

Abstract
AIM: To construct a hepatitis B virus (HBV)-based vector with a reporter gene and to establish an HBV infection system to evaluate the availability of the vector.
METHODS: The HBV-based vectors with green fluorescence protein (GFP) were packaged into the liver of immunodeficient mice through transfer and helper plasmid using hydrodynamic technology. Wild type HBV (wt HBV) was provided by plasmid MC2009. Primary human hepatocytes (PHH) were isolated and infected with recombinant HBV (rHBV) or wt HBV. GFP expression was monitored by confocal and flow cytometry. HBV DNA and HBV surface antigen (HBSAg) were analyzed by PCR and ELISA.
RESULTS: 3 × 107 wt HBV copies/mL and 5 × 106 rHBV copies/mL were collected from mice serum. In the wt HBV infected group, HBV progeny was 2 × 107 copies/mL and HBSAg was 770 ng/mL. In the rHBV infected group, GFP fluorescence was detected on d 3 post-infection and over 85% of the parenchymal cells expressed green fluorescence on d 12 post-infection. Compared with wt HBV in the PHH infection system, no rHBV DNA or HBSAg were detected in PHH culture media.
CONCLUSION: An effective HBV based vector was developed, which proved to be a useful HBV infection system. This vector and infection system can be applied to develop a therapeutic vector and study the HBV life cycle and viral pathogenesis.
Keywords: Hepatitis B virus, Primary human hepatocyte, Transfer plasmid, Helper plasmid, Infection system
INTRODUCTION
The hepatitis B virus (HBV) belongs to the family Hepadnaviridae and is the smallest DNA virus known[1].The HBV genome consists of a partially double-stranded, relaxed circular DNA, which has a compact organization. It employs widely overlapping open reading frames and regulatory sequences[2].
Despite the availability of a safe and efficient HBV vaccine, HBV is still a major cause of infectious liver disease throughout the world. While the majority of acutely infected adults recover from this disease, chronic HBV infections remains a major public health problem, affecting more than 350 million people worldwide[3]. Systemic administration of interferon has been the best available therapy for chronic HBV infections for the past 2 decades; however, a sustained response is achieved in only one-third of affected patients[4]. Currently, nucleoside analogues, such as lamivudine and adefovir, are new treatment options that result in a rapid decrease in serum HBV DNA, as well as an improvement of liver histology. Unfortunately, these agents are associated with a rapid rate of relapse and selection of resistant viral variants[5,6].
As such, novel therapeutic alternatives are continually being explored. One of the more attractive therapeutic approaches involves the development of methods that permit the selective delivery of genes into parenchymal cells of the liver. While hydrodynamic injection can result in a high level of transgene expression in the liver, the combined effect of a large injection volume and high injection speed can result in irregular hearty function[7]. Further, few of the established viral vectors can specifically target hepatocytes[8]. Because HBV is capable of specifically targeting hepatocytes after inoculation into the bloodstream and efficiently infects quiescent hepatocytes, human HBV as liver-directed gene transfer has been pursued for more than one decade. Recently, the ability of recombinant HBV (rHBV) and duck HBV to serve as vectors for a hepatocyte-specific gene transfer has been demonstrated[9,10].
An appropriate and effective in vitro infection system that permits evaluation of antiviral drugs and the study of the kinetics of the hepadenaviral infection, such as replication and viral resistance, is very important yet unavailable to date. This stems from the inability to grow HBV in culture and the absence of a convenient and reliable in vitro system to efficiently study different mutations in the various stages of HBV replication and their influence on viral pathogenesis.
Hepadnaviruses infect only the well-differentiated primary hepatocytes of their specific hosts in cell culture[11]. Several cell modes based on HBV-related hepadnaviruses, such as Pekin duck and Tupaia primary hepatocytes, are presently available to assess antiviral drugs, and to provide information about factors involved in the establishment of HBV infection. Duck HBV (DHBV) is a well characterized model system of hepadnaviral infection, and cultures of primary duck hepatocytes (PDHs) can be readily established and efficiently infected[12]. There are, however, significant differences between duck and human hepatitis viruses. Most importantly perhaps is the fact that, human HBV envelope polypeptides, the likely mediators of entry, are N-glycosylated, whereas DHBV envelope polypeptides are not. Thus, the degree to which information from DHBV applies to human HBV infection may be limited[13].
Primary tupaia hepatocytes are susceptible to infection with serum-derived HBV. The HBV infection of primary tupaia hepatocytes develops slowly, and the virus life cycle is not the same as HBV in human hepatocytes[14,15]. Hepatoma cell lines such as Huh7 and HepG2.2.15, and the HepaRG cell line are known to be non-susceptible to HBV infection but permit viral replication after artificial import of the viral genome (e.g., transfection of cloned HBV DNA)[16-18]. Drawbacks to these transient transfections, inefficient transfection and thereby produce low virus yields that unavoidably vary from experiment to experiment, since only a fraction of the cells are transfected. This leaves a high background of non-producer cells, and virus expression ceases within a few days[19]. Although they are difficult to maintain in culture and become non-permissive for HBV very quickly after plating, primary human hepatocytes (PHH) are susceptible to infection with HBV[20].
In light of the above-described challenges and clinical importance of studying HBV, the purpose of this study was to establish an effective HBV infection system including standard virus origin, and to construct the HBV based vector with a reporter gene capable of targeting hepatocytes naturally with high expression without expressing viral DNA and protein. Ultimately, this study will lay the foundation for the construction of a therapeutic HBV vector and will permit the intensive investigation of the HBV life cycle and viral pathogenesis.
MATERIALS AND METHODS
Animals
NOD/SCID mice were purchased from the Animal center of Sun Yat-Sen University (GuangZhou, China). All animals were maintained under specific pathogen-free conditions on alternating 12 h light/dark cycles. Food and water were available ad libitum.
Isolation and culture of primary human hepatocytes (PHH)
PHH were isolated from fresh surgical specimens of patients undergoing partial hepatectomy, with the informed consent of each patient and the local ethics committee. In all cases, patients were negative for HBV, hepatitis C virus serologic markers and specific HBV-DNA sequences (determined by PCR). Healthy human liver tissue samples (3 cm × 3 cm × 2 cm) were placed on ice in Dulbecco's
modified Eagle's medium (DMEM) solution with 1% Gentamycin (Sigma). Immediately after collection, the liver tissue was treated by a two-step collagenase perfusion procedure[21]. Briefly, perfusion was initiated with the preperfusion solution of D-Hanks balanced salt solution (Sigma) containing 0.5 mmol/L ethylene-glycoltetraacetic acid (EGTA, Sigma) and 50 mmol/L HEPES heated to 37°C for 15 to 25 min at a flow rate of 20-40 mL/min, then, continued with perfusion solution of F12 medium-DMEM (Invitrogen) containing 0.05% collagenase type IV (Gibco) and 5 mmol/L CaCl2 for 15-25 min at a flow rate of 25-50 mL/min. After gentle mechanical dissociation, the PHH were washed and separated by three successive centrifugations at 50 × g, for 5 min each. The PHH were subsequently resuspended in DMEM medium and viability was assessed by trypan blue exclusion. After isolation, 1.0 × 105 viable cells/cm2 were seeded onto collagen-coated culture plates (collagen type I, sigma) in medium containing 8% fetal calf serum (FCS) and maintained at 37°C with 5% CO2. Cell culture media were supplemented with L-glutamine (5 mmol/L), HEPES (23 mmol/L, pH 7.4), gentamycin (50 mg/mL), penicillin (50 IU/mL, sigma), streptomycin (50 mg/mL, sigma), hydrocortisone (4.8 mg/mL, sigma) and insulin (1 mg/mL, sigma). Culture medium was changed at 3 h, then 12 to 16 h after seeding. Medium was changed every 24 to 48 h thereafter, but without the addition of serum[20].
Plasmid constructs
The DNA vector MC2009 contains 2 tandem copies of the HBV genome by head to tail connection (Figure 1B), This vector will produce wild-type (wt) HBV virus capable of infection after injection into mouse liver, or delivered to cultures of human hepatocytes. This vector can be used to generate serum with a high titer of HBV to serve as a standard for this project.
Figure 1.
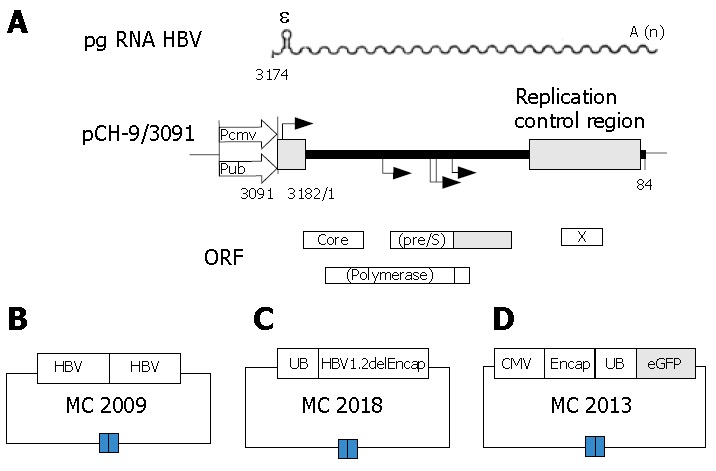
Schematic representation of plasmid constructs. A: Plasmid constructs used for the production of recombinant hepatitis B virus (rHBV)[10], (pgRNA HBV = sinusoidal line; ε = encapsidation signal; A (n) = poly A tail; CMV = cytomegalovirus; UB= ubiquitin); B: The vector MC2009 contains 2 tandem copies of HBV genome; C: The transfer plasmid DNA vector MC2013 obtained by replacing a DNA fragment containing the small envelope gene (HBV position 1446 to 2347) with a PCR fragment encoding a fluorescence-enhanced green fluorescence protein (eGFP), meanwhile, a DNA fragment encompassing the HBV preS2/S promoter was substituted by a PCR fragment encoding UB promoter which could drive transgene expression. A CMV promoter was introduced to drive the expression of the UB.eGFP reporter expression cassette with the HBV encapsidation signal, and premature stop codons were introduced into all remaining HBV open reading frames; D: The helper plasmid MC2018 contains only one copy of the HBV genome with a deletion of the encapsidation signal ε or direct repeats necessary for reverse transcription. The UB promoter was introduced to drive expression of HBV 1.2delEncap.
The DNA vector MC2018, the helper plasmid (Figure 1C), is capable of expressing all HBV proteins, including the large, middle, and small envelope proteins, the core protein, and the polymerase. The DNA sequence is the same as that in MC2009, except that it contains only one copy of the HBV genome with a deletion of the encapsidation signal ε and direct repeats necessary for reverse transcription and be derived by an ubiquitin promoter (UB). Thus, the DNA vector MC2018 can only provide HBV proteins needed for packaging in trans, but is not able to generate a complete HBV virus[22].
DNA vector MC2013, the transfer plasmid (Figure 1D), was obtained by replacing a DNA fragment containing the small envelope gene (HBV position 1446 to 2347) with a PCR fragment encoding a fluorescence-enhanced green fluorescence protein (eGFP)[9]. The DNA fragment encompassing the HBV preS2/S promoter was substituted by a PCR fragment encoding a ubiquitin promoter to drive transgene expression[23]. A cytomegalovirus (CMV) promoter was introduced to drive the expression of UB, eGFP reporter expression cassette with the HBV encapsidation signal and premature stop codons were introduced into all remaining HBV open reading frames[10].
Generation of HBV virions
Five μg of plasmid MC2009 was dissolved in a volume of saline equivalent to 8% of the body mass of the target mouse (i.e. 1.6 mL for a 20 g mouse) to obtain a working solution. It was then injected into the tail vein of 6- to 9-wk-old NOD/SCID mice within 5-8 s[24]. After 1 d, blood (and ultimately serum) was collected from the mice by cutting the mouse tail. The HBV DNA copies in the mouse serum were determined by quantitative PCR assay, Mouse serum containing wild-type HBV virus was maintained at -20°C until use.
Preparation of recombinant HBV virions
Five μg of plasmids MC2013 and MC2018 (each) was combined and diluted with saline to a volume equivalent to 8% of the body mass of the mouse. This was then injected into the tail vein of 6- to 9-wk-old NOD/SCID mice within 5-8 s[24], The number of recombinant HBV (rHBV) DNA copes was determined by quantitative PCR. The rHBV was then purified from the mouse blood and measured by a quantitative PCR assay on d 3 to 6 after injection, and stored until use.
Infection of PHH with HBV and rHBV
On d 2 post-seeding, PHH cultures were incubated with wt HBV or rHBV. After infecting cultures overnight, the PHH cells were washed three times, every third day, and the culture medium was exchanged and collected for virus product analysis to test the HBV infection of PHH in vitro, To detect the susceptibility of PHH to HBV, PHH cultures were treated on d 12 post-seeding (using the same method).
Detection of transgene expression
GFP expression in PHH incubated with rHBV or wt HBV were detected by confocal microscopy (Leica, Germany) using a standard fluorescein isothiocyanate (FITC) filter set with excitation by blue light (488 nm) on d 3, 6 and 12 post-infection.
Evaluation of the efficiency of transgene expression
To determine the efficiency of transgene delivery, PHH cultures were analyzed by flow cytometry (BD FACSAriaTM US) on d 12 after seeding. Liver parenchymal cells with transgene expression were identified by GFP, Liver sinusoidal endothelial cells were identified by acetylated low-density lipoprotein (Alexa Fluor®594 AcLDL, Invitrogen), and kupffer cells were identified by Dextran conjugates (Carboxy-Q-Rhodamine, Invitrogen).
Analysis of viral DNA and proteins
HBV DNA in the culture medium was quantified using a quantitative PCR assay (HBV-DNA detection kit, Da'an company, China) according to the manufacturer's in-structions. HBsAg in the supernatant of wt HBV-infected or rHBV-infected PHH was determined using an ELISA kit and the AxSYM system (Abbott Diagnostics, Chicago, USA) according to the manufacturer's instructions.
RESULTS
Isolation and culture of PHH
PHH were isolated from fresh surgical specimens of liver tissue. Mean cell yield was 5 × 105 cells/g liver tissue with 80% mean viability. Morphology of typical PHH cultures on d 30 post-seeding is shown in Figure 2A. PHH growth was inhibited following the addition of DMSO, but continued to grow when the DMSO was withdraw (data not shown).
Figure 2.
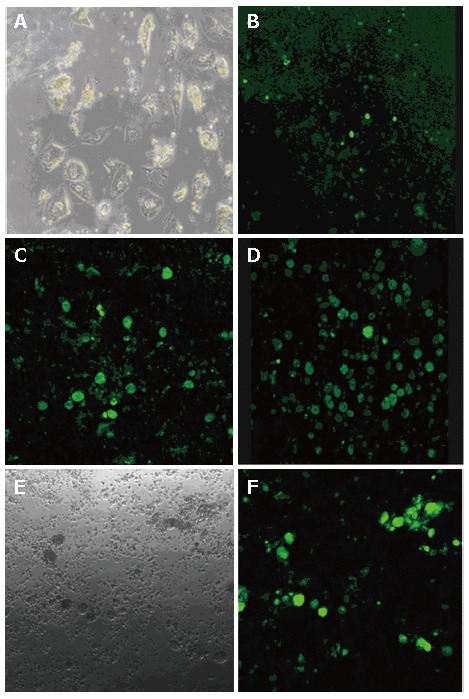
Transduction of primary human hepatocytes (PHH) by the HBV-based vector. PHH were infected with rHBV-GFP, a recombinant HBV that carries a GFP gene under control of the UB promoter (described previously in Figure 1). A: Cell morphology of PHH on d 30; B: GFP expression on d 3 post-infection; C: GFP expression on d 6 post-infection; D: on d 12 post-infection; E: Transduction of PHH demonstrated by an overlay of the fluorescence; F: Phase contrast of the same field.
Production of Recombinant HBV and Wild type HBV
MC2009 DNA vector was delivered to NOD/SCID mice using the hydrodynamic technique. On d 1 through 3 post-injection, no HBV DNA could be detected, whereas after d 3 post-injection, HBV DNA levels were 3 × 107 copies/mL.
MC2018 and MC2013 were co-injected into NOD/SCID mouse liver using hydrodynamic technology. It was subsequently determined that rHBV DNA could be detected only on d 3 to d 6 post-injection. The average amount of rHBV DNA detected was 5 × 106 copies/mL.
Gene transfer in PHH by rHBV vector
Infectivity of the rHBV particles was demonstrated by incubating primary human hepatocytes with rHBV at a multiplicity of infection (moi) of 10 copies/cell for 24 h on d 2 after plating. GFP fluorescence was clearly detected on d 3 post-infection (Figure 2B), and cells expressing GFP reached its maximum on d 12 post-infection (Figure 2D). This finding indicates that the HBV-based vector that was generated can target liver cells and deliver the genetic information in a 'natural' way.
The efficiency of transgene expression
In PHH cultures, 75.2% of the cells were GFP positive (Figure 3B), 2% of the cells were identified as Kupffer cells (Figure 3C), and 10.4% of the cells were liver sinusoidal endothelial cells (Figure 3D). In addition, cultures contained small amounts of red cells and debris. Thus, nonparenchymal liver cells, which cannot be infected by HBV, accounted for only 12.4% of the total number of cells up to d 12 after seeding. Of the 75.2% GFP-expressing cells, over 85% of parenchymal liver cells were infected by the rHBV, and stable GFP expression was observed for up to 4 wk. These results strongly suggest that the HBV-based vector has high infectivity to PHH and a high efficiency of transgene expression.
Figure 3.
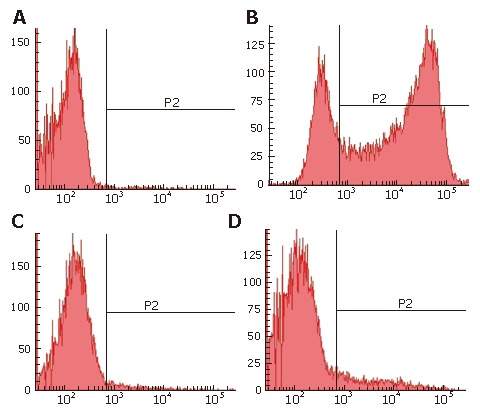
PHH were detected by FACS. The purity of PHH cultures was analyzed by identification of nonparenchymal and parenchymal liver cells on d 6 after seeding. A: Control; B: FACS analysis of parenchymal liver cells with GFP; C: FACS analysis of kupffer cells with Dextran conjugates; D: FACS analysis of liver sinusoidal endothelial cells (LSEC) with acetylated low-density lipoprotein. The population consisted of 10.4% AcLDL positive cells, 2% Carboxy-Q-Rhodamine positive cells and 75.2% GFP positive cells, meaning that total parenchymal cells is about 87.6%, approximately 85% of it were infected by HBV based vector with GFP.
Analysis the kinetics of HBV or rHBV infection in PHH
PHH cultures were incubated with mouse serum containing wt HBV or rHBV at a moi of 10 copies/cell on d 2 after plating. The cell culture media was collected every 3 d post-infection. In the wt HBV infected group, wt HBV DNA copies were 1 × 103 to 2 × 107 copies/mL (Figure 4A) and HBV surface antigen (HBSAg) was 175 to 770 ng/mL (Figure 4B). In contrast, rHBV DNA copies were 1 × 101 to 0 (Figure 4A) and HBSAg was undetectable at all times (Figure 4B). Collectively, these results suggest that the PHH system can be infected by wt HBV (with a time constraint), and that the rHBV vector cannot express HBV products in PHH.
Figure 4.
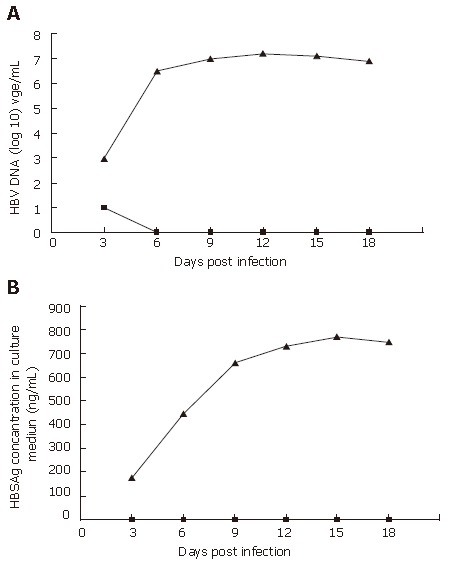
The efficiency of PHH infected with wt HBV or rHBV. PHH were infected at a multiplicity of infection of 10 DNA-containing HBV particles/cell on d 1 post-seeding, HBV DNA (A), and HBSAg (B) secreted into cell culture medium were determined every 3 d after infection. The triangles denote PHH cells infected with wt HBV, and squares denote PHH cells infected with rHBV. In the supernatant from PHH infected with rHBV, no HBV DNA and HBsAg can be detected, meaning that all viral gene were knocked out.
DISCUSSION
Although PHH are difficult to maintain in culture for HBV infection, PHH are undoubtedly the ideal tool for studying HBV infection and hepatotropic vectors. In the present report, it was demonstrated that: (1) PHH cultures, maintained under optimized PHH culture conditions, can serve as a useful infection system for HBV in vitro; (2) HBV from MC2009 are useful for HBV infection studies; and (3) the constructed HBV-based vectors described herein is highly efficient and safe.
The fundamental prerequisites for the viral life-cycle include the ability to replicate the genome, and to produce infectious progeny virions. At present, the major limitation in studying HBV entry is the lack of an in vitro infection system capable of supporting the entire life cycle of HBV. Several systems derived from hepatoma cells have previously been used to study HBV infections in vitro[16-18]. Unfortunately, none of the described systems have proven ideal. HPP cultures derived from liver explants have shown to be susceptible to HBV infection, but infection occurs in only 10% to 20% of the cells[25]. Further, infection of PHH cultures is only for a limited amount of time following explantation[11,13].
In this study, PHH were isolated from small pieces of healthy liver tissue from patients and maintained under optimized culture conditions. The PHH cultures survived up to 10 wk. wt HBV DNA reached the level of 2 × 107 copies/mL, and HBSAg reached 770 ng/mL in PHH cultures infected with wt HBV. Over 85% of parenchymal liver cells were infected by rHBV. In previous PHH systems and some hepatoma cell systems[18,20], high dose DMSO (1.5%-2%) was used to enhance HBV infection in vitro. In the present study, it was found that growth of PHH cultures was seriously inhibited with addition of DMSO, but continued to grow once the DMSO was withdrawn. In addition, cell survival in the culture was reduced by the addition of complement inactivated serum. In our PHH system, cells lost their susceptibility to HBV on d 12 after seeding. This suggests that the PHH system has a time constraint and is consistent with prior reports[11].
In previously described HBV infection systems[22,26], HBV virion was primarily prepared from the serum of chronic HBV patients, because this serum always carried variant virus, HBV proteins and cccDNA[27]. Since these products could falsify study results, the serum of chronic HBV patients was unsuitable for research on the HBV life cycle. The construct MC2009 could solve this problem by providing standard wild type HBV without accessory viral products.
The goal of this study was to generate hepatocyte-specific viral vectors, which only expressed the transgene at a high level of expression. In addition, we wished to completely eliminate HBV viral gene expression, and to insert foreign promoter and functional transgenes of at least 800 base pairs. In the transfer plasmid MC2013, all viral genes were knocked out and endogenous HBV preS2/S promoter was substituted by a UB promoter to drive reporter gene eGFP expression. The expression of UB. eGFP reporter expression cassette with the HBV encapsidation signal was driven by a CMV promoter. This helper plasmid can provide HBV proteins needed for packaging in trans so that the mRNA is packed into a recombinant HBV virion.
In PHH incubated with this rHBV virion, nearly 85% of parenchyma cells expressed GFP on d 12 post-infection. The data from this study demonstrated that the HBV based vector can specifically infect liver cells, and provides direct evidence that gene transfer into PHH by a HBV vector is possible. The data presented here go much beyond earlier studies reporting the efficacy of transgene expression: the use of the UB promoter leads to higher GFP expression than the original preS2/S-and CMV-promoters[9,10]. In the vector created in this study, a greater than 800 bp eGFP gene was inserted, This gene can potentially be replaced with useful effector genes coding, for example, specific antisense-RNAs or RNAi, or a large number of immunomodulatory cytokines. This may result in novel means of treating HBV infections in the future. Given the high expression efficacy and larger inserts, this study lays the foundation for construction of a therapeutic HBV vector.
If rHBV is to be used as a gene transfer vector, the presence of wt HBV by homologous recombination is a major safety issue. In the HBV system created here, safety did not appear to be of concern: no HBSAg was detected, only 1 × 101 HBV DNA were detected on d 3 post-infection, and no HBV DNA was detected on or after d 6 post-infection. The identification of 1 × 101 HBV DNA on d 3 post-infection might simply be residual from the sample that was added. In the HBV system, homologous recombination was possible either between transfer and helper plasmids or in between the redundant HBV sequences of the helper plasmid[10,22]. To avoid this, all viral ORFs were knocked out in the transfer, and the terminal redundant sequences were deleted to minimize the possibility of transfer-helper recombination and to abolish helper-helper recombination.
Despite the fact that the early steps of HBV infection determine the virus-related pathogenesis, the molecular basis of the steps remains poorly understood. Infection begins with cell attachment, followed by entry and delivery of the viral genetic information to the host cell's nucleus[13]. In the absence of ideal cell and/or animal models for studying HBV infection, data concerning the early, post-attachment steps in hepadnaviral entry are largely based on studies performed with DHBV in primary duck liver hepatocytes[28]. In the infection system developed by this research group, PHH can be infected naturally by HBV, and the HBV-based vector with reporter gene GFP can also easily enter the cell. As a result, this infection system may provide a novel means of elucidating the molecular basis of hepadnavirus infection.
In conclusion, we have established an HBV infection system using PHH cultures, and constructed a HBV-based vector with reporter gene. This HBV-based vector and HBV infection system may lay the foundation for developing a therapeutic vector and an effective tool for studying HBV life cycle and viral pathogenesis.
ACKNOWLEDGMENTS
We are grateful to Professor Zhi-Yin Chen (Departments of Pediatrics and Genetics, Stanford University School of Medicine, Stanford, California, USA) for his excellent technical support. We would also like to thank Bao-Hua Hou and Xuan Huang for their co-operation in obtaining human liver tissue samples.
Footnotes
Supported by the grants from the National Science Foundation of China, No. 30271177 and No. 39870676, and the Natural Science Foundation of Guangdong Province, No. 021903
S- Editor Liu Y L- Editor Alpini GD E- Editor Che YB
References
- 1.Tiollais P, Pourcel C, Dejean A. The hepatitis B virus. Nature. 1985;317:489–495. doi: 10.1038/317489a0. [DOI] [PubMed] [Google Scholar]
- 2.Büscher M, Reiser W, Will H, Schaller H. Transcripts and the putative RNA pregenome of duck hepatitis B virus: implications for reverse transcription. Cell. 1985;40:717–724. doi: 10.1016/0092-8674(85)90220-x. [DOI] [PubMed] [Google Scholar]
- 3.Lok AS. Chronic hepatitis B. N Engl J Med. 2002;346:1682–1683. doi: 10.1056/NEJM200205303462202. [DOI] [PubMed] [Google Scholar]
- 4.Tillmann HL. Antiviral therapy and resistance with hepatitis B virus infection. World J Gastroenterol. 2007;13:125–140. doi: 10.3748/wjg.v13.i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcellin P. Advances in therapy for chronic hepatitis B. Semin Liver Dis. 2002;22 Suppl 1:33–36. doi: 10.1055/s-2002-35698. [DOI] [PubMed] [Google Scholar]
- 6.Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, et al. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;354:1807–1812. doi: 10.1056/NEJMoa051214. [DOI] [PubMed] [Google Scholar]
- 7.Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, Dean DA, Liu D. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004;11:675–682. doi: 10.1038/sj.gt.3302210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nash KL, Jamil B, Maguire AJ, Alexander GJ, Lever AM. Hepatocyte-specific gene expression from integrated lentiviral vectors. J Gene Med. 2004;6:974–983. doi: 10.1002/jgm.591. [DOI] [PubMed] [Google Scholar]
- 9.Protzer U, Nassal M, Chiang PW, Kirschfink M, Schaller H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc Natl Acad Sci USA. 1999;96:10818–10823. doi: 10.1073/pnas.96.19.10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Untergasser A, Protzer U. Hepatitis B virus-based vectors allow the elimination of viral gene expression and the insertion of foreign promoters. Hum Gene Ther. 2004;15:203–210. doi: 10.1089/104303404772680001. [DOI] [PubMed] [Google Scholar]
- 11.Gripon P, Diot C, Guguen-Guillouzo C. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology. 1993;192:534–540. doi: 10.1006/viro.1993.1069. [DOI] [PubMed] [Google Scholar]
- 12.Tuttleman JS, Pugh JC, Summers JW. In vitro experimental infection of primary duck hepatocyte cultures with duck hepatitis B virus. J Virol. 1986;58:17–25. doi: 10.1128/jvi.58.1.17-25.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu X, Block T. Study of the early steps of the Hepatitis B Virus life cycle. Int J Med Sci. 2004;1:21–33. doi: 10.7150/ijms.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luscombe C, Pedersen J, Uren E, Locarnini S. Long-term ganciclovir chemotherapy for congenital duck hepatitis B virus infection in vivo: effect on intrahepatic-viral DNA, RNA, and protein expression. Hepatology. 1996;24:766–773. doi: 10.1053/jhep.1996.v24.pm0008855174. [DOI] [PubMed] [Google Scholar]
- 15.Köck J, Baumert TF, Delaney WE, Blum HE, von Weizsäcker F. Inhibitory effect of adefovir and lamivudine on the initiation of hepatitis B virus infection in primary tupaia hepatocytes. Hepatology. 2003;38:1410–1418. doi: 10.1016/j.hep.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Rabe B, Glebe D, Kann M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J Virol. 2006;80:5465–5473. doi: 10.1128/JVI.02303-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y, Guo H, Zhang L, Xie H, Zhao X, Wang F, Li Z, Wang Y, Ma S, Tao J, et al. Genomic analysis of anti-hepatitis B virus (HBV) activity by small interfering RNA and lamivudine in stable HBV-producing cells. J Virol. 2005;79:14392–14403. doi: 10.1128/JVI.79.22.14392-14403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA. 2002;99:15655–15660. doi: 10.1073/pnas.232137699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nassal M, Dallmeier K, Schultz U, Sun D. Phenotyping hepatitis B virus variants: from transfection towards a small animal in vivo infection model. J Clin Virol. 2005;34 Suppl 1:S89–S95. doi: 10.1016/s1386-6532(05)80017-4. [DOI] [PubMed] [Google Scholar]
- 20.Schulze-Bergkamen H, Untergasser A, Dax A, Vogel H, Büchler P, Klar E, Lehnert T, Friess H, Büchler MW, Kirschfink M, et al. Primary human hepatocytes--a valuable tool for investigation of apoptosis and hepatitis B virus infection. J Hepatol. 2003;38:736–744. doi: 10.1016/s0168-8278(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 21.Mahieu-Caputo D, Allain JE, Branger J, Coulomb A, Delgado JP, Andreoletti M, Mainot S, Frydman R, Leboulch P, Di Santo JP, et al. Repopulation of athymic mouse liver by cryopreserved early human fetal hepatoblasts. Hum Gene Ther. 2004;15:1219–1228. doi: 10.1089/hum.2004.15.1219. [DOI] [PubMed] [Google Scholar]
- 22.Klöcker U, Oberwinkler H, Kürschner T, Protzer U. Presence of replicating virus in recombinant hepadnavirus stocks results from recombination and can be eliminated by the use of a packaging cell line. J Virol. 2003;77:2873–2881. doi: 10.1128/JVI.77.5.2873-2881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Udagawa T, Puder M, Wood M, Schaefer BC, D'Amato RJ. Analysis of tumor-associated stromal cells using SCID GFP transgenic mice: contribution of local and bone marrow-derived host cells. FASEB J. 2006;20:95–102. doi: 10.1096/fj.04-3669com. [DOI] [PubMed] [Google Scholar]
- 24.Tada M, Hatano E, Taura K, Nitta T, Koizumi N, Ikai I, Shimahara Y. High volume hydrodynamic injection of plasmid DNA via the hepatic artery results in a high level of gene expression in rat hepatocellular carcinoma induced by diethylnitrosamine. J Gene Med. 2006;8:1018–1026. doi: 10.1002/jgm.930. [DOI] [PubMed] [Google Scholar]
- 25.Walter E, Keist R, Niederöst B, Pult I, Blum HE. Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology. 1996;24:1–5. doi: 10.1002/hep.510240101. [DOI] [PubMed] [Google Scholar]
- 26.Glebe D, Aliakbari M, Krass P, Knoop EV, Valerius KP, Gerlich WH. Pre-s1 antigen-dependent infection of Tupaia hepatocyte cultures with human hepatitis B virus. J Virol. 2003;77:9511–9521. doi: 10.1128/JVI.77.17.9511-9521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong DK, Yuen MF, Yuan H, Sum SS, Hui CK, Hall J, Lai CL. Quantitation of covalently closed circular hepatitis B virus DNA in chronic hepatitis B patients. Hepatology. 2004;40:727–737. doi: 10.1002/hep.20353. [DOI] [PubMed] [Google Scholar]
- 28.Stoeckl L, Funk A, Kopitzki A, Brandenburg B, Oess S, Will H, Sirma H, Hildt E. Identification of a structural motif crucial for infectivity of hepatitis B viruses. Proc Natl Acad Sci USA. 2006;103:6730–6734. doi: 10.1073/pnas.0509765103. [DOI] [PMC free article] [PubMed] [Google Scholar]