

Abstract
AIM: To investigate whether increased intestinal permeability contributes to the pathogenesis and progress of nonalcoholic steatohepatitis by observing its dynamic change in rat models.
METHODS: Rat models of nonalcoholic steatohepatitis were established by giving a fat-rich diet. The rats were sacrificed at wk 8, 12 and 16 during the study. Rats fed with normal diet were taken as control. Plasma D-lactate, plasma diamine oxidase, serum lipids and liver transaminases were measured in blood of the femoral artery. Hepatic steatosis and inflammation were assessed by haematoxylin-eosin staining.
RESULTS: A rat model of nonalcoholic steatohepatitis was established successfully. Plasma D-lactate level in model group at wk 8, 12 and 16 and diamine oxidase level in model group at wk 12, 16 increased significantly compared with those in control group. There were notable differences of D-lactate and diamine oxidase level in model group between wk 8 and 12 as well as between wk 12 and 16. Serum lipids, liver transaminases and liver injury also increased with disease development.
CONCLUSION: Increased intestinal permeability caused by intestinal bacterial overgrowth and endotoxin-induced intestinal destruction exists in rats with nonalcoholic steatohepatitis, which may partially explain the pathogenesis and progress of this disease.
Keywords: Nonalcoholic steatohepatitis, Intestinal permeability, D-lactate, Diamine oxidase
INTRODUCTION
The pathogenesis and progress of nonalcoholic steato-hepatitis (NASH) remain unclear and the most advocated theory is “two-hit hypothesis”[1]. Briefly, the first hit is the deposition of fatty acid in hepatocytes triggered by different factors which further down-regulate their flexibility towards other stress changes while the second hit is the concomitant liver damage induced by oxidative stress and lipid peroxidation. Nowadays, it is well accepted that endotoxin from intestinal Gram-negative bacteria is responsible for the second hit by releasing inflammatory cytokines and oxygen-free radicals but the underlying mechanism is still unclear[2-4]. Several researches demonstrated that the increased intestinal permeability which may be due to endotoximia and ensuing intestinal bacterial translocation plays a critical role in different liver diseases such as alcoholic liver disease[5-8] and liver cirrhosis[9-11]. Therefore, it seems plausible that despite of various etiologies, change of intestinal permeability may be involved in the pathogenesis and progress of liver diseases.
As NASH shares histological similarity to alcoholic hepatitis, we hypothesize that increased intestinal permeability is also involved in its pathogenesis and progress, and allows endotoxin to escape into the portal circulation and liver where it can initiate a hepatic necroinflammatory cascade. Wigg and colleagues[12] reported that there is no significant change of intestinal permeability in patients with NASH and control subjects without liver disease. Riordan and coworkers[13] demonstrated that intestinal permeability in NASH patients is much higher than that in controls. However, both studies investigated the static intestinal permeability at certain stage of NASH instead of the serial change in intestinal permeability at different stages of NASH. The conflicting results may be due to the discrepancy of NASH stage between the selected patients. Furthermore, the dual lactulose-rhamnose sugar test used to measure intestinal permeability has its own limitations in the process of urine collection.
In recognition of the deficiency of above mentioned experiments, the present study was to test this hypothesis by evaluating intestinal permeability in rat model of NASH, on which it is much easier to observe serial change in intestinal permeability and obtain data on hepatic histology. Here we used plasma D-lactate and diamine oxidase (DAO), two well established markers, to estimate the intestinal permeability. D-lactate is a product of bacterial fermentation and decomposition in the gastrointestinal tract and its concentration is low in plasma[14]. During liver cirrhosis, alcoholic hepatitis, gut failure or other liver diseases, increased intestinal permeability leads to efflux of bacteria and their metabolic products into the circulation, including D-lactate, which cannot be metabolized in the liver. Therefore, plasma level of D-lactate is a useful predictor for evaluation of intestinal permeability[15]. Nevertheless, D-lactate level may be influenced by the overgrowth of gastrointestinal bacteria in the absence of increased intestinal permeability. DAO, a highly active intracellular enzyme existing in approximately 95% intestinal mucosa and ciliated epithelial cells but little in plasma of mammals[16], catalyzes and metabolizes histamine, putrescine and cadaverine. When small intestinal barrier is injured, intestinal mucosal cells exfoliate into the bowel lumen and DAO enters the lymphatic vessel and blood stream in the intracellular space, thus increasing plasma DAO level.
MATERIALS AND METHODS
Experimental animal model
A total of 61 Sprague-Dawley (SD) rats at the age of 12 wk, weighing 160-170 g were purchased from the Medical Science Institution of Zhejiang Province (Hangzhou, China). All rats were maintained in a 12-h light/dark cycle at controlled room temperature and randomly divided into model group and control group. Control group (n = 34) was given normal diet (8% rice bran, 50% maize, 30% soybean powder, 3% bone powder, 1.3% multi-vitamin and 6.7% mineral) while model group (n = 27) was established by giving fat-rich diet containing 80.5% normal diet, 2% cholesterol, 7% lard, 10% yolk powder and 0.5% bile salt. Twelve, 11 and 11 rats from control group and 12, 6 and 9 rats from model group were sacrificed by femoral exsanguination at wk 8, 12 and 16 during the study. This study was approved by the University and Hospital Animal Review Board.
Reagents and methods
Body weight, food and water intake were recorded weekly. Femoral blood, liver wet weight and hepatic index (liver weight/body weight) reflecting liver hypertrophy were obtained and estimated. Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglyceride (TG), total cholesterol (TCh) and albumin (ALB) were measured using an automatic biochemical analyzer 7600-110E (Hitachi). D-lactate and DAO were measured as previously described[17,18]. D-lactic dehydrogenase, D-lactate standard solution, O-dianisidine, cadaverine dihydrochioride, DAO standard solution and horseradish peroxidase were purchased from Sigma Chemical Company, USA.
Livers were fixed in 10% formalin, dehydrated with ethanol, and embedded in paraffin. Two μm thick sections were cut, collected sequentially onto glass slides, and stained with haematoxylin-eosin (HE). Steatosis, inflammation and necrosis of hepatic cells were observed under Olympus microscope while the severity of hepatic injury was estimated according to the extent of hepatic steatosis (divided as “-, +, ++, +++”) and histological activation index (HAI)[19,20]. Briefly, HAI was divided into perisinusoidal inflammation (P), lobular inflammation (L), patch necrosis (PN) and bridge necrosis (BN), each of which was scored as 1 (mild), 3 (moderate), 4 (severe). The average score was calculated according to the equation: (P + L + 2PN + 2BN)/4.
Statistical analysis
Normally distributed data were expressed as mean ± SD. Student’s t test between two groups and one-way ANOVA among more than two groups were used. Nonparametric data were presented as the median and the range comprising the lowest and highest values observed. Statistical analyses were performed using SPSS software (version 11.5) and P < 0.05 was considered statistically significant.
RESULTS
Establishment of NASH rat model
NASH rat model was established successfully eight weeks after the rat was fed with fat-rich diet. During the experiment, the weights of rats in the two groups increased gradually and no unexpected death occurred. Compared with the control group, the body weight and hepatic index in model group were markedly higher (P < 0.05, Table 1). During the whole study, H-E stained liver tissues from model group rats demonstrated that simple fatty liver involved 66% of the whole hepatic cells (predominantly macrovesicular) at the 8th wk. At the 12th wk, steatosis became worse with obvious ballooning of hepatocytes (predominantly in zone 3). Mild to moderate chronic inflammation of portal and intra-acinar cells was also noted. At the 16th wk, significant progress in steatosis, hepatocellular ballooning, lobular and portal inflammation was observed (Table 1).
Table 1.
Change of body, hepatic and serum lipid parameters in rats
| Time (wk) | Group | Number | Weight | Hepatic index (%) | TG (mmol/L) | TCh (mmol/L) |
Degree of Hepatic
Steatosis |
HAI mark | |||
| - | + | ++ | +++ | ||||||||
| 8 | Control | 12 | 307.58 ± 17.04 | 2.64 ± 0.09 | 0.51 ± 0.07 | 1.62 ± 0.16 | 12 | 0 | 0 | 0 | 0.67 ± 0.33 |
| Model | 12 | 404.50 ± 13.18a | 3.88 ± 0.53a | 0.35 ± 0.05b | 2.49 ± 0.36c | 0 | 3 | 6 | 3 | 0.68 ± 0.27 | |
| 12 | Control | 11 | 411.05 ± 16.84 | 2.81 ± 0.09 | 0.48 ± 0.10 | 1.38 ± 0.24 | 11 | 0 | 0 | 0 | 0.71 ± 0.12 |
| Model | 6 | 506.16 ± 15.99a | 4.58 ± 0.07a | 0.49 ± 0.10 | 6.20 ± 2.14d,e,f | 0 | 0 | 2 | 4 | 3.95 ± 0.83h,i | |
| 16 | Control | 11 | 453.55 ± 49.11 | 2.77 ± 0.12 | 0.51 ± 0.07 | 1.96 ± 0.45 | 11 | 0 | 0 | 0 | 0.69 ± 0.15 |
| Model | 9 | 554.22 ± 28.37a | 4.74 ± 0.13a | 0.47 ± 0.10 | 2.93 ± 0.55g | 0 | 0 | 1 | 9 | 5.25 ± 1.39h,j | |
Values are expressed as mean ± SD. a: P < 0.001 vs corresponding control group; b: t = 6.31, P < 0.001, vs control group at the 8th wk; c: t = 7.67, P < 0.001, vs control group at the 8th wk; d: t = 5.49, P = 0.003, vs control group at the 12th wk; e: t = 3.60, P = 0.012, vs model group at the 16th wk; f: t = 4.21, P = 0.008, vs model group at the 8th wk; g: t = 3.53, P = 0.002, vs control group at the 16th wk; h: P < 0.05, vs control group at the same stage of time; i: P < 0.05, vs model group at the 8th wk; j: P < 0.05, vs model group at the 12th wk.
TCh in rats of the model group began to increase from the 8th wk, reached its peak at the 12th wk, and decreased until the 16th wk. Nevertheless, significant difference was observed at different time points (8th, 12th, 16th wk) in the model group compared with the control group. TG in rats of the model group was markedly decreased at the 8th wk, which may be attributed to the deposition of TG in liver. However, there was no significant difference between the two groups at the 12th and 16th wk (Table 1). Serum ALT and AST levels in rats of the model group were remarkably increased from the 8th wk (P < 0.05 vs control group) and there were also certain differences in the model group at different time points. ALB was much lower in the model group at the 8th wk (P < 0.05, Table 2).
Table 2.
Change of hepatic function and intestinal permeability in rats
| Time (wk) | Group | Number | ALT (mmol/L) | AST (mmol/L) | ALB (g/L) | D-lactate (mmol/L) | DAO (U/L) |
| 8 | Control | 12 | 54.75 ± 7.05 | 200.33 ± 14.49 | 30.39 ± 2.33 | 0.19 ± 0.04 | 0.74 ± 0.11 |
| Model | 12 | 125.75 ± 34.21a | 270.91 ± 45.38d | 26.91 ± 2.37g | 0.36 ± 0.11h | 0.77 ± 0.09 | |
| 12 | Control | 11 | 56.73 ± 8.46 | 215.64 ± 31.53 | 31.09 ± 1.30 | 0.17 ± 0.03 | 0.65 ± 0.15 |
| Model | 6 | 149.67 ± 30.81b | 311.33 ± 53.33e | 29.83 ± 1.72 | 0.54 ± 0.04i | 0.92 ± 0.10k | |
| 16 | Control | 11 | 51.59 ± 7.54 | 195.10 ± 18.72 | 29.12 ± 2.51 | 0.20 ± 0.05 | 0.77 ± 0.17 |
| Model | 9 | 166.56 ± 43.53c | 314.22 ± 54.41f | 28.98 ± 2.10 | 0.86 ± 0.12j | 1.28 ± 0.19l |
Values are expressed as mean ± SD. a: t = 7.04, P < 0.001, vs control group at the 8th wk; b: t = 7.24, P < 0.001, vs control group at the 12th wk; c: t = 7.83, P < 0.001, vs control group at the 16th wk; d: t = 5.13, P < 0.001, vs control group at the 8th wk; e: t = 4.70, P < 0.001, vs control group at the 12th wk; f: t = 6.82, P < 0.001, vs control group at the 16th wk; g: t = 5.24, P < 0.001, vs control group at the 8th wk.; h: t = 5.50, P < 0.001, vs control group at the 8th wk ; i: t = 9.70, P < 0.001, vs control group at the 12th wk; j: t = 7.22, P < 0.001, vs control group at the 16th wk; k: t = 3.91, P = 0.001, vs control group at the 12th wk; l: t = 6.27, P < 0.001, vs control group at the 16th wk.
Change in intestinal permeability
Plasma D-lactate levels in the model group were significantly higher than those in the control group at the 8th, 12th and 16th wk. Plasma DAO levels in the model group were markedly increased compared with the control group at the 12th and 16th wk. Both D-lactate and DAO had a tendency to increase, suggesting that D-lactate and DAO levels in rats of the model group were much higher at the 12th wk and 16th wk than at the 8th and 12th wk (Figure 1, Table 2).
Figure 1.
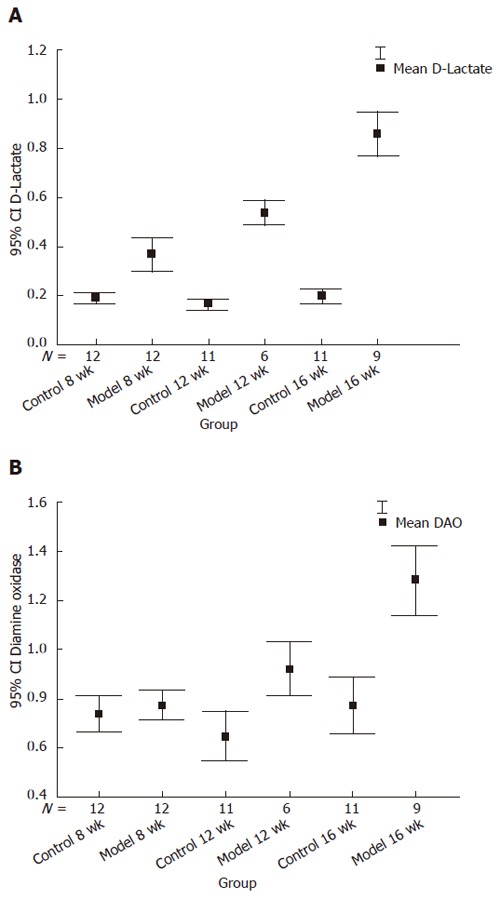
Level of D-lactate (A) and DAO (B) between the model and control groups at different time points. Y axis represents the value of D-lactate and DAO with 95% confidence. A: Model at the 12th wk vs model at the 8th wk, t = 3.67, P = 0.002; model at the 16th wk vs model at the 12th wk, t = 6.29, P < 0.001; B: Model 12th wk vs model at the 8th wk, t = 3.13, P = 0.006; model at the 16th wk vs model at the 12th wk, t = 4.28, P = 0.001.
DISCUSSION
In 1980, Ludwig et al[21] coined the term NASH to describe the morphologic pattern of liver injury in 20 patients who had histological evidence for alcoholic hepatitis on liver biopsy but no history of alcohol abuse. Since then, researches on this disease have been booming. However, the pathogenesis of NASH is still poorly understood and endotoxin is actively involved.
In this study, we used plasma D-lactate and DAO to evaluate the intestinal permeability in rats with NASH and to explore the association of intestinal permeability with NASH. Eight weeks after the rats were fed with fat-rich diet, we successfully established the rat model of NASH, and observed its serial change in intestinal permeability at the 8th, 12th and 16th wk. The plasma DAO level was normal while the plasma D-lactate level increased significantly in the model group at the 8th wk, suggesting that this increase may be attributed to the overgrowth of intestinal bacteria, thus leading to the increased absorption of D-lactate, instead of increased intestinal permeability. This finding is consistent with the result of Wigg[12]. Meanwhile, H-E staining also confirmed that rats in the model group mainly stayed at the stage of simple fatty liver, implying occurrence of the first hit. The results obtained at the 12th and 16th wk indicate that impairment of intestinal barrier and high intestinal permeability occurred at or even before the 12th wk during which the histological evidence showed occurrence of the second hit and stability at the stage of steatohepatitis. The degree of necrosis and inflammation as indicated by ALT, AST, HAI and steatosis was paralleled with the change in intestinal permeability that aggravated from the 12th wk to the 16th wk. The serial change in D-lactate and DAO showed that intestinal barrier dysfunction and high intestinal permeability play an important role in the progress from simple fatty liver to nonalcoholic steatohepatitis.
The mechanism of increased intestinal permeability in NASH is still unknown. Normally, it may be influenced by genetic and/or environmental factors such as ecological equilibrium of the intestinal lumen flora[22], mechanical barrier of intestinal mucosa[23], immune response in the intestinal tract[24] and the gut-liver feedback axis. Cofactors for NASH including obesity, insulin resistance and hyperlipidemia can lead to intestinal dysbacteriosis and further proliferation of the intestinal flora, thus impairing mesenteric cell architecture and shortening or exfoliating intestinal villi[25]. Endotoxins produced by Gram-negative bacteria pass through intestinal mucosa into blood and increase intestinal permeability due to hypoxia and acidosis of the intestinal mucosa, which would create a vicious cycle that perpetuates a necroinflammatory cascade and causes liver damage[26,27]. Intestinal wall edema secondary to hypertension or hypoalbuminemia might be another explanation[28]. The ALB level in the 8th wk was significantly lower in model group, suggesting that hypoalbuminemia is involved in pathogenesis of NASH at the starting stage. The underlying mechanism might be the increased portal pressure which reduces the absorptive capacity of small intestine and the occurrence of malnutrition or hypoalbuminemia[29]. However, as hypoalbuminemia is usually a laboratory feature of advanced cirrhosis and seldom observed in patients with NASH, reduced ALB level at the early stage of NASH needs further verification in large scale experiments. Moreover, further research on portal tension and morphologic change in intestinal mucosa is required.
In conclusion, increased intestinal permeability is associated with the pathogenesis and progress of NASH in rats. Our study is consistent with the conclusions of several reports that endotoximia and translocation of intestinal bacteria may develop a vicious circle in the liver damage[2,5,6]. Our finding is of clinical significance for developing a means of reversing increased intestinal permeability in NASH patients. Though the NASH rat model established by acute over feeding of high fat diet may not be fully representative of human NASH mainly caused by insulin resistance, it can be used in research of nonalcoholic fatty liver disease[30]. Further studies on the correlation between intestinal permeability, circulating levels of endotoxin and portal tension are required.
ACKNOWLEDGMENTS
The authors thank Ms Qiaojuan Shi for her help in the establishment of NASH model and Mr Guocai Lv for his instructions on testing serum D-lactate and DAO. We are also grateful to Mr Lei Xu and Mr Hangbin Jin for collecting serum.
COMMENTS
Background
The pathogenesis and progress of NASH remain unclear. The most advocated theory is the “two-hit hypothesis” mainly consisting of hepatic fat deposition as the first hit and concomitant hepatic inflammation as the second hit. Several potential pathogenic factors including insulin resistance, mitochondrial dysfunction, lipid peroxidation, intestinal permeability, have been intensely investigated.
Research frontiers
Many hot spots on NASH have been studied. The search for non-invasive diagnosis and effective treatment of NASH has been under intensive investigation. Nevertheless, we still call for the prospective, double blind and placebo-controlled clinical trials to verify the effect of treatment and the pathogenesis of NASH.
Innovations and breakthroughs
Since previous studies on the intestinal permeability in NASH remain controversial, the conflicting results may be due to the discrepancy in NASH stage between the selected patients. In our study, the serial change in intestinal permeability in rat model of NASH was found to be associated with NASH
Applications
Since increased intestinal permeability may contribute to the development of NASH, drugs can be used to prevent the increase in intestinal permeability and to further alleviate NASH.
Peer review
This is an experimental dynamic study conducted on rats. Intestinal permeability was assessed by plasma levels of D-lactate and diamine oxidase. Results of this study provide evidence for increased intestinal permeability in parallel with degree of steatohepatitis. This interesting experimental study merits publication in WJG.
Footnotes
S- Editor Liu Y L- Editor Wang XL E- Editor Zhou T
References
- 1.Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 2.Nolan JP. Intestinal endotoxins as mediators of hepatic injury--an idea whose time has come again. Hepatology. 1989;10:887–891. doi: 10.1002/hep.1840100523. [DOI] [PubMed] [Google Scholar]
- 3.Tiegs G, Wolter M, Wendel A. Tumor necrosis factor is a terminal mediator in galactosamine/endotoxin-induced hepatitis in mice. Biochem Pharmacol. 1989;38:627–631. doi: 10.1016/0006-2952(89)90208-6. [DOI] [PubMed] [Google Scholar]
- 4.Nagakawa J, Hishinuma I, Hirota K, Miyamoto K, Yamanaka T, Tsukidate K, Katayama K, Yamatsu I. Involvement of tumor necrosis factor-alpha in the pathogenesis of activated macrophage-mediated hepatitis in mice. Gastroenterology. 1990;99:758–765. doi: 10.1016/0016-5085(90)90965-4. [DOI] [PubMed] [Google Scholar]
- 5.Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 6.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 7.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 8.Keshavarzian A, Fields JZ, Vaeth J, Holmes EW. The differing effects of acute and chronic alcohol on gastric and intestinal permeability. Am J Gastroenterol. 1994;89:2205–2211. [PubMed] [Google Scholar]
- 9.Ersöz G, Aydin A, Erdem S, Yüksel D, Akarca U, Kumanlioglu K. Intestinal permeability in liver cirrhosis. Eur J Gastroenterol Hepatol. 1999;11:409–412. doi: 10.1097/00042737-199904000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Ruan P, Gong ZJ, Zhang QR. Changes of plasma D(-)-lactate, diamine oxidase and endotoxin in patients with liver cirrhosis. Hepatobiliary Pancreat Dis Int. 2004;3:58–61. [PubMed] [Google Scholar]
- 11.Zhao H, Li XO, Wang P, Lou GQ, Chen WP, Tu SX, Zhao GG. Research on intestine permeability in patients with post-hepatitis cirrhosis. Zhonghua Chuanranbing Zazhi. 2002;20:105–107. [Google Scholar]
- 12.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riordan SM, Duncombe VM, Thomas MC, Nagree A, Bolin TD, McIver CJ, Williams R. Small intestinal bacterial overgrowth, intestinal permeability, and non-alcoholic steatohepatitis. Gut. 2002;50:136–138. doi: 10.1136/gut.50.1.136-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcos MA, Vila J, Gratacos J, Brancos MA, Jimenez de Anta MT. Determination of D-lactate concentration for rapid diagnosis of bacterial infections of body fluids. Eur J Clin Microbiol Infect Dis. 1991;10:966–969. doi: 10.1007/BF02005455. [DOI] [PubMed] [Google Scholar]
- 15.Murray MJ, Gonze MD, Nowak LR, Cobb CF. Serum D(-)-lactate levels as an aid to diagnosing acute intestinal ischemia. Am J Surg. 1994;167:575–578. doi: 10.1016/0002-9610(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 16.Takagi K, Nakao M, Ogura Y, Nabeshima T, Kunii A. Sensitive colorimetric assay of serum diamine oxidase. Clin Chim Acta. 1994;226:67–75. doi: 10.1016/0009-8981(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 17.Brandt RB, Siegel SA, Waters MG, Bloch MH. Spectrophotometric assay for D-(-)-lactate in plasma. Anal Biochem. 1980;102:39–46. doi: 10.1016/0003-2697(80)90314-0. [DOI] [PubMed] [Google Scholar]
- 18.Li JY, Yu Y, Hao J, Jin H, Xu HJ. Detection of the diamine oxidase (DAO) activation of blood and intestine by spectrophotometric method. Anjisuan he Shengwu Ziyuan. 1996;18:28–30. [Google Scholar]
- 19.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 20.Wang TL, Liu X, Zhou YP, He JW, He Z, Li NZ. A semiquantitative scoring system for assessment of hepatic inflammation and fibrosis in chronic viral hepatitis. Zhonghua Ganzangbing Zazhi. 1998;6:195–197. [Google Scholar]
- 21.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 22.Liévin V, Peiffer I, Hudault S, Rochat F, Brassart D, Neeser JR, Servin AL. Bifidobacterium strains from resident infant human gastrointestinal microflora exert antimicrobial activity. Gut. 2000;47:646–652. doi: 10.1136/gut.47.5.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter EM, van Dam E, Valore EV, Ganz T. Broad-spectrum antimicrobial activity of human intestinal defensin 5. Infect Immun. 1997;65:2396–2401. doi: 10.1128/iai.65.6.2396-2401.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagnoff MF. Mucosal immunology: new frontiers. Immunol Today. 1996;17:57–59. doi: 10.1016/0167-5699(96)80579-2. [DOI] [PubMed] [Google Scholar]
- 25.Aldersley MA, Howdle PD. Intestinal permeability and liver disease. Eur J Gastroenterol Hepatol. 1999;11:401–403. doi: 10.1097/00042737-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Zhao LF, Li H, Han DW. Intestinal endotoxemia and liver diseases. Shijie Huaren Xiaohua Zazhi. 2000;8:1145–1149. [Google Scholar]
- 27.Li XH, Gong JP, Shi YJ, Liu CA, Peng Y. In vitro expression of CD14 protein and its gene in Kupffer cells induced by lipopolysaccharide. Hepatobiliary Pancreat Dis Int. 2003;2:571–575. [PubMed] [Google Scholar]
- 28.Quigley EM. Gastrointestinal dysfunction in liver disease and portal hypertension. Gut-liver interactions revisited. Dig Dis Sci. 1996;41:557–561. doi: 10.1007/BF02282341. [DOI] [PubMed] [Google Scholar]
- 29.Taylor RM, Bjarnason I, Cheeseman P, Davenport M, Baker AJ, Mieli-Vergani G, Dhawan A. Intestinal permeability and absorptive capacity in children with portal hypertension. Scand J Gastroenterol. 2002;37:807–811. [PubMed] [Google Scholar]
- 30.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]