
Figure 7.
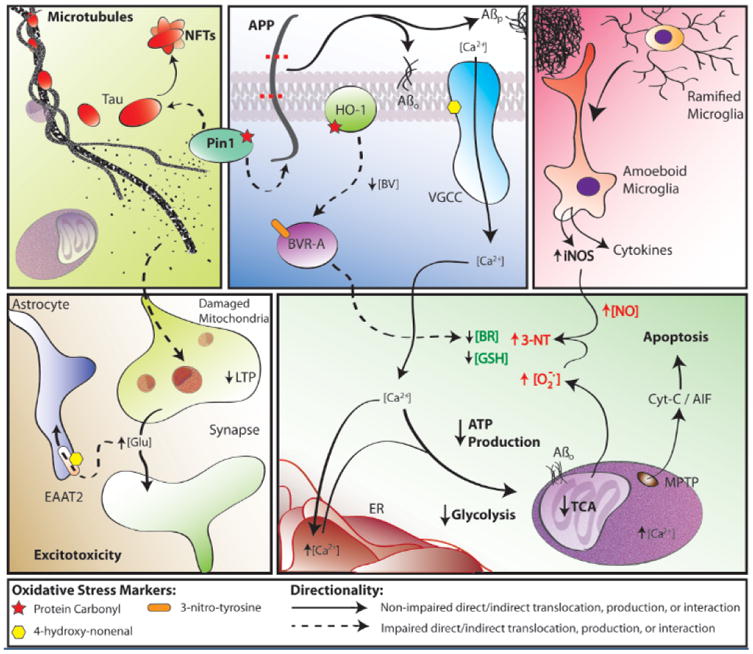
Some findings and discoveries related to Aβ(1-42) oligomer-induced oxidative stress and its sequela published from the Butterfield laboratory that contribute to a unifying oxidative stress centric hypothesis for neuronal death in AD that is consistent with the pathology, biochemical alterations, and clinical presentation in this disorder. Amyloid β-peptide, produced by β- and γ-secretase (top, middle panel), aggregates into extracellular fibrils forming the core of SP or aggregates into hydrophobic oligomers that insert into the plasma membrane. One-electron oxidation of the S-atom of Met-35 of Aβ(1-42) [see text] initates the chain reaction of lipid peroxidation (LPO), greatly amplyfing the damage of the initial free radical on the peptide. HNE, produced by LPO, binds to and causes dysfunction of key proteins in the PM and cytosol. Among the former are ion-motive ATPases, e.g., Na,K-ATPase and Ca-ATPase. The resultant loss of cell potential opens voltage gated Ca2+ channels, leading to a massive influx of Ca2+ to the cytosol (bottom, right panel), and subsequent attempts to sequester this Ca2+ in ER and mitochondria. However, this massive overload of Ca2+ causes ER to undergo unfolded protein response and damage and decreases protein synthesis. Mitochondrial Ca2+ overload causes swelling of mitochondria and opening of the MPTP with release of cytochrome c to initiate the intrinsic pathway for apoptosis (bottom, right panel). The intracellular Ca2+ also activates numerous degradative enzymes such as calpains, PLA2, endonucleases, etc. causing necrotic mechanisms to engage (see text). The oxidative stress in cytosol and mitochondria lead to damage to many glycolytic, TCA, and ETC enzymes or complexes, resulting in dramatic loss of ATP production (bottom, right panel), which leads to loss of many important neuronal functions, ranging from axonal transport to neurotransmission (bottom, left panel), as well as maintaining the cellular and organelle potentials. This oxidative stress also damages Pin1, which regulates both APP and Tau (top, left/middle panels), but also a key tau kinase (GSK-3β) and a tau phosphatase (PP2A). The resultant hyperphosphorylated tau, falls off microtubules (MT) [top, left panel], leading to cessation of anterograde and retrograde transport. Among other determimental consequences of this, synaptic-resident mitochondria are no longer able to produce the required ATP to maintain presynaptic function, including loss of LTP, needed for learning and memory (bottom, left panel). Note that Pin1 is involved in three major neuropathological hallmarks of AD: SP, NFT, and synapse loss. The intracellular detritus resulting from all these aberrant processes is not removed, since the proteostasis network of the ubiquitin-proteasome system (UPS), ER, and autophagy are all damaged by oxidative stress (see text). The Aβ(1-42)-initiated oxidative stress also leads to damage to heme oxygenase -1 (HO-1) and biliverdin reductase-A (BVR-A), which decreases intracellular antioxidant bilirubin (top, middle panel; bottom, right panel). Moreover, glutathione (GSH) is decreased because of elevated oxidative stress and since its rate limiting synthetic enzyme, γ-Glu-Cys ligase, is damaged by oxidative stress. There are many other processes associated with oxidative stress on which we and others have published. However, this schematic diagram outlines some of the key processes involved in neuronal death in AD that fit the pathology, biochemical alterations, and clinical presentations observed in AD that were published from our laboratory based on neurochemical, oxidative stress, and proteomics studies of AD and its earlier forms.