For the model group, the right middle cerebral artery was occluded for 2 hours, followed by reperfusion for 24, 48, or 72 hours (n = 13 per time point). For the electric stimulation group, the left cerebellar fastigial nucleus was electrically stimulated for 1 hour one day before cerebral artery ischemia (2 hours), followed by reperfusion for 24, 48 or 72 hours (n = 13 per time point). For each time point, brain tissue from the right side was taken from eight rats and used for immunohistochemistry, double immunofluorescence labeling, the apoptosis assay, and western blot analysis. The remaining five rats were used for triphenyltetrazolium chloride staining and to measure cerebral infarction size.
Fastigial nucleus stimulation reduced brain infarct volume
Compared with the model group, brain infarct size was significantly decreased (by approximately 40%) at each time point after electric stimulation of the fastigial nucleus (P < 0.05; Table 1). However, the infarct size in the two groups was not significantly changed at reperfusion time (Table 1).
Table 1.
Effect of fastigial nucleus stimulation on cerebral infarction volumes (mm3), expression of Ku70 and Bax (absorbance) of ischemia/reperfusion in rats
Ku70 in the ischemia/reperfusion area increased following fastigial nucleus stimulation
Immunohistochemical staining showed that Ku70 was mainly located in the nucleolus and cytoplasm (Figure 1A, B). Compared with control group, the number of Ku70-positive cells was significantly higher 24 hours after brain ischemia/reperfusion injury (P < 0.05) increased further at 48 hours, and peaked at 72 hours (Figures 1, 2). A similar effect was seen for the electric stimulation group, and furthermore, the number and intensity of Ku70 at each time point after stimulation were significantly higher than those of the model group (P < 0.05; Figures 1, 2). Western blot analysis further confirmed this effect, particularly at 48 and 72 hours (P < 0.05; Table 1, Figure 3A).
Figure 1.
Immunohistochemical staining of Ku70 and Bax, as well as terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, in the ischemia/reperfusion cortex following fastigial nucleus-stimulation.
Fastigial nucleus-stimulation was given to rats 72 hours before cerebral ischemia/reperfusion injury. Ku70 is mainly localized in the nucleus and cytoplasm, and Bax is present in the cytoplasm. TUNEL staining occurs in the nucleus. Scale bars: 50 μm.
Figure 2.
Cell counts of Ku70-, TUNEL- and Bax-positive cells in the ischemia/reperfusion area of rats following fastigial nucleus stimulation.
Data are expressed as mean ± SD (n = 8 per group). Data were analyzed using the one-way analysis of variance followed by the Student-New-man-Keuls post hoc test. aP < 0.05, vs. model group; bP < 0.05, vs. previous one time point. TUNEL: Terminal deoxynucleotidyl transferase dUTP nick end labeling.
Figure 3.
Western blot analysis of Ku70 and Bax in the brain of cerebral ischemia/reperfusion rats following fastigial nucleus stimulation.
Lanes 1–3 indicate 24, 48, 72 hours of reperfusion in the model group and lanes 4–6 point to 24, 48, 72 hours of reperfusion in the electric stimulation group.
Expression of Ku70 and apoptosis following fastigial nucleus stimulation
In the model group, the number of cells positive for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) increased 24 hours after cerebral ischemia/reperfusion injury, reached a peaked at 48 hours and declined at 72 hours. However, in the electric stimulation group, the number of TUNEL-positive cells was significantly lower than that of the model group (P < 0.05), particularly at 72 hours (Figure 1C, D and Figure 2). Immunofluorescence double staining of Ku70 and TUNEL showed that Ku70-positive cells co-localized with TUNEL-positive cells (Figure 4A).
Figure 4.
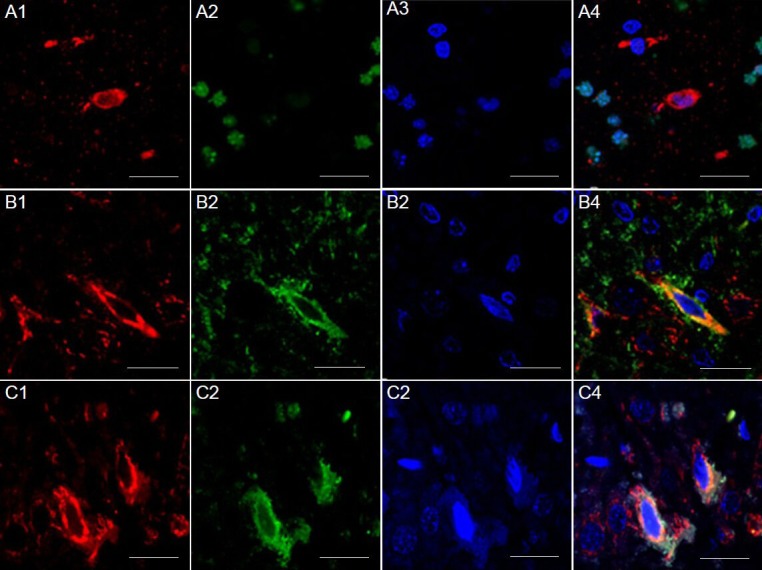
Presence of Ku70 and TUNEL/Bax in the cortex of rats (immunofluorescence double staining).
(A) Presence of Ku70 and TUNEL in the cortex of rats of the electric stimulation group. Ku70-positive cells (red, A1), TUNEL (green, A2) and 4’,6-diamidino-2-phenylindole (DAPI) nuclear staining (blue, A3), and all three merged (A4). Ku70 did not co-localize with TUNEL-positive cells. (B) Presence of Ku70 and Bax in the cortex 72 hours after cerebral ischemia/reperfusion injury in the model group. Immunofluorescence staining for Bax (red, B1) and Ku70 (green, B2), DAPI nuclear staining (blue, B3), and all three merged (B4). Ku70 co-localized with Bax in the cytoplasm. (C) Presence of Ku70 and Bax in the cortex 72 hours after cerebral ischemia/reperfusion injury in the electric-stimulating group. Immunofluorescence staining for Bax (red, C1) and Ku70 (green, C2), DAPI nuclear staining (blue, C3), and all three merged (C4). Ku70 co-localized with Bax in the cytoplasm. Fastigial nucleus-stimulation was given to rats 72 hours before cerebral ischemia/reperfusion injury. Scale bars: 20 μm.
Ku70 and Bax expression following fastigial nucleus stimulation
Bax was mainly expressed in the cytoplasm. The number of Bax-positive cells in the electric stimulation group was not significantly different to that of the model group (Figure 2). Western blot analysis further confirmed this result Table 1, Figure 3B). Double immunostaining of Ku70 and Bax showed that Ku70-positive cells co-localized with Bax in the cytoplasm (Figure 4B, C). A significantly higher proportion of Bax/Ku70 co-localized cells were found in the fastigial nucleus-stimulated group compared with the model group at 72 hours (P < 0.05; Figure 5).
Figure 5.
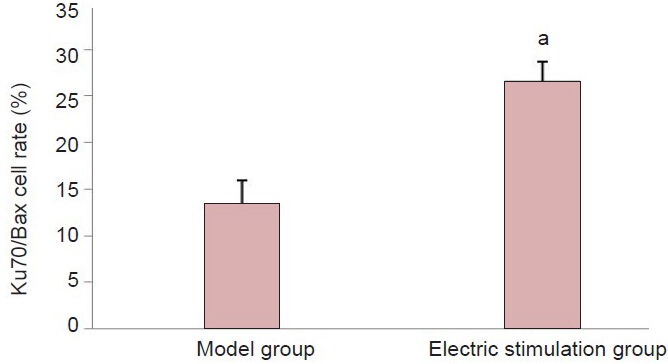
Count of cells double-labeled for Ku70 and Bax 72 hours following cerebral ischemia/reperfusion injury.
Data are expressed as mean ± SD (n = 8 per group). Data were analyzed using the one-way analysis of variance followed by the Student-New-man-Keuls post hoc test. aP < 0.05, vs. model group.
Discussion
Apoptosis is the process of programmed cell death, and is regulated by multiple genes. The exogenous death receptor pathway and endogenous mitochondrial pathway[32,33] are involved in this process. Mitochondria sense and amplify death signals and play an unquestionable role in regulating apoptosis despite the involvement of numerous other mechanisms[12,34]. Moreover, many Bcl-2 family proteins, including Bcl-2, Bax, and Bcl-xl, are located in the mitochondrial intermembrane space[12].
Bcl-2 family proteins promote the release of cytochrome c and change the permeability of the mitochondrial membrane[34]. Bax is a pro-apoptotic protein in the Bcl-2 family, playing a key role in the induction of apoptosis[12]. In non-apoptotic cells, Bax resides in the cytoplasm in an inactive form, and translocates from the cytoplasm to mitochondria (by forming oligomers to insert itself deeply into the outer mitochondrial membrane) after receiving an apoptotic stimulus, where it activates caspases by promoting the release of mitochondrial pro-apoptotic factors[35,36].
Bax insertion increases the mitochondrial permeability leading to a collapse of the mitochondrial membrane potential and the release of pro-apoptotic factors to the cytosol[37]. In neurons, the translocation of Bax from the cytosol to mitochondria represents one of the first steps in programmed cell death. The pro-apoptotic activity of Bax is related to its location, thus blocking the translocation of Bax from the cytoplasm to mitochondria may inhibit the release of mitochondrial cytochrome c, thereby inhibiting the Bax-mediated mitochondrial apoptotic pathway. Recent studies have demonstrated that Ku70 is an important factor that can regulate the pro-apoptotic activity of Bax[29,30,38,42].
Ku70 is mainly distributed in the nucleus and cytoplasm. In the nucleus, Ku70 repairs impaired double-stranded DNA. Several recent studies have found that the anti-Bax activity of Ku70 is independent from its DNA repair activity in the nucleus[43,44,45,46]. The cell permeable, five amino-acid peptide, Bax-inhibiting peptide (designed from the Bax binding domain of Ku70), protects cells against Bax-dependent apoptotic stimuli in multiple cell types[47], demonstrating the inhibition of Bax-induced death at the initial stage of activation. These results suggest that binding of Ku70 and Bax in the cytoplasm can inhibit Bax-mediated apoptosis by preventing the translocation of Bax from the cytoplasm to mitochondria[29,30].
Accumulating evidence has also confirmed decreased expression of Ku70 before the degradation of DNA following cerebral hypoxia/ischemia in neonatal rats, and its increase after ischemic preconditioning[19,46]. Therefore, Ku70 may play a role in the DNA repair mechanism during cerebral ischemia/reperfusion[19,46]. In the present study, the expression of Ku70 at different time points following cerebral ischemia/reperfusion was significantly higher in the fastigial nucleus-stimulated group than in the model group. This response was accompanied by a decrease in the number of apoptotic cells and reduced infarct volumes. Moreover, Ku70 did not co-localize with TUNEL-positive cells. Therefore, these results suggested that Ku70 may exhibit anti-apoptotic activity, and thus be a protective factor during cerebral ischemia/reperfusion injury after electrical stimulation of the fastigial nucleus. However, the specific anti-apoptotic pathway involved remains unknown. Zhou et al.[48] have shown that electrical stimulation of the fastigial nucleus reduces drug-induced translocation of Bax. However, this translocation does not relate to a change in the expression levels of Bax[5]. A possible explanation for this discrepancy is that electrical stimulation of the fastigial nucleus alters the distribution of Bax but does not change its overall expression levels. The translocation of Bax and its insertion into the outer mitochondrial membrane is a critical step in the initiation of apoptosis. Once an apoptotic signal is received, Bax is oligomerized and inserted into the outer mitochondrial membrane. This effect increases the permeability of the mitochondrial membrane and causes the release of mitochondrial intermembrane space proteins, including cytochrome c, which indirectly activates caspases[37,39]. In the current study, the expression level of Bax protein did not change after electrical stimulation of the fastigial nucleus. However, the number of cells with co-localized Ku70 and Bax significantly increased 72 hours following electrical stimulation. Furthermore, co-localization of Ku70 and Bax was increased in the cytoplasm. Our results also showed that Ku70 did not co-localize with TUNEL-positive cells. Overall, these findings suggest an involvement of Ku70 with Bax in the cytoplasm, which may provide an understanding into the anti-apoptotic activity of Ku70 in rats exposed to electrical stimulation of the cerebellar fastigial nucleus. In the cytoplasm, Ku70 may bind to Bax, thereby suppressing the Bax-mediated mitochondrial apoptotic pathway. This response may be a characteristic of Ku70 in addition to its effect on DNA repair. In conclusion, to our knowledge, this study is the first showing that preconditioning via fastigial nucleus stimulation may result in Ku70-mediated inhibition of Bax translocation.
Materials and Methods
Design
A randomized, controlled animal study.
Time and setting
This study was performed in the Neuropharmacology Laboratory of Anhui Medical University, China, from November 2009 to August 2010. Laser confocal microscopy and image analysis were performed at the Experimental Center of Life Sciences, University of Science and Technology of China.
Materials
Healthy male adult Wistar rats (n = 78), of clean grade, weighing 250 ± 30 g, were provided by the Experimental Animal Center of Guangxi Medical University, Nanning, China (license No. SCXK (Gui) 2009-0002). All rats were maintained at room temperature and given free access to food and water. Animals were treated humanely, conforming to the current ethical standards. All experiments were approved by the Animal Ethnics Committee of the First Hospital Affiliated to Guangxi Medical University, China. Every effort was made to minimize the number of rats used and their suffering.
Methods
Electrical stimulation of fastigial nucleus
Electrical stimulation of the fastigial nucleus was performed according to a previously described method[2], but with slight modifications. Rats were fasted for 8–12 hours, then anesthetized with 3.5% (w/v) chloral hydrate (10 mL/kg, intraperitoneally), followed by immobilization in a brain stereotactic endoscope (Japan Mau Science Equipment Research Institute, Tokyo, Japan). After a calvarium thoracic incision was made, the fastigial nucleus was accurately positioned using a stereotaxic atlas[49] of the rat brain. With the posterior border of the anterior fontanelle set as the zero point, an 11.1-mm incision was made, with a 1.1-mm incision on the left side of the median line. A hole (5.6 mm deep) was made in the skull for the attachment of electrodes. A YC-2 programmed electrical stimulation instrument (Chengdu Instrument Factory, Chengdu, Sichuan Province, China) was used, in which a 70-µA direct-current square-wave pulse (50 Hz) was applied. Each electrical stimulation lasted 1 hour during light anesthesia.
Establishment of cerebral artery ischemia/reperfusion model
The middle cerebral artery ischemia/reperfusion model was established using the nylon monofilament occlusion method[50]. A neck median incision was made to expose the common carotid artery bifurcation by applying a suture from the external carotid artery into the internal carotid artery to a depth of 18.5 ± 0.5 mm. In the sham surgery group, the suture was only inserted to a depth of 10 mm and was then withdrawn. The scoring system described by Vemuganti et al.[51] was used to evaluate neurological impairment after rats were recovered from anesthesia. Following cerebral ischemia, rats were also tested for neurological deficits using a 5-point scale: 0, no observable neurological deficits (normal); 1, failure to extend right forepaw (mild); 2, circling to the contralateral side (moderate); 3, falling to the right (severe); 4, inability to walk spontaneously; 5, depressed level of consciousness (very severe). Rats with a score of “0” or “4” were excluded. Successful ischemia/reperfusion was confirmed by triphenyltetrazolium chloride staining, which indicated the infarct focus (foci).
Measurement of cerebral infarct volumes by triphenyltetrazolium chloride staining
Rats were anesthetized with chloral hydrate and brains removed 24, 48, and 72 hours after cerebral ischemia/reperfusion injury. Coronal sections (2 μμm thickness) from brains were made, then placed in 2% (w/v) triphenyltetrazolium chloride (SANGON Biological Engineering Co., Ltd., Shanghai, China) solution, and staining was developed for 30 minutes at 37°C in the dark. Normal and damaged tissue was stained red and white, respectively. Sections were further fixed in 10% (v/v) formalin and then photographed (Olympus, Tokyo, Japan). The stained infarct area of each brain slice was assessed using a LUZEX-F image analyzer (Fuji Photo Film Co., Tokyo, Japan). The infarct volume was calculated using the following formula: V =t(A1+A2+...An)–(A1+An)t/2, in which “t” was the thickness of the section and “A” was the infarct area.
Immunohistochemical staining
Animals were sacrificed 24, 48, and 72 hours after cerebral ischemia/reperfusion injury, the brains were collected and then embedded in paraffin. Sections (8 μm thickness) at the level of the mid-thalamus were deparaffinized. Antigen retrieval was performed by microwave irradiation of sections in 10 mmol/L citrate buffer (pH 6.0). Sections were incubated in 3% (v/v) hydrogen peroxide solution at 37°C for 10 minutes, followed by incubation in normal sheep serum for 10 minutes at 37°C. The sections were incubated with mouse anti-rat Ku70 monoclonal antibody (1:100; Abcam, London, UK) or rabbit anti-rat Bax monoclonal antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C overnight, and re-warmed to 37°C for 20 minutes in the next morning. Sections were exposed to biotinylated sheep anti mouse/rabbit IgG (1:200; Beijing Zhongshan Golden Bridge Biological Engineering Co., Ltd., Beijing, China) solution at 37°C for 30 minutes and then incubated with horseradish peroxidase-labeled streptavidin at 37°C for 30 minutes. After several washes with PBS, color was developed using diaminobenzidine (Beijing Zhongshan Golden Bridge Biological Engineering Co., Ltd) for 5–10 minutes. Five randomly selected microscopic fields were captured using a 400 × light microscope (CX21, Olympus). The number of Ku70- and Bax-positive cells was counted, and the mean values were calculated.
TUNEL staining
Sections at 24, 48, and 72 hours were processed according to the manufacturer's directions of the TUNEL kit (Roche, Mannheim, Germany). Positively labeled nuclei (brown color) were identified from negatively unstained nuclei (blue color). The number of positive nuclei was determined by counting all the positively labeled nuclei present in five random visual fields under a microscope (400 × magnification; Olympus). The percentage of TUNEL-positive nuclei to all nuclei counted was used as the apoptotic index. Apoptotic cells were characterized as single cells with no inflammatory phenotypes, a curling of the cell membrane, and brown particulate or fragmented nuclei.
Immunofluorescence double labeling
Ku70 and TUNEL were used for immunofluorescence double labeling 24, 48, and 72 hours after cerebral ischemia/reperfusion injury. Sections were incubated with mouse anti-rat Ku70 monoclonal antibody (1:100) at 4°C overnight and then re-warmed to 37°C for 30 minutes. Enzyme solution and TUNEL solution were mixed (1:9) and goat anti-mouse Cy5 polyclonal IgG (1:400; Abcam) was added to the sections at 37°C for 1 hour. The enzyme/label solution was added to the sections at 37°C for 60 minutes. The nucleolus was stained by incubating sections with 4′,6 diamidino-2-phenylindole (1:40) for 5 minutes. After anti-fluorescence quenching mounting agent (Abcam) was added, the staining was observed using a laser confocal microscope (Zeiss LSM510 META, Zeiss, Frankfurt, Germany).
For double labeling of Ku70 and Bax, sections were simultaneously incubated with mouse anti-rat Ku70 monoclonal antibody (1:100; Abcam) and rabbit anti-rat Bax monoclonal antibody (1:100; Santa Cruz Biotechnology) at 4°C overnight. Sections were then re-warmed for 30 minutes at 37°C. Goat anti-mouse fluorescein isothiocyanate secondary antibody (1:200; Santa Cruz Biotechnology) and goat anti-rabbit IgG/red fluorescein Rhodamine (1:200; Santa Cruz Biotechnology) was added to the sections at 37°C for 60 minutes. The nucleolus was stained with DAPI (1:40; Santa Cruz Biotechnology) for 5 minutes. After anti-fluorescence quenching mounting agent was added, the staining was observed using a confocal microscope (Zeiss). Five randomly selected microscopic fields were captured using a 400 × light microscope (Olympus). Images were collected using a confocal microscope and the numbers of double-labeled cells were counted.
Western blot analysis
Total protein was isolated from the 2-mm cortical tissue using radio-immunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts (20 µg) of protein were loaded and separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, then transferred to immobilon polyvinylidenefluoride membranes (Millipore Corp, Plymouth, MA, USA). Membranes were blocked with 5% non-fat milk in PBS with 0.05% Tween 20 for 1 hour and then incubated overnight on ice with mouse anti-rat Ku70 monoclonal antibody (1:200; Abcam) or mouse anti-rat Bax monoclonal antibody (1:1,000; Santa Cruz Biotechnology). Mouse anti-rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal antibody (1:1,000; Beyotime Institute of Biotechnology) served as the house-keeping protein. Membranes were then washed and incubated with anti-mouse antibody (1:10,000; KPL, Los Angeles, USA) for 1 hour. Immunoreactive bands were visualized using the LICOR Odyssey infrared imaging system (LI-COR, Lincoln, USA) according to the manufacturer's protocol. Densitometric analysis of bands was performed using LICOR Odyssey software (LI-COR). Ku70 and Bax band absorbance intensity was normalized to GAPDH.
Statistical analysis
Data are expressed as mean ± SD and were analyzed by the one-way analysis of variance followed by the Student-Newman-Keuls post hoc test. Statistical analysis was performed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Differences were considered significant at P < 0.05.
Acknowledgments:
We greatly appreciate the colleagues from the Neuropharmacology Laboratory of Anhui Medical University and Experimental Center of Life Sciences, University of Science and Technology of China for their help in this study.
Footnotes
Funding: This study was financially supported by the National Natural Science Foundation of China, No. 30860291.
Conflicts of interest: None declared.
Peer review: The present study showed that, Ku70 expression at different time points after electrical stimulation in the rats with cerebral ischemia/reperfusion injury was increased compared with model group, but the number of apoptotic cells was reduced. No co-localization of Ku70 with TUNEL was found. Our findings indicate that Ku70 is a protective factor against fastigial nucleus stimulation in the brain of cerebral ischemia/reperfusion rats, and the protective effect may be related to anti-apoptosis activity of Ku70. However, the specific anti-apoptotic pathway is still unknown. Interestingly we found that anti-apoptotic effects of Ku70 after fastigial nucleus stimulation is closely linked to the co-localization with Bax in the cytoplasm, where Ku70 may inhibit Bax-mediated mitochondrial apoptosis pathway by binding to Bax.
Copyedited by Mark F, Norman C, He YS, Xu YS, Yu J, Yang Y, Li CH, Song LP, Zhao M
References
- [1].Reis DJ, Golanov EV, Galea E, et al. Central neurogenic neuroprotection: central neural systems that protect the brain from hypoxia and ischemia. Ann N Y Acad Sci. 1997;835:168–186. doi: 10.1111/j.1749-6632.1997.tb48628.x. [DOI] [PubMed] [Google Scholar]
- [2].Golanov EV, Liu F, Reis DJ. Stimulation of cerebellum protects hippocampal neurons from global ischemia. Neuroreport. 1998;9(5):819–824. doi: 10.1097/00001756-199803300-00010. [DOI] [PubMed] [Google Scholar]
- [3].Reis DJ, Kobylarz K, Yamamoto S, et al. Brief electrical stimulation of cerebellar fastigial nucleus conditions long-lasting salvage from focal cerebral ischemia: conditioned central neurogenic neuroprotection. Brain Res. 1998;780(1):159–163. [PubMed] [Google Scholar]
- [4].Glickstein SB, Golanov EV, Reis DJ. Intrinsic neurons of fastigial nucleus mediate neurogenic neuroprotection against excitotoxic and ischemic neuronal injury in rat. J Neurosci. 1999;19(10):4142–4154. doi: 10.1523/JNEUROSCI.19-10-04142.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou P, Qian L, Glickstein SB, et al. Electrical stimulation of cerebellar fastigial nucleus protects rat brain, in vitro, from staurosporine-induced apoptosis. J Neurochem. 2001;79(2):328–338. doi: 10.1046/j.1471-4159.2001.00585.x. [DOI] [PubMed] [Google Scholar]
- [6].Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348(14):1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- [7].Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14(4):469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- [9].Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- [10].Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90(6):1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- [11].Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- [12].Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15(6):691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- [13].Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- [14].Ott M, Norberg E, Zhivotovsky B, et al. Mitochondrial targeting of tBid/Bax: a role for the TOM complex? Cell Death Differ. 2009;16(8):1075–1082. doi: 10.1038/cdd.2009.61. [DOI] [PubMed] [Google Scholar]
- [15].Plesnila N. Role of mitochondrial proteins for neuronal cell death after focal cerebral ischemia. Acta Neurochir Suppl. 2004;89:15–19. doi: 10.1007/978-3-7091-0603-7_3. [DOI] [PubMed] [Google Scholar]
- [16].Robertson CL, Scafidi S, McKenna MC, et al. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218(2):371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hetz C, Vitte PA, Bombrun A, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280(52):42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- [18].Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5(5):367–378. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- [19].Sugawara T, Noshita N, Lewén A, et al. Neuronal expression of the DNA repair protein Ku 70 after ischemic preconditioning corresponds to tolerance to global cerebral ischemia. Stroke. 2001;32(10):2388–2393. doi: 10.1161/hs1001.097109. [DOI] [PubMed] [Google Scholar]
- [20].Kim GW, Noshita N, Sugawara T, et al. Early decrease in DNA repair proteins, Ku70 and Ku86, and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 2001;32(6):1401–1407. doi: 10.1161/01.str.32.6.1401. [DOI] [PubMed] [Google Scholar]
- [21].Chechlacz M, Vemuri MC, Naegele JR. Role of DNA-dependent protein kinase in neuronal survival. J Neurochem. 2001;78(1):141–154. doi: 10.1046/j.1471-4159.2001.00380.x. [DOI] [PubMed] [Google Scholar]
- [22].Fewell JW, Kuff EL. Intracellular redistribution of Ku immunoreactivity in response to cell-cell contact and growth modulating components in the medium. J Cell Sci. 1996;109(Pt 7):1937–1946. doi: 10.1242/jcs.109.7.1937. [DOI] [PubMed] [Google Scholar]
- [23].Bakalkin G, Yakovleva T, Hurd YL, et al. Autoantigen Ku in the brain. Developmentally regulated expression and subcellular localization. Neuroreport. 1998;9(9):2147–2151. doi: 10.1097/00001756-199806220-00044. [DOI] [PubMed] [Google Scholar]
- [24].Rathaus M, Lerrer B, Cohen HY. DeubiKuitylation: a novel DUB enzymatic activity for the DNA repair protein, Ku70. Cell Cycle. 2009;8(12):1843–1852. doi: 10.4161/cc.8.12.8864. [DOI] [PubMed] [Google Scholar]
- [25].Zou H, Volonte D, Galbiati F. Interaction of caveolin-1 with Ku70 inhibits Bax-mediated apoptosis. PLoS One. 2012;7(6):e39379. doi: 10.1371/journal.pone.0039379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sawada M, Sun W, Hayes P, et al. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5(4):320–329. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- [27].Liu JL, Dong WW, Li JP. Effect of fastigial nucleus electrical stimulation on Ku expression in rat with focal cerebral ischemia and reperfusion. Zhongguo Linchuang Shenjing Kexue. 2003;11(2):117–120. [Google Scholar]
- [28].Gomez JA, Gama V, Yoshida T, et al. Bax-inhibiting peptides derived from Ku70 and cell-penetrating pentapeptides. Biochem Soc Trans. 2007;35(Pt 4):797–801. doi: 10.1042/BST0350797. [DOI] [PubMed] [Google Scholar]
- [29].Amsel AD, Rathaus M, Kronman N, et al. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc Natl Acad Sci U S A. 2008;105(13):5117–5122. doi: 10.1073/pnas.0706700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Han B, Wang Q, Cui G, et al. Post-treatment of Bax-inhibiting peptide reduces neuronal death and behavioral deficits following global cerebral ischemia. Neurochem Int. 2011;58(2):224–233. doi: 10.1016/j.neuint.2010.12.008. [DOI] [PubMed] [Google Scholar]
- [31].Iriyama T, Kamei Y, Kozuma S, et al. Bax-inhibiting peptide protects glutamate-induced cerebellar granule cell death by blocking Bax translocation. Neurosci Lett. 2009;451(1):11–15. doi: 10.1016/j.neulet.2008.12.021. [DOI] [PubMed] [Google Scholar]
- [32].Mattson MP, Kroemer G. Mitochondria in cell death: novel targets for neuroprotection and cardioprotection. Trends Mol Med. 2003;9(5):196–205. doi: 10.1016/s1471-4914(03)00046-7. [DOI] [PubMed] [Google Scholar]
- [33].Mishra NC, Kumar S. Apoptosis: a mitochondrial perspective on cell death. Indian J Exp Biol. 2005;43(1):25–34. [PubMed] [Google Scholar]
- [34].Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- [35].Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun. 2003;304(3):437–444. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- [36].Trougakos IP, Lourda M, Antonelou MH, et al. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin Cancer Res. 2009;15(1):48–59. doi: 10.1158/1078-0432.CCR-08-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cohen HY, Lavu S, Bitterman KJ, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell. 2004;13(5):627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- [38].Gama V, Yoshida T, Gomez JA, et al. Involvement of the ubiquitin pathway in decreasing Ku70 levels in response to drug-induced apoptosis. Exp Cell Res. 2006;312(4):488–499. doi: 10.1016/j.yexcr.2005.11.016. [DOI] [PubMed] [Google Scholar]
- [39].Gama V, Gomez JA, Mayo LD, et al. Hdm2 is a ubiquitin ligase of Ku70-Akt promotes cell survival by inhibiting Hdm2-dependent Ku70 destabilization. Cell Death Differ. 2009;16(5):758–769. doi: 10.1038/cdd.2009.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vishnudas VK, Miller JB. Ku70 regulates Bax-mediated pathogenesis in laminin-alpha2-deficient human muscle cells and mouse models of congenital muscular dystrophy. Hum Mol Genet. 2009;18(23):4467–4477. doi: 10.1093/hmg/ddp399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Subramanian C, Jarzembowski JA, Opipari AW, Jr, et al. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia. 2011;13(8):726–734. doi: 10.1593/neo.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lee SJ, Jang H, Park C. Maspin increases Ku70 acetylation and Bax-mediated cell death in cancer cells. Int J Mol Med. 2012;29(2):225–230. doi: 10.3892/ijmm.2011.833. [DOI] [PubMed] [Google Scholar]
- [43].Liu J, Naegele JR, Lin SL. The DNA-PK catalytic subunit regulates Bax-mediated excitotoxic cell death by Ku70 phosphorylation. Brain Res. 2009;1296:164–175. doi: 10.1016/j.brainres.2009.07.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li JJ, Gu QH, Li M, et al. Role of Ku70 and Bax in epigallocatechin-3-gallate-induced apoptosis of A549 cells in vivo. Oncol Lett. 2013;5(1):101–106. doi: 10.3892/ol.2012.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Subramanian C, Hada M, Opipari AW, Jr, et al. CREB-binding protein regulates Ku70 acetylation in response to ionization radiation in neuroblastoma. Mol Cancer Res. 2013;11(2):173–181. doi: 10.1158/1541-7786.MCR-12-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meng S, Lin L, Lama S, et al. Cerebral expression of DNA repair protein, Ku70, and its association with cell proliferation following cerebral hypoxia-ischemia in neonatal rats. Int J Dev Neurosci. 2009;27(2):129–134. doi: 10.1016/j.ijdevneu.2008.12.002. [DOI] [PubMed] [Google Scholar]
- [47].Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol. 2003;5(4):352–357. doi: 10.1038/ncb955. [DOI] [PubMed] [Google Scholar]
- [48].Zhou P, Qian L, Zhou T, et al. Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. J Neurochem. 2005;95(1):221–229. doi: 10.1111/j.1471-4159.2005.03358.x. [DOI] [PubMed] [Google Scholar]
- [49].Paxinos G, Watson C. 5th ed. London: Academic Press; 2005. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- [50].Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- [51].Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke. 2004;35(1):179–184. doi: 10.1161/01.STR.0000106479.53235.3E. [DOI] [PubMed] [Google Scholar]