

Abstract
Obesity is a substantial risk factor for developing asthma, but the molecular mechanisms underlying this relationship are unclear. We tested the role of insulin in airway responsiveness to nerve stimulation using rats genetically prone or resistant to diet-induced obesity. Airway response to vagus nerve stimulation and airway M2 and M3 muscarinic receptor function were measured in obese-prone and -resistant rats with high or low circulating insulin. The effects of insulin on nerve-mediated human airway smooth muscle contraction and human M2 muscarinic receptor function were tested in vitro. Our data show that increased vagally mediated bronchoconstriction in obesity is associated with hyperinsulinemia and loss of inhibitory M2 muscarinic receptor function on parasympathetic nerves. Obesity did not induce airway inflammation or increase airway wall thickness. Smooth muscle contraction to acetylcholine was not increased, indicating that hyperresponsiveness is mediated at the level of airway nerves. Reducing serum insulin with streptozotocin protected neuronal M2 receptor function and prevented airway hyperresponsiveness to vagus nerve stimulation in obese rats. Replacing insulin restored dysfunction of neuronal M2 receptors and airway hyperresponsiveness to vagus nerve stimulation in streptozotocin-treated obese rats. Treatment with insulin caused loss of M2 receptor function, resulting in airway hyperresponsiveness to vagus nerve stimulation in obese-resistant rats, and inhibited human neuronal M2 receptor function in vitro. This study shows that it is not obesity per se but hyperinsulinemia accompanying obesity that potentiates vagally induced bronchoconstriction by inhibiting neuronal M2 muscarinic receptors and increasing acetylcholine release from airway parasympathetic nerves.
Keywords: hyperinsulinemia, obesity, asthma, airway responsiveness, neural M2 muscarinic receptor
Clinical Relevance
Obesity-induced asthma does not respond well to traditional anti-inflammatory therapies, suggesting that it is a unique asthma phenotype. Here we show that hyperinsulinemia causes airway hyperresponsiveness to vagus nerve stimulation in obese rats. In human trachea and in rats, we demonstrate that insulin inhibits M2 muscarinic receptors on airway parasympathetic nerves, resulting in increased acetylcholine release and increased airway contraction. Because hyperinsulinemia is greater and more prevalent in obese individuals, these data may explain why obese individuals are prone to asthma exacerbations and suggest that anticholinergic drugs may be effective in this specific phenotype of asthma.
In the United States, 31% of adults and 15% of children are obese. In obese and overweight individuals, the prevalence of asthma (1–3) and the rate of new-onset asthma have increased (4–6). Obese patients with asthma also have increased severity of illness and reduced effectiveness of steroids compared with nonobese patients with asthma (1, 6, 7). The mechanisms by which obesity predisposes to asthma are unclear, limiting our ability to prevent and treat obesity-related asthma.
There is a lack of agreement in epidemiological studies regarding the relationship between asthma, body mass index, cytokines (e.g., leptin), and hormones (e.g., insulin). One study suggests that insulin resistance may predict asthma in obese individuals dependent upon body mass index (4). However, insulin resistance may also predispose to asthma independent of body mass index (5, 8–12). The frequency of asthma increases with increasing insulin resistance (as reflected by acanthosis nigricans [10] or by homeostatic model assessment of insulin resistance [11, 12]) in children independent of body mass. Thus, insulin resistance and compensatory hyperinsulinemia may be one of the mechanisms that increase asthmatic symptoms in obese individuals. Three additional studies also suggest a direct role of insulin in airway hyperresponsiveness. Inhaled insulin causes a significant decline in FEV1 when used to treat patients with diabetes during the initial 15 to 90 days of treatment (13). This fall in FEV1 is significantly greater in patients with diabetes and asthma compared with patients with diabetes without asthma (14, 15).
In the lungs, M2 muscarinic receptors limit acetylcholine (ACh) release from parasympathetic nerves (16–18), thereby limiting vagally induced bronchoconstriction (19, 20). Dysfunctional M2 muscarinic receptors on airway parasympathetic nerves result in airway hyperresponsiveness in humans with asthma (21–23) and in animal models of asthma (24–26). Here we used a polygenic model of diet-induced obesity to test whether obesity increases insulin that subsequently decreases neuronal M2 receptor function, resulting in airway hyperresponsiveness to vagus nerve stimulation. Furthermore, we used human tracheas to test whether exogenous insulin can directly reduce neuronal M2 receptor function and potentiate neurally mediated airway smooth muscle contraction.
Materials and Methods
Animals
Obese-prone and obese-resistant rats were purchased from Charles River Labs (Seattle, WA) (details are provided in the online supplement) and were fed a high-fat or a low-fat diet for 5 weeks. To determine the role of insulin in airway hyperresponsiveness to vagus nerve stimulation, some rats were treated with streptozotocin (STZ) to reduce insulin secretion (27, 28) (Figure 1), and some of these STZ-treated rats were given supplemental insulin (27, 28) once (acute insulin) or daily for 4 weeks (chronic insulin) (Figure 1). Body fat, plasma insulin, and glucose were measured. Additional details are provided in the online supplement.
Figure 1.

Obese-prone rats were originally developed by selectively breeding the rat pairs who gained the most weight with each other; the offspring became obese when fed a high-fat diet. Obese-resistant rats, the control for obese-prone rats, were developed by selectively breeding rat pairs who gained the least weight with each other, and the offspring did not become obese despite being fed a high-fat diet. Obese-prone and obese-resistant rats (male, 5 wk old) were fed a high-fat diet (60% of calories come from fat, Test Diet 58126) or a low-fat diet (13% of calories come from fat, Test Diet 58124) for 5 weeks. Some obese-prone and obese-resistant rats were treated with streptozotocin (STZ) (65 mg/kg, intraperitonelly, given once in the first week) to reduce insulin secretion from β cells in the pancreas. Some STZ-treated rats were given supplemental insulin (Lantus; Sanofi US, Bridgewater, NJ) 3 units/rat subcutaneously) daily starting 7 days after STZ and continuing until 24 hours before physiological measurements were made (chronic insulin). Other STZ-treated rats were given supplemental insulin (Lantus, 3 units/rat subcutaneously) once 24 hours before physiological measurements were made (acute insulin). Experiment groups were listed in the table right below the graph. HFD, high-fat diet; LFD, low-fat diet; i.p., intraperitoneally; s.c., subcutaneously.
Measuring Bronchoconstriction and M2 and M3 Muscarinic Receptor Function In Vivo
To investigate parasympathetic nerve–mediated airway hyperresponsiveness, bronchoconstriction in response to electrical stimulation of the vagus nerves was measured as previously described (29). Briefly, rats were anesthetized, paralyzed, ventilated, chemically sympathectomized, and vagotomized. Electrical stimulation of both vagus nerves produced frequency-dependent bronchoconstriction measured as the increase of pulmonary inflation pressure (in mm H2O). Because neuronal M2 muscarinic receptor function and M3 muscarinic receptor function on airway smooth muscle can affect parasympathetic nerve–mediated airway hyperresponsiveness, their functions were measured. Neural M2 muscarinic receptor function was tested by measuring the ability of pilocarpine (P6628; Sigma-Aldrich, St. Louis, MO), an M2 receptor agonist, to inhibit vagally induced bronchoconstriction. M3 muscarinic receptor function on airway smooth muscle was tested by measuring intravenous ACh-induced bronchoconstriction in vagotomized rats. Additional details are provided in the online supplement.
Bronchoalveolar Lavage and Morphologic Study of Lung
Inflammatory cells recovered from bronchoalveolar lavage were counted as previous described (29). To test whether airway remodeling contributed to the airway hyperresponsiveness to vagus nerve stimulation in obese rats, lungs from animals were fixed in zinc formalin, sectioned, and stained with hemotoxylin-eosin. The ratio between airway smooth muscle area and airway basement membrane was calculated as shown in Figure E1 in the online supplement. Additional details are provided in the online supplement.
Human Tracheal Responsiveness and M2 and M3 Muscarinic Receptor Function
To test the direct effect of insulin on parasympathetic nerve–mediated airway smooth muscle contraction and M3 muscarinic receptor function, human tracheal smooth muscle strips were suspended in organ baths in the absence and presence of insulin. Contractions were induced by electrical field stimulation (EFS) or by increasing concentrations of methacholine (MCh) as previously described (30). Neuronal M2 muscarinic receptor function was tested in human tracheal smooth muscle by measuring the ability of gallamine, a selective antagonist, to potentiate EFS-induced contractions (19). Additional details are provided in the online supplement.
Statistical Analysis
In vivo bronchoconstriction to electrical stimulation of the vagus nerves and to intravenous ACh dose-response curves to pilocarpine and in vitro airway smooth muscle contraction to EFS dose-response curves to gallamine were analyzed using repeated-measures ANOVA. Appropriate statistical analyses were performed for other in vivo and in vitro data, and details are provided in the online supplement. A P value < 0.05 was considered significant.
Results
Effect of Obesity on Airway Responsiveness
In anesthetized rats, electrical stimulation of the vagus nerves produced frequency-dependent bronchoconstriction in all animals (Figure 2). The bronchoconstriction in response to vagus nerve stimulation was significantly potentiated only in obese-prone rats fed a high-fat diet versus all other groups (Figures 2A and 2B). In contrast, bronchoconstriction in response to intravenous ACh, which stimulates smooth muscle directly in vagotomized animals, was not different between obese-prone and obese-resistant rats regardless of diet (Figures 2C and 2D). The bronchoconstriction in response to intravenous ACh was also not statistically different in rats fed a low- or high-fat diet regardless of whether they were genetically obese prone or obese resistant (Figures 2C and 2D). Thus, M3 muscarinic receptor–mediated contraction of airway smooth muscle is unlikely to be responsible for the increased bronchoconstriction in response to vagus nerve stimulation. The cholinergic nature of vagally induced bronchoconstriction was demonstrated by blocking contractions with the muscarinic antagonist atropine at the end of each experiment (data not shown).
Figure 2.

A high-fat diet increased vagally induced bronchoconstriction in obese-prone rats. In anesthetized rats, bronchoconstriction (measured as an increase in pulmonary inflation pressure [Ppi]) was induced by stimulating both vagus nerves (2–50 Hz, 10 V, 0.2-ms pulse duration, for 5 s at 45-s duration) (A and B) or by administering acetylcholine (ACh) intravenously to vagotomized rats (C and D). Vagally induced bronchoconstriction was significantly greater in obese-prone rats fed a high-fat diet (A) than in any other group (A and B). Intravenous ACh–induced bronchoconstriction was not significantly different between obese-prone and obese-resistant rats on high-fat (C) or low-fat (D) diets. Each point represents the mean, and vertical bars show SEM. *P < 0.05.
Effect of Diet on Body Weight, Body Fat, Insulin and Glucose Levels, Airway Inflammation, and Airway Wall Thickness
On a high-fat diet, the average food intake was 21 ± 1.9 g/d or 104.7 ± 1.2 calories/d for obese-prone rats and 16 ± 0.3 g/d or 80.6 ± 1.4 calories/d for obese-resistant rats. On a low-fat diet, the average food intake was 27 ± 1.9 g/d or 109.5 ± 7.4 calories/d for obese-prone rats and 22 ± 1.2 g/d or 88.1 ± 4.9 calories/d for obese-resistant rats.
After 5 weeks, obese-prone rats weighed significantly more than obese-resistant rats regardless of whether they were fed a high- or low-fat diet (Figure 3A). Additionally, obese-prone rats fed a high-fat diet had significantly greater weight gain compared with obese-prone rats on a low-fat diet (Figure 3A). Body fat (measured by nuclear magnetic resonance) was significantly greater in obese-prone rats than in obese-resistant rats regardless of diet and was greatest in obese-prone rats on a high-fat diet (Figure 3B). After 5 weeks on a high- or low-fat diet, there were no differences in baseline pulmonary inflation pressure, heart rate, or blood pressure in obese-prone versus obese-resistant rats regardless of diet (Table 1).
Figure 3.

Obese-prone rats (closed bars) had significantly heavier body weight (A) and body fat (B) than obese-resistant rats (open bars) regardless of whether they were on a high- or low-fat diet. STZ (gray bars) significantly attenuated weight gain (C) and increased body fat (D) induced by a HFD. Insulin administered daily for 5 weeks (chronic insulin [cINS]) did not restore weight (C) but increased body fat in obese-resistant rats (D, last two bars on right). Each bar represents mean ± SEM (n = 6–14). *P < 0.05.
Table 1.
Pulmonary Inflation Pressure and Cardiovascular Parameters Are Not Changed by Obesity, Diet, or Treatment with Streptozotocin or Insulin
| Treatment Group | Ppi (mm H2O) | Heart Rate (beats/min) | Systolic BP (mm Hg) | Diastolic BP (mm Hg) | Animal Number |
|---|---|---|---|---|---|
| OP/HFD | 108 ± 9 | 328 ± 24 | 85 ± 3 | 53 ± 3 | 11 |
| OR/HFD | 110 ± 6 | 363 ± 22 | 70 ± 6 | 47 ± 7 | 12 |
| OP/LFD | 101 ± 5 | 337 ± 28 | 74 ± 11 | 46 ± 7 | 11 |
| OR/LFD | 106 ± 3 | 355 ± 17 | 73 ± 5 | 51 ± 6 | 9 |
| OP/HFD/STZ | 108 ± 2 | 418 ± 20 | 75 ± 7 | 46 ± 1 | 5 |
| OR/HFD/STZ | 118 ± 7 | 313 ± 20 | 89 ± 3 | 49 ± 5 | 6 |
| OP/LFD/STZ | 108 ± 8 | 318 ± 23 | 77 ± 5 | 50 ± 6 | 6 |
| OR/LFD/STZ | 124 ± 10 | 344 ± 44 | 71 ± 8 | 41 ± 5 | 5 |
| OP/HFD/STZ/INS | 104 ± 4 | 452 ± 22 | 88 ± 17 | 62 ± 6 | 6 |
| OR/HFD/STZ/INS | 108 ± 7 | 410 ± 34 | 77 ± 6 | 51 ± 5 | 6 |
Definition of abbreviations: BP, blood pressure; HFD, high-fat diet; INS, insulin; LFD, low-fat diet; OP, obese prone; OR, obese resistant; Ppi, pulmonary inflation pressure; STZ, streptozotocin.
Fasting glucose was also similar across all groups of animals regardless of their predisposition to obesity or their diet (Figure 4C). However, fasting insulin in obese-prone rats on a high-fat diet was higher than in other groups (Figure 4A). Treatment with STZ, which destroys pancreatic β cells, decreased circulating fasting insulin levels in obese-prone and obese-resistant rats on a high-fat diet (Figure 4A) and increased fasting glucose levels (Figure 4C). Insulin replacement restored the nonfasting insulin levels within a physiological range in STZ-treated rats (Figure 4B). The nonfasting insulin levels were significantly greater after chronic insulin treatment in obese-resistant rats than in obese-prone rats (Figure 4B). However, nonfasting glucose was not different between obese-prone and obese-resistant rats after chronic or acute supplemental insulin (Figure 4D).
Figure 4.

Changes of insulin and glucose in response to diet, STZ treatment, and supplemental insulin. (A) Obese-prone rats on a high-fat diet demonstrate the highest fasting insulin levels among all animal groups. STZ clearly reduces fasting insulin levels in obese-prone and obese-resistant rats on a high-fat diet. (B) Supplemental insulin causes a significant increase (when administered chronically) or a trend of increase (when administered acutely) in nonfasting insulin levels in obese-resistant rats as compared with those in obese-prone rats. (C) Fasting glucose levels are increased after STZ in obese-prone and obese-resistant rats on a high-fat diet. Fasting (C) and nonfasting (D) glucose levels are not different between obese-prone and obese-resistant rats regardless of diet, STZ treatment, or supplemental insulin. Each symbol represents an individual rat; horizontal bar = mean ± SEM. *P < 0.05. OP, obese prone; OR, obese resistant.
Most of the cells recovered by bronchoalveolar lavage were macrophages, and there were no significant differences between the total number of macrophages recovered (for obese-prone rats, 2.4 ± 0.7 million on a high-fat diet and 1.0 ± 0.1 million on a low-fat diet; for obese-resistant rats, 1.0 ± 0.2 million on a high-fat diet and 1.4 ± 0.2 million on a low-fat diet). Airway smooth muscle area was not different among groups (Figure E1). These data show that neither inflammation nor smooth muscle hypertrophy likely contribute to airway hyperresponsiveness in obese animals on a high-fat diet.
Role of Insulin in Airway Hyperresponsiveness
STZ reduced insulin in obese-prone and obese-resistant rats (Figure 4A). STZ-treated animals, whether obese prone or obese resistant, gained much less weight (Figure 3C) and had a lower % body fat (Figure 3D) than animals that did not receive STZ. Restoring insulin to STZ-treated animals did not prevent weight loss (Figure 3D).
Suppressing insulin (Figure 1) significantly reduced vagally induced bronchoconstriction in obese-prone rats on a high-fat diet (Figure 5A, open circles with solid line), so that bronchoconstriction was no longer potentiated compared with obese-resistant rats (Figure 5B, closed triangles with solid line). In contrast, STZ did not affect vagally induced bronchoconstriction in obese-resistant rats (Figure 5B, open triangles with solid line).
Figure 5.
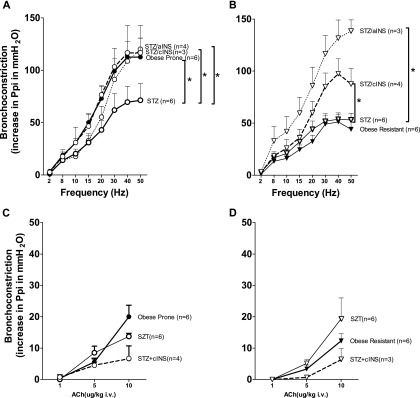
Insulin mediated increased vagally induced bronchoconstriction in obese-prone and obese-resistant rats on a high-fat diet. Bronchoconstriction in obese-prone rats was potentiated (A, closed circles) compared with obese-resistant rats (B, closed triangles). These data were from Figure 2A and were regraphed here. Reducing insulin with STZ significantly reduced bronchoconstriction in response to parasympathetic nerve stimulation in obese-prone rats on a high-fat diet (A, empty circles with solid line) but had no effect on bronchoconstriction in obese-resistant rats (B, empty triangles with solid line). Restoring insulin, either acutely (acute insulin [aINS], once 24 h before physiological measurements) or chronically (chronic insulin [cINS], daily for 4 wk) blocked the effect of STZ in obese-prone rats (A, dashed lines). However, in obese-resistant rats, acute and chronic supplemental insulin significantly potentiated bronchoconstriction above STZ alone (B, dashed lines). ACh-induced bronchoconstriction in vagotomized rats was not significantly changed by STZ (open circles and open triangles) or by insulin (dashed lines) in obese-prone (C) or obese-resistant (D) rats on a high-fat diet. Each point represents the mean, and vertical bars show SEM. *P < 0.05.
Restoring insulin to STZ-treated rats, either chronically over 27 days or acutely 24 hours before physiological responses were measured (Figure 1), restored the potentiation of vagally induced bronchoconstriction in obese rats on a high-fat diet to preSTZ levels (Figure 5A, open circles with dashed lines). In obese-resistant rats, insulin significantly potentiated vagally induced bronchoconstriction to the same level as obese-prone rats on a high-fat diet (Figure 5B, dashed lines).
Bronchoconstriction to intravenous ACh was not affected by STZ (Figures 5C and 5D, open circles and triangles with solid line) or supplemental insulin (Figures 5C and 5D, open circles and triangles with dashed lines).
Effect of Insulin on Inhibitory M2 Muscarinic Receptors on Vagus Nerves in Obese Rats
The M2 agonist pilocarpine inhibited vagally induced bronchoconstriction in a dose-dependent manner in obese-prone and obese-resistant rats on a high-fat diet (Figure 6A), demonstrating the inhibitory effect of neuronal M2 muscarinic receptors. The inhibitory effect of pilocarpine was significantly attenuated in obese-prone rats (Figure 6A, closed circles) versus obese-resistant rats (Figure 6A, closed triangles), demonstrating dysfunction of neuronal M2 muscarinic receptors in obese-prone rats on a high-fat diet. In these rats, STZ markedly enhanced the ability of pilocarpine to inhibit vagally induced bronchoconstriction (Figure 6B, closed circles with solid line), so that it was no longer different from the effect of pilocarpine in obese-resistant rats on a high-fat diet, indicating restored M2 receptor function (Figures 6A and 6C, closed triangle with solid line). However, replacing insulin in these STZ-treated animals reversed the effect of STZ, so that M2 receptors were again dysfunctional (Figure 6B, open circles with dashed line).
Figure 6.

Insulin decreased neuronal M2 muscarinic receptor function in obese-prone and obese-resistant rats on a high-fat diet. Neuronal M2 muscarinic receptor function was measured as the ability of pilocarpine, a selective muscarinic agonist, to inhibit vagally induced bronchoconstriction. Pilocarpine inhibited vagally induced bronchoconstriction in a dose-related manner, but the effect was significantly less in obese-prone (A, closed circles) than in obese-resistant (A, closed triangles) rats on a high-fat diet, demonstrating decreased function of inhibitory neuronal M2 muscarinic receptor in obese-prone rats on a high-fat diet. Reducing insulin with STZ in obese-prone rats (B, open circles with solid line) significantly enhanced the ability of pilocarpine to inhibit vagally induced bronchoconstriction. Acute administration of insulin to STZ-treated rats (B, dashed line) reversed the effect of STZ. In obese-resistant rats, reducing insulin with STZ (C, open triangles with solid line) did not change the ability of pilocarpine to inhibit vagally induced bronchoconstriction. However, in these rats, supplemental insulin significantly reduced the ability of pilocarpine to inhibit vagally induced bronchoconstriction (C, dashed line), similar to obese-prone rats on a high-fat diet (A, closed circle). Each point represents the mean, and vertical bars show SEM. *P < 0.05.
In contrast to the obese-prone rats on a high-fat diet, STZ had no additional effect on the already robust response to pilocarpine in obese-resistant rats (Figure 6C, open triangles with solid line). However, insulin administration to obese-resistant rats significantly reduced the ability of pilocarpine to inhibit vagally induced bronchoconstriction (Figure 6C, open triangles with dashed line), so that it was similar to obese-prone rats on a high-fat diet (compare open triangles/dashed line in Figure 6C with closed circles/solid line in Figures 6A or 6B).
Effect of Insulin on Contraction of Human Trachea
EFS of isolated human tracheal smooth muscle caused frequency-dependent contraction, which was significantly potentiated by 10−6 M insulin (Figure 7A). EFS-induced contractions in the absence or presence of insulin were blocked by 1 μM tetrodotoxin or by 1 μM atropine (data not shown), confirming that contraction was mediated via ACh release from nerves onto muscarinic receptors in the absence and presence of insulin.
Figure 7.
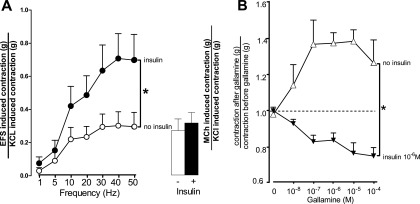
In human airway smooth muscle, insulin potentiated contraction induced by electrical field stimulation (EFS) but not by methacholine (MCh) (A). Human tracheal smooth muscle strips were incubated in organ baths in the absence and presence of insulin (10−6 M). Contraction was measured in response to EFS (100 V, 0.2 ms, for 5 s duration, 1–50 Hz at 1-min intervals). Insulin significantly potentiated EFS-induced contractions (closed circles). The maximum EFS-induced contraction at 50 Hz in the absent of insulin (open circle) was matched by 3 μM MCh (open bar). Insulin did not potentiate MCh-induced contraction (closed bar). Data are expressed as the EFS- or MCh-induced contraction normalized to KCl-induced contraction. Insulin inhibited human airway neuronal M2 muscarinic receptor function in vitro (B). Neuronal M2 muscarinic receptor function was measured as the ability of the selective M2 receptor antagonist gallamine to potentiate contraction induced by EFS. Human tracheal smooth muscle strips were incubated in organ baths in the absence and presence of insulin (10−6 M). Contraction was measured in response to EFS (30 Hz, 100 V, 0.2-ms pulse duration, for 5 s) at 1-minute intervals. Once reproducible contractions were obtained (2.0 ± 1.2 g in the absence of insulin and 1.4 ± 0.5 g in the presence of insulin), the M2 muscarinic antagonist gallamine was added to the baths. In the absence of insulin, gallamine potentiated EFS-induce contractions in a dose-related manner, demonstrating functional M2 muscarinic receptors on nerves in human trachea (open triangles). Insulin completely blocked the ability of gallamine to potentiate EFS-induced contractions (closed triangles), demonstrating that neuronal M2 muscarinic receptor function in human trachea was blocked by insulin. Data were expressed as the ratio of contraction after gallamine divided by contraction before gallamine. Each point represents the mean, and vertical bars show SEM (n = 5 donors). *P < 0.05.
In the absence of insulin, contraction induced by the muscarinic receptor agonist MCh (3 μM) (Figure 7A, open bar) was approximately equal to the maximum contractions induced by EFS of isolated human tracheal smooth muscle (Figure 7A, open circles). Although insulin significantly potentiated contractions induced by EFS (Figure 7A, closed circles), it had no significant effect on MCh-induced contraction (Figure 7A, closed bar). Thus, M3 muscarinic receptor–mediated contraction of airway smooth muscle was not responsible for the increased contraction induced by EFS; nor did insulin change the resting baseline tension of human trachea in the absence of MCh or EFS (in the absence of insulin, 0.64 ± 0.07 g; in the presence of insulin, 0.63 ± 0.08 g [n = 5]). Insulin did not change the contraction induced by histamine (10−7–10−4 M) or serotonin (10−8–10−2 M) (data not shown).
Effect of Insulin on Inhibitory M2 Muscarinic Receptors on Nerves in Human Trachea
In the absence of insulin, the M2 antagonist gallamine potentiated EFS-induced contractions of human trachea in a concentration-dependent manner (Figure 7B, open triangles) by blocking inhibitory M2 muscarinic receptors on the parasympathetic nerves. Insulin blocked the ability of gallamine to potentiate EFS-induced contractions (Figure 7B, closed triangles). Thus, insulin inhibited neuronal M2 muscarinic receptor function in human trachea.
Discussion
We have used a polygenic model of diet-induced obesity to investigate the role of insulin in obesity-related airway hyperresponsiveness to vagus nerve stimulation. We demonstrated in vivo that bronchoconstriction in response to electrical stimulation of both vagus nerves was dramatically increased in obese-prone animals fed a high-fat but not in obese-prone rats on a low-fat diet or obese-resistant rats (Figure 2). Obese-prone rats fed a low-fat diet gained significant weight, approaching that of the obese-prone rats on a high-fat diet (Figure 3), but did not become hyperresponsive, showing that airway hyperresponsiveness to vagus nerve stimulation was independent of weight. The obese-prone rats on a low-fat diet also had increased body fat but did not have increased airway responsiveness to vagus nerve stimulation, showing that airway hyperresponsiveness to vagus nerve stimulation was independent of body fat. Obese-resistant rats on a high-fat diet did not have increased airway response to vagus nerve stimulation, showing that airway hyperresponsiveness to vagus nerve stimulation was independent of diet. The only factor that was unique to the obese-prone rats on a high-fat diet was increased insulin (Figure 4).
In sharp contrast to the dramatic increase in vagally induced bronchoconstriction in obese-prone rats on a high-fat diet (Figure 2A), bronchoconstriction in response to intravenous ACh was not significantly different among any of the groups (Figures 2C and 2D). Bronchoconstriction induced by intravenous ACh was mediated only via activation of M3 muscarinic receptors on airway smooth muscle since both vagus nerves were cut to remove the vagal reflex (31, 32). It is unclear whether the function of M3 receptors on airway smooth muscle is different between obese-prone and obese-resistant rats because we were unable to use a full concentration of ACh. The maximum dose of ACh used in vivo was 10 μg/kg (intravenously); higher doses caused lethal polymorphic ventricular tachycardia in the obese rats on a high-fat diet. Thus, although our data clearly show loss of function of inhibitory M2 muscarinic receptors, which increases ACh release, we cannot conclusively exclude an additional effect of altered M3 receptors on the smooth muscle in obese rats in vivo. In this context it is worth noting that in human airways treatment with insulin caused M2 receptor dysfunction and an increased response to nerve stimulation without changing M3 receptor function on the smooth muscle (Figure 7A).
Release of ACh from parasympathetic nerves is normally locally controlled by inhibitory neuronal M2 muscarinic receptors (16–20), which were initially described on nerves supplying lungs in guinea pigs (19) and have subsequently been described in the parasympathetic nerves supplying the lungs of all species studied thus far (22, 33–38), including humans. Loss of M2 receptor function has been described in asthma (21, 23) and is a common feature of many different animal models of airway hyperresponsiveness, including acute infection with parainfluenza virus (24), sensitization and challenge with antigen (26, 33), acute exposure to ozone (25), and acute exposure to organophosphate pesticides (39). M2 muscarinic receptor function is measured in vivo in this study using the selective agonist pilocarpine. It has been shown that in the presence of functional M2 receptors, pilocarpine inhibits parasympathetic nerve–mediated bronchoconstriction in a dose-related manner (19). Our data show that pilocarpine inhibits vagally induced bronchoconstriction in obese-resistant rats fed a high-fat diet (Figure 6), demonstrating that they have functional M2 muscarinic receptors. In contrast, pilocarpine did not inhibit vagally induced bronchoconstriction in obese-prone rats fed a high-fat diet (Figure 6), demonstrating that neuronal M2 receptors can no longer be stimulated with exogenous agonists. The airways of obese-prone rats fed a high-fat diet also had increased response to vagus nerve stimulation (Figure 2), suggesting that, similar to antigen challenge and other models of airway hyperresponsiveness, increased bronchoconstriction in obese-prone rats on a high-fat diet is accompanied by loss of neuronal M2 receptor function.
The ability of endogenous ACh to stimulate neuronal M2 muscarinic receptors and inhibit subsequent transmitter release can be measured in vitro using the selective M2 antagonist gallamine (19). In the presence of functional M2 receptors, gallamine blocks the ability of neuronal M2 muscarinic receptors to inhibit ACh (19), thus potentiating airway smooth muscle contraction induced by nerve stimulation (Figure 7B). However, in the presence of insulin, gallamine lost its ability to potentiate neurally induced muscle contraction (Figure 7B). These data demonstrate that insulin caused dysfunction of neuronal M2 muscarinic receptors, thus removing local inhibitory control of endogenous ACh release and resulting in increased smooth muscle contraction. Therefore, EFS-induced contraction of human tracheal smooth muscle appears to be potentiated by the presence of insulin, in comparison to when insulin is absent (Figure 7A). These data indicate that, in the presence of insulin, endogenous ACh does not effectively stimulate inhibitory neuronal M2 muscarinic receptors, resulting in potentiation of nerve-induced contraction of human tracheal smooth muscle.
In vivo, insulin was significantly increased in obese-prone rats on a high-fat diet (Figure 4). Our data show that suppressing insulin with STZ completely reversed airway hyperresponsiveness to vagus nerve stimulation (Figure 5) and prevented the loss of neuronal M2 muscarinic receptor function (Figure 6) in obese-prone rats on a high-fat diet. Replacing insulin to physiological levels (Figure 4B), either chronically or acutely, not only restored airway hyperresponsiveness to vagus nerve stimulation in obese rats but also induced hyperresponsiveness to vagus nerve stimulation de novo in lean rats (Figure 5). Supplementing insulin reduced neuronal M2 muscarinic receptor function, measured using pilocarpine, in STZ-treated obese and lean rats (Figure 6). Thus, increased insulin induced neuronal M2 receptor dysfunction and airway hyperresponsiveness to vagus nerve stimulation even in the absence of obesity.
Our data that insulin can inhibit M2 receptors and that STZ reverses this effect are consistent with our previous studies showing that insulin depletion increases M2 muscarinic receptor function and expression (30, 40). In these previous studies, done in nonselected Sprague Dawley rats on a noncontrolled diet, increased function of M2 receptor on airway nerves decreased the bronchoconstriction response to vagal stimulation, an effect we did not see in obese-resistant rats on a controlled diet. Likewise, radioligand binding studies show increased M2 receptors in the vas deferens of rats after depleting insulin with STZ (28), and again this effect was reversed by exogenous insulin. M2 receptors are the dominant muscarinic receptor in the heart and expression of M2 receptors in cultured rat atrial myocytes was also reduced by insulin (41). Our data show that insulin reduced M2 receptor function rapidly; we were able to decrease M2 receptor function after only 2 hours incubation with insulin.
Circulating insulin is doubled in obese-prone versus obese-resistant rats on a high-fat diet (Figure 4). Insulin might interact directly with M2 muscarinic receptors or via insulin receptor pathways. However, for the latter, the nerves must remain insulin sensitive, whereas other tissues are insulin resistant. This is possible because insulin resistance is tissue specific (42). This was demonstrated in obese mice, which developed insulin resistance in liver before white adipose tissue, whereas skeletal muscle cells of these mice retained insulin sensitivity (43). Thus, it is possible that parasympathetic nerves, like skeletal muscle, remain responsive to high insulin levels, whereas liver cells and adipocytes develop insulin resistance in obese individuals.
Although asthma is usually associated with inflammation of the lungs, human studies have shown that obesity-related asthma is independent of airway inflammation (44–46). For example, Dixon and colleagues reported that weight loss by bariatric surgery reduces airway hyperresponsiveness in obese patients with asthma but does not decrease the proportion of lymphocytes in bronchoalveolar lavage fluid or T-cell production of inflammatory cytokines (46). Furthermore, sputum eosinophils are not higher in obese patients with asthma than in nonobese patients with asthma (7, 45). Leptin, a proinflammatory adipokine, is also reported to be caused by obesity-related airway hyperresponsiveness because airway hyperresponsiveness is potentiated in obese mice depleted of leptin (ob/ob mice) or leptin receptors (db/db mice) (47). This may be explained by the finding that leptin deficiency in the central neural system increases parasympathetic nerve activity (48) regardless of whether the bronchi are inflamed. This is consistent with our data that vagally induced bronchoconstriction was increased in obese rats without a concomitant increase in inflammatory cells compared with nonobese rats. Thus, obesity-induced airway hyperresponsiveness is independent of inflammation, which may explain why patients with this specific phenotype of asthma respond poorly to anti-inflammatory treatments (1).
The incidence and severity of asthma are increased in obese patients. Previous studies show that dysfunctional M2 receptor is the mechanism of airway hyperresponsiveness in animals that are antigen challenged (26, 33), exposed to ozone (25), infected with virus (24), or exposed to organophosphate pesticides (39) and in humans with asthma (21–23). Here we show that insulin also inhibits neuronal M2 receptor function, resulting in airway hyperresponsiveness to nerve stimulation. Because the parasympathetic nerves provide the dominant autonomic control of airway smooth muscle (49), any decrease in neuronal M2 receptor function may predispose individuals to increased reflex-mediated bronchoconstriction, contributing to asthma in obesity.
Asthma is a heterogeneous disease with complex phenotypes (50). Obese adult-onset asthma is characterized by less allergy and less inflammation and has been identified as a “Th2-low” asthma phenotype (50). Our data demonstrate that, in the absence of inflammation, hyperinsulinemia potentiates vagally induced bronchoconstriction by decreasing the function of inhibitory M2 muscarinic receptors on parasympathetic nerves, resulting in increased ACh release and airway hyperresponsiveness. If this mechanism is functional in obese individuals, our data suggest that anticholinergic drugs may be more effective than anti-inflammatory drugs in the treatment of this unique asthma phenotype.
Acknowledgments
Acknowledgments
The authors thank the members of the Pacific Northwest Transplant Bank, Portland, Oregon, for procuring and supplying tracheas from organ donors and Dr. Dan Marks for body fat measurement.
Footnotes
This work was supported by a grant from The Oregon Health and Science University Medical Research Foundation (Z.N.) and by National Institutes of Health grants HL71795 (D.B.J.), AI92210 (D.B.J.), HL113023 (D.B.J.), AR061567 (D.B.J.), HL55543 (A.D.F.), and ES14601 (A.D.F.).
Originally Published in Press as DOI: 10.1165/rcmb.2013-0452OC on March 8, 2014
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Mosen DM, Schatz M, Magid DJ, Camargo CA., JrThe relationship between obesity and asthma severity and control in adults J Allergy Clin Immunol 2008122507–511.e506 [DOI] [PubMed] [Google Scholar]
- 2.Nystad W, Meyer HE, Nafstad P, Tverdal A, Engeland A. Body mass index in relation to adult asthma among 135,000 Norwegian men and women. Am J Epidemiol. 2004;160:969–976. doi: 10.1093/aje/kwh303. [DOI] [PubMed] [Google Scholar]
- 3.Romieu I, Avenel V, Leynaert B, Kauffmann F, Clavel-Chapelon F. Body mass index, change in body silhouette, and risk of asthma in the e3n cohort study. Am J Epidemiol. 2003;158:165–174. doi: 10.1093/aje/kwg131. [DOI] [PubMed] [Google Scholar]
- 4.Assad N, Qualls C, Smith LJ, Arynchyn A, Thyagarajan B, Schuyler M, Jacobs DR, Jr, Sood A. Body mass index is a stronger predictor than the metabolic syndrome for future asthma in women: the longitudinal Cardia study. Am J Respir Crit Care Med. 2013;188:319–326. doi: 10.1164/rccm.201303-0457OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brumpton BM, Camargo CA, Jr, Romundstad PR, Langhammer A, Chen Y, Mai XM. Metabolic syndrome and incidence of asthma in adults: the HUNT study. Eur Respir J. 2013;42:1495–1502. doi: 10.1183/09031936.00046013. [DOI] [PubMed] [Google Scholar]
- 6.Black MH, Zhou H, Takayanagi M, Jacobsen SJ, Koebnick C. Increased asthma risk and asthma-related health care complications associated with childhood obesity. Am J Epidemiol. 2013;178:1120–1128. doi: 10.1093/aje/kwt093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lessard A, Turcotte H, Cormier Y, Boulet LP. Obesity and asthma: a specific phenotype? Chest. 2008;134:317–323. doi: 10.1378/chest.07-2959. [DOI] [PubMed] [Google Scholar]
- 8.Thuesen BH, Husemoen LL, Hersoug LG, Pisinger C, Linneberg A. Insulin resistance as a predictor of incident asthma-like symptoms in adults. Clin Exp Allergy. 2009;39:700–707. doi: 10.1111/j.1365-2222.2008.03197.x. [DOI] [PubMed] [Google Scholar]
- 9.Husemoen LL, Glumer C, Lau C, Pisinger C, Morch LS, Linneberg A. Association of obesity and insulin resistance with asthma and aeroallergen sensitization. Allergy. 2008;63:575–582. doi: 10.1111/j.1398-9995.2007.01613.x. [DOI] [PubMed] [Google Scholar]
- 10.Cottrell L, Neal WA, Ice C, Perez MK, Piedimonte G. Metabolic abnormalities in children with asthma. Am J Respir Crit Care Med. 2011;183:441–448. doi: 10.1164/rccm.201004-0603OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arshi M, Cardinal J, Hill RJ, Davies PS, Wainwright C. Asthma and insulin resistance in children. Respirology. 2010;15:779–784. doi: 10.1111/j.1440-1843.2010.01767.x. [DOI] [PubMed] [Google Scholar]
- 12.Al-Shawwa BA, Al-Huniti NH, DeMattia L, Gershan W. Asthma and insulin resistance in morbidly obese children and adolescents. J Asthma. 2007;44:469–473. doi: 10.1080/02770900701423597. [DOI] [PubMed] [Google Scholar]
- 13.Terzano C, Morano S, Ceccarelli D, Conti V, Paone G, Petroianni A, Graziani E, Carnovale A, Fallarino M, Gatti A, et al. Effect of insulin on airway responsiveness in patients with type 2 diabetes mellitus: a cohort study. J Asthma. 2009;46:703–707. doi: 10.1080/02770900903056203. [DOI] [PubMed] [Google Scholar]
- 14.Mudaliar S, Henry RR. Inhaled insulin in patients with asthma and chronic obstructive pulmonary disease. Diabetes Technol Ther. 2007;9:S83–S92. doi: 10.1089/dia.2007.0217. [DOI] [PubMed] [Google Scholar]
- 15.Ceglia L, Lau J, Pittas AG. Meta-analysis: efficacy and safety of inhaled insulin therapy in adults with diabetes mellitus. Ann Intern Med. 2006;145:665–675. doi: 10.7326/0003-4819-145-9-200611070-00009. [DOI] [PubMed] [Google Scholar]
- 16.Jacoby DB, Xiao HQ, Lee NH, Chan-Li Y, Fryer AD. Virus- and interferon-induced loss of inhibitory m2 muscarinic receptor function and gene expression in cultured airway parasympathetic neurons. J Clin Invest. 1998;102:242–248. doi: 10.1172/JCI1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacoby DB, Yost BL, Kumaravel B, Chan-Li Y, Xiao HQ, Kawashima K, Fryer AD. Glucocorticoid treatment increases inhibitory m(2) muscarinic receptor expression and function in the airways. Am J Respir Cell Mol Biol. 2001;24:485–491. doi: 10.1165/ajrcmb.24.4.4379. [DOI] [PubMed] [Google Scholar]
- 18.Baker DG, Don HF, Brown JK. Direct measurement of acetylcholine release in guinea pig trachea. Am J Physiol. 1992;263:L142–L147. doi: 10.1152/ajplung.1992.263.1.L142. [DOI] [PubMed] [Google Scholar]
- 19.Fryer AD, Maclagan J. Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol. 1984;83:973–978. doi: 10.1111/j.1476-5381.1984.tb16539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaber LC, Fryer AD, Maclagan J. Neuronal muscarinic receptors attenuate vagally-induced contraction of feline bronchial smooth muscle. Br J Pharmacol. 1985;86:723–728. doi: 10.1111/j.1476-5381.1985.tb08951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol. 1989;67:2461–2465. doi: 10.1152/jappl.1989.67.6.2461. [DOI] [PubMed] [Google Scholar]
- 22.Minette PA, Barnes PJ. Prejunctional inhibitory muscarinic receptors on cholinergic nerves in human and guinea pig airways. J Appl Physiol. 1988;64:2532–2537. doi: 10.1152/jappl.1988.64.6.2532. [DOI] [PubMed] [Google Scholar]
- 23.Ayala LE, Ahmed T. Is there loss of protective muscarinic receptor mechanism in asthma? Chest. 1989;96:1285–1291. doi: 10.1378/chest.96.6.1285. [DOI] [PubMed] [Google Scholar]
- 24.Fryer AD, Jacoby DB. Parainfluenza virus infection damages inhibitory m2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol. 1991;102:267–271. doi: 10.1111/j.1476-5381.1991.tb12164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultheis AH, Bassett DJ, Fryer AD. Ozone-induced airway hyperresponsiveness and loss of neuronal m2 muscarinic receptor function. J Appl Physiol. 1994;76:1088–1097. doi: 10.1152/jappl.1994.76.3.1088. [DOI] [PubMed] [Google Scholar]
- 26.Fryer AD, Wills-Karp M. Dysfunction of m2-muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol. 1991;71:2255–2261. doi: 10.1152/jappl.1991.71.6.2255. [DOI] [PubMed] [Google Scholar]
- 27.Belmonte KE, Fryer AD, Costello RW. Role of insulin in antigen-induced airway eosinophilia and neuronal m2 muscarinic receptor dysfunction. J Appl Physiol. 1998;85:1708–1718. doi: 10.1152/jappl.1998.85.5.1708. [DOI] [PubMed] [Google Scholar]
- 28.Kamai T, Fukumoto Y, Gousse A, Yoshida M, Davenport TA, Weiss RM, Latifpour J. Diabetes-induced alterations in the properties of muscarinic cholinergic receptors in rat vas deferens. J Urol. 1994;152:1017–1021. doi: 10.1016/s0022-5347(17)32646-0. [DOI] [PubMed] [Google Scholar]
- 29.Nie Z, Scott GD, Weis PD, Itakura A, Fryer AD, Jacoby DB. Role of tnf-alpha in virus-induced airway hyperresponsiveness and neuronal m(2) muscarinic receptor dysfunction. Br J Pharmacol. 2011;164:444–452. doi: 10.1111/j.1476-5381.2011.01393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coulson FR, Jacoby DB, Fryer AD. Insulin regulates neuronal m2 muscarinic receptor function in the ileum of diabetic rats. J Pharmacol Exp Ther. 2004;308:760–766. doi: 10.1124/jpet.103.057570. [DOI] [PubMed] [Google Scholar]
- 31.Wagner EM, Jacoby DB. Methacholine causes reflex bronchoconstriction. J Appl Physiol. 1999;86:294–297. doi: 10.1152/jappl.1999.86.1.294. [DOI] [PubMed] [Google Scholar]
- 32.Costello RW, Evans CM, Yost BL, Belmonte KE, Gleich GJ, Jacoby DB, Fryer AD. Antigen-induced hyperreactivity to histamine: role of the vagus nerves and eosinophils. Am J Physiol. 1999;276:L709–L714. doi: 10.1152/ajplung.1999.276.5.L709. [DOI] [PubMed] [Google Scholar]
- 33.Larsen GL, Fame TM, Renz H, Loader JE, Graves J, Hill M, Gelfand EW. Increased acetylcholine release in tracheas from allergen-exposed ige-immune mice. Am J Physiol. 1994;266:L263–L270. doi: 10.1152/ajplung.1994.266.3.L263. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto S, Nagayama T, Kanno T, Yamasaki M, Shimizu T. Evidence for the presence of function of the inhibitory m2 receptors in the rabbit airways and lungs. J Auton Nerv Syst. 1995;53:126–136. doi: 10.1016/0165-1838(94)00168-j. [DOI] [PubMed] [Google Scholar]
- 35.Wang ZW, Yu MF, Robinson NE, Derksen FJ. Acetylcholine release from airway cholinergic nerves in horses with heaves, an airway obstructive disease. Am J Respir Crit Care Med. 1995;151:830–835. doi: 10.1164/ajrccm/151.3_Pt_1.830. [DOI] [PubMed] [Google Scholar]
- 36.Aas P, Maclagan J. Evidence for prejunctional m2 muscarinic receptors in pulmonary cholinergic nerves in the rat. Br J Pharmacol. 1990;101:73–76. doi: 10.1111/j.1476-5381.1990.tb12091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito Y, Yoshitomi T. Autoregulation of acetylcholine release from vagus nerve terminals through activation of muscarinic receptors in the dog trachea. Br J Pharmacol. 1988;93:636–646. doi: 10.1111/j.1476-5381.1988.tb10321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes LB, Fryer AD, Hirshman CA. M2 muscarinic receptors inhibit isoproterenol-induced relaxation of canine airway smooth muscle. J Pharmacol Exp Ther. 1992;262:119–126. [PubMed] [Google Scholar]
- 39.Fryer AD, Lein PJ, Howard AS, Yost BL, Beckles RA, Jett DA. Mechanisms of organophosphate insecticide-induced airway hyperreactivity. Am J Physiol Lung Cell Mol Physiol. 2004;286:L963–L969. doi: 10.1152/ajplung.00343.2003. [DOI] [PubMed] [Google Scholar]
- 40.Belmonte KE, Jacoby DB, Fryer AD. Increased function of inhibitory neuronal m2 muscarinic receptors in diabetic rat lungs. Br J Pharmacol. 1997;121:1287–1294. doi: 10.1038/sj.bjp.0701274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pathak A, Smih F, Galinier M, Verwaerde P, Rouet P, Philip-Couderc P, Montastruc JL, Senard JM. Insulin downregulates m(2)-muscarinic receptors in adult rat atrial cardiomyocytes: a link between obesity and cardiovascular complications. Int J Obes (Lond) 2005;29:176–182. doi: 10.1038/sj.ijo.0802751. [DOI] [PubMed] [Google Scholar]
- 42.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16:144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Kleemann R, van Erk M, Verschuren L, van den Hoek AM, Koek M, Wielinga PY, Jie A, Pellis L, Bobeldijk-Pastorova I, Kelder T, et al. Time-resolved and tissue-specific systems analysis of the pathogenesis of insulin resistance. PLoS ONE. 2010;5:e8817. doi: 10.1371/journal.pone.0008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farah CS, Kermode JA, Downie SR, Brown NJ, Hardaker KM, Berend N, King GG, Salome CM. Obesity is a determinant of asthma control, independent of inflammation and lung mechanics. Chest. 2011;140:659–666. doi: 10.1378/chest.11-0027. [DOI] [PubMed] [Google Scholar]
- 45.van Veen IH, Ten Brinke A, Sterk PJ, Rabe KF, Bel EH. Airway inflammation in obese and nonobese patients with difficult-to-treat asthma. Allergy. 2008;63:570–574. doi: 10.1111/j.1398-9995.2007.01597.x. [DOI] [PubMed] [Google Scholar]
- 46.Dixon AE, Pratley RE, Forgione PM, Kaminsky DA, Whittaker-Leclair LA, Griffes LA, Garudathri J, Raymond D, Poynter ME, Bunn JY, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation J Allergy Clin Immunol 2011128508–515.e502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnston RA, Zhu M, Rivera-Sanchez YM, Lu FL, Theman TA, Flynt L, Shore SA. Allergic airway responses in obese mice. Am J Respir Crit Care Med. 2007;176:650–658. doi: 10.1164/rccm.200702-323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arteaga-Solis E, Zee T, Emala CW, Vinson C, Wess J, Karsenty G. Inhibition of leptin regulation of parasympathetic signaling as a cause of extreme body weight-associated asthma. Cell Metab. 2013;17:35–48. doi: 10.1016/j.cmet.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nadel JA, Barnes PJ. Autonomic regulation of the airways. Annu Rev Med. 1984;35:451–467. doi: 10.1146/annurev.me.35.020184.002315. [DOI] [PubMed] [Google Scholar]
- 50.Wenzel SE. Complex phenotypes in asthma: current definitions. Pulm Pharmacol Ther. 2013;26:710–715. doi: 10.1016/j.pupt.2013.07.003. [DOI] [PubMed] [Google Scholar]