

Background: Bacterial toxins, including P. aeruginosa exotoxin A (PE), are valuable tools to dissect biological processes.
Results: A genome-wide genetic screen identifies several novel host factors used by PE, including GPR107.
Conclusion: Bacterial toxins can help identify novel host components involved in key intracellular trafficking steps.
Significance: GPR107 may be a receptor that associates with G-proteins at the Golgi to regulate membrane transport.
Keywords: Bacterial Toxin, Furin, G-protein-coupled Receptor (GPCR), Intracellular Trafficking, Sortase A, GPR107, KBM7, Pseudomonas aeruginosa Exotoxin A, Haploid Genetic Screen
Abstract
A number of toxins, including exotoxin A (PE) of Pseudomonas aeruginosa, kill cells by inhibiting protein synthesis. PE kills by ADP-ribosylation of the translation elongation factor 2, but many of the host factors required for entry, membrane translocation, and intracellular transport remain to be elucidated. A genome-wide genetic screen in human KBM7 cells was performed to uncover host factors used by PE, several of which were confirmed by CRISPR/Cas9-gene editing in a different cell type. Several proteins not previously implicated in the PE intoxication pathway were identified, including GPR107, an orphan G-protein-coupled receptor. GPR107 localizes to the trans-Golgi network and is essential for retrograde transport. It is cleaved by the endoprotease furin, and a disulfide bond connects the two cleaved fragments. Compromising this association affects the function of GPR107. The N-terminal region of GPR107 is critical for its biological function. GPR107 might be one of the long-sought receptors that associates with G-proteins to regulate intracellular vesicular transport.
Introduction
Several bacterial pathogens cause disease through the activities of secreted toxin proteins with targets located within the eukaryotic cells. Although cellular receptors and the biochemical reactions leading to cell death have been elucidated for a number of such toxins, the precise steps of their transport from the cell surface to the target sites of action have not been well defined. In eukaryotic cells, transport of proteins between the various membranous compartments is mediated by cargo vesicles that bud off from the donor organelle and fuse with the appropriate acceptor organelle (1). These processes serve not only to maintain cellular homeostasis but also allow cells to communicate with the outside environment (2). Bacterial toxins exploit these very same pathways where they rely on receptor-mediated endocytosis and vesicular trafficking to reach their final destinations (3). Consequently, such toxins are useful probes to explore the cell biology that underlies membrane trafficking.
Pseudomonas aeruginosa exotoxin A (PE)2 is a polypeptide of 66 kDa that contains three structural subdomains (4, 5). After entering host cells via receptor-mediated endocytosis, PE is processed by furin and exerts its cytotoxicity by virtue of its ADP-ribosyltransferase activity; it ADP-ribosylates the diphthamide residue of eukaryotic translation elongation factor 2 (eEF2). This causes a block in protein synthesis and leads to cell death (6). Although PE must cross a biological membrane to reach the cytosol and its substrates (7, 8), only a partial list of the host proteins involved in this process is known.
Vesicular transport is a process that involves several classes of proteins such as SNAREs, the GARP complex, cytoskeletal proteins, and GTPases (9). Members of the small GTPases of the Rab superfamily localize to various intracellular compartments and regulate many aspects of membrane trafficking (10, 11). The other class of GTPases are the heterotrimeric G-proteins, which also contribute to vesicular trafficking (12). Membrane vesiculation (13, 14) and cargo trafficking (15) at the TGN are regulated by Gβγ subunits through activation of the serine/threonine protein kinase D (PKD) (16). Intracellular transport and secretion of heparan sulfate proteoglycan by epithelial cells involve the pertussis toxin-sensitive Gαi3, localized to the Golgi apparatus (17). No Golgi-resident GPCRs associated with these G-proteins have been identified.
A haploid genetic screen was performed in KBM7 cells, a myeloid leukemia cell line with a haploid karyotype except for chromosome 8, to identify host factors required for entry and trafficking of PE. Several host factors not previously implicated in intoxication by PE were identified, including GPR107, an orphan GPCR. GPR107 localizes to the TGN and is cleaved by furin, also identified as a hit in the screen. GPR107 is involved in retrograde protein transport and may be a long-sought receptor that associates with G-proteins to regulate intracellular membrane trafficking.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit anti-TGN46 and rabbit anti-Giantin were from Abcam. Rabbit anti-furin was from Santa Cruz Biotechnology. The rat monoclonal anti-HA-coupled beads were from Roche Applied Science, and anti-HA-Alexa488 was from MBL. Streptavidin-HRP was from Fisher. Fluorophore-conjugated secondary antibodies were from Invitrogen.
Cloning, Expression, and Purification of Exotoxin A
The coding sequence for PE (GenBankTM accession number AAB59097) was amplified by PCR from P. aeruginosa genomic DNA (18) and cloned into pMMB67H vector using HindIII and EcoRI restriction sites. Alternatively, PE that carries a sortase recognition motif, LPETG, near its C terminus followed by His6 was cloned into pMMB67H vector using the same restriction enzymes. The plasmids were then introduced into PA103-EA, a nonvirulent P. aeruginosa strain that is deficient in endogenous PE production. PA103-EA carrying the plasmids were grown at 37 °C in LB media supplemented with 1% glycerol and 200 μg/ml ampicillin until the A600 reached 0.6. Protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 18 h at 37 °C, and cells were pelleted by centrifugation. The supernatants that contain the toxin were centrifuged again at 10,000 rpm for 2 h, filtered to further remove any remaining debris, and concentrated using the 30K Amicon concentrator (Millipore). For the toxin that carries the sortase recognition motif (PE-LPETG-His6) (19, 20), the concentrate was applied to a nickel-nitrilotriacetic acid column equilibrated with 50 mm Tris-HCl, 150 mm NaCl, and 10 mm imidazole, pH 8.0. The column was washed with 10 column volumes of buffer, and the protein was eluted with 50 mm Tris-HCl, 150 mm NaCl, and 500 mm imidazole, pH 8.0. The proteins were further purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare) and eluted with 50 mm Tris-HCl, 150 mm NaCl, and 10% glycerol, pH 7.4. Fractions containing the correct toxin were pooled, concentrated, and stored at −80 °C.
Sortase-mediated Labeling of PE
Sortase A was expressed and purified as described previously (19, 20). Synthesis of the nucleophiles G3K(Atto647)-RDELK, G3K-(biotin)-RDELK, and G3K-biotin were done as described previously (21). Sortase-labeling reaction mix was prepared as follows: 25 μm PE-LPETG-His6, 50 μm sortase A, and 1 mm nucleophile in 50 mm Tris-HCl, 150 mm NaCl, and 10 mm CaCl2. The reaction was incubated at 37 °C for 16 h. The whole reaction mix was then loaded onto a nickel-nitrilotriacetic acid column to remove unreacted toxin and the enzyme (sortase A carries His6 at its C terminus). The flow-through that contains the PE conjugated with the nucleophile was then concentrated and stored at 4 °C for short term storage or at −80 °C for long term storage.
Haploid Genetic Screen and Isolation of the Mutant Clone
The construction of gene-trap (22) viral vectors, generation of mutant KBM7 libraries, mapping of GT integration sites, and general screening approach were done as described previously (23). Briefly, about 100 million mutagenized KBM7 cells were exposed to 50 ng/ml PE for 10 days. The survivors were pooled and expanded for a few days. Genomic DNA was isolated, and inverse PCR was performed using primer sequences flanking the retroviral insertion sites followed by Illumina sequencing. The statistical significance of insertions at a given gene in the PE-treated population was calculated by comparing the number of inactivating insertions to those in the untreated control data set. To isolate the GPR107GT clone, cells were FACS-sorted in 96-well dishes and grown until confluent. Genomic DNA from the individual clones was extracted using a genomic DNA isolation kit (Qiagen). Genomic insertions were identified by inverse PCR using a forward primer located within the gene-trap (5′-CTCGGTGGAACCTCCAAAT-3′) and a reverse primer designed to target the GPR107 gene. The gene-trap insertions were mapped by sequencing of the PCR product using the forward primer. RT-PCR analysis was performed to determine the absence of the GPR107 transcript in the isolated mutant cell line using SuperScriptTM III first-strand synthesis kit (Invitrogen). The following primers were used: 5′-ATGGCCGCTCTGGCGCCCGTCGGCT-3 and (5′-GGCCTTCTTGGTCATCAGTGC-3′). As a positive control, RT-PCR analysis of the GAPDH gene was performed.
Cell Culture and Virus Transduction
KBM7 and HeLa cells were grown in Iscove's modified Dulbecco's medium or DMEM supplemented with 10% heat-inactivated fetal serum, respectively, at 37 °C and 5% CO2. Cell lines stably overexpressing various versions of GPR107 constructs were generated by infecting with retroviruses expressing the corresponding cDNAs and were selected for G418 (0.8 mg/ml for HeLa and 1.2 mg/ml for GPR107GT cells). Of the three reported splice variants of GPR107 (24), we detected only the expression of isoform 2 (UniProt accession number Q5VW38-2).
Designing CRISPR Target Sequence and Prediction of Off-target Effects
Target sequences for CRISPR interference were designed as detailed in Ref. 25. The target sequence preceding the PAM motif was obtained from the region of the exon of the indicated genes (Table 1). Potential off-target effects of the target sequence were confirmed using the NCBI Homo sapiens Nucleotide BLAST.
TABLE 1.
CRISPR target sequences
| Gene | Target sequence |
|---|---|
| GPR107 | GGTGCCATCCTCTTCCCAG |
| FURIN | CGAGCCCAACCACATCACT |
| VPS53 | GTAAGAGGTCAGACGAACG |
| KDELR1 | AGTTCAAAGCTACTTACGA |
| TMEM110 | TCTTCCTGCAGGGGCTGCT |
| AP1M1 | CGGAACTACCGTGGCGACG |
| SCFD1 | TAGTTGATTTCGAAGATCC |
| OSTC | CCGACGGCATGTGCAACCA |
Procedure for Generating CRISPR RNA Lentivirus Vector
CRISPR gBlock was designed to incorporate into the restriction enzymatic site NheI/BamHI of CMV promoter-deleted pCDH-EF1-Hygro (SBI; CD515B-1) as follows: cacagtcagacagtgactcaGTGTCACAgctagcTTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCTGTTAGAGAGATAATTAGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAATACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGnnnnnnnnnnnnnnnnnnnGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTggatccTGTGCACAgtcagtcacagtcagtctac (where n is CRISPR target sequence). The gBlock was digested by NheI and BamHI restriction enzymes and incorporated into the pCDH vector linearized with the same restriction enzyme.
Genotyping of the CRISPR/Cas9-generated Mutant Cell Populations Using Surveyor Assay
Surveyor assay was performed as described previously (26, 27). Genomic DNA from treated and control crude HeLa cells was extracted. PCR was performed using specific primers (Table 2) under the following conditions: 94 °C for 2 min; 35× (98 °C for 10 s, 60 °C for 30 s, and 68 °C for 30 s); 68 °C for 2 min; hold at 4 °C. PCR product was loaded onto an ethidium bromide-stained agarose gel (3%) and purified. 500 ng of the purified PCR product were treated with Surveyor nuclease (Transgenomic) for 30 min and resolved using 3% agarose gel.
TABLE 2.
Primers used for the Surveyor assay
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GPR107 | AGCACGTGGGACTGGAAAGAATGC | TTAGAACTCTGTTAAATGCCAGCTCATGGTG |
| FURIN | TGCATCATCGACATCCTCACCGAGC | ATTCCTGCAACATGGGACAGTCCC |
| VPS53 | CAACAAGGCGATGCATCAGCCTGC | TGAGCGGTGCCACAGTTCACGG |
| KDELR1 | TCTGGACTCCTGCGTCTGAGGG | GCAGAAGGAAGCCTGTGAGAGGGT |
| TMEM110 | TACCGGCTCAGGCTTGGGATCC | TCACTCCTGTCTGGAGGCCACAG |
| AP1M1 | ATTCACACTGGGAGATGCTCTTCTCC | TCGTGCATTGTCTGCCTGTCTCCG |
| SCFD1 | CAGAACAGTTGAAGATCGTGTCTGCC | AACAATTCTGTTGTTCTGGTCATCTGTC |
| OSTC | ACCTCCGAGCTTTACGGATCTGCG | TGAGAACACGAATTCTCTTAAGACAGGGC |
Pulse-Chase Experiments, Furin, and Glycosidase Digestion
Pulse-chase experiments were performed as described previously (28). Briefly, HeLa cells stably expressing C-terminally HA-tagged GPR107WT, GPR107R182A, GPR107C109A/C228A, or GPR107Δ40–182 were grown in 10-cm culture dishes. For a given time point, 1 × 10-cm dish was used in all experiments. Cells were starved in methionine- and cysteine-free DMEM for 45 min, pulse-labeled with [35S]methionine/cysteine at 0.77 mCi/ml for 30 min, and chased for different time points in complete media containing 1 mm cold methionine/cysteine. Where indicated, cells were treated with 100 μg/ml brefeldin A (BFA) or 100 nm concanamycin A for 1 h prior to labeling and chased in the continuous presence of the drugs. At different time points during the chase, cells were harvested, lysed in Tris buffer (150 mm NaCl, 5 mm MgCl2, 25 mm Tris-HCl, pH 7.4) containing 0.5% Nonidet P-40 followed by immunoprecipitation with anti-HA-coupled beads. Typically, the immunoprecipitates were eluted with 100 μl of PBS containing 1% SDS. Another 900 μl of Tris buffer was added to get a 0.1% final concentration of SDS. Samples were then re-immunoprecipitated using anti-HA-coupled beads. Where indicated, immunoprecipitates were subjected to Endo H, PNGase F, or furin digestion according to the manufacturer's instructions (New England Biolabs). Immunoprecipitates were eluted with SDS sample buffer and resolved by SDS/-PAGE. Samples were visualized with autoradiography using DMSO/2,5-diphenyloxazole and exposed to Kodak XAR-5 film.
Microscopy
HeLa cells grown on coverslips were fixed with 4% paraformaldehyde in PBS for 30 min. Fixation was stopped by incubating the coverslips with 50 mm NH4Cl for 10 min. Samples were then blocked with binding buffer (0.1% saponin and 0.2% BSA in PBS) for 30 min and incubated with the first antibodies followed by secondary antibodies conjugated with Alexa Fluor. Images were captured using a confocal microscope with a 63× 1.40 N.A. of the Carl Zeiss Plan Apo oil objective and processed using Velocity and Adobe Photoshop software.
Generation of GPR107-depleted HeLa Cells
Lentiviral plasmids containing the shRNA against human GPR107 and the control (shGFP) were purchased from Open Biosystems. To generate lentiviruses, low passage HEK293T cells were transfected with these plasmids using Lipofectamine 2000 following the manufacturer's protocol. GPR107-knockdown HeLa cell lines were generated by infecting the cells with the lentiviruses and then selecting them in the presence of puromycin (1 μg/ml) for 1 week. In parallel, we transduced HeLa cells that overexpress HA-tagged GPR107 with the same viruses. Because there are no good antibodies, we determined the efficiency of the knockdown by Western blot analysis using anti-HA HRP.
Cell Viability Assay and Flow Cytometry
About 25 × 103 cells per well were seeded in 96-well dishes with clear flat bottoms (Costar) and treated with different concentrations of PE for 18 h in the case of HeLa cells or 48 h for KBM7 cells. Cell viability assay was performed using the CellTiter-Glo® luminescent cell viability assay kit (Promega) according to the manufacturer's protocol. This method determines cell viability based on quantitation of the ATP present, an indicator of metabolically active cells. ATP-based bioluminescence levels were measured using an EnVision plate reader (PerkinElmer Life Sciences).
For flow cytometry, cells were treated for 18 h with CDTs of different bacterial origin. The cells were harvested, washed with cold PBS, fixed with ethanol, stained with propidium iodide, and subjected to cytofluorometry (FACSCalibur, BD Biosciences). FlowJo was used to analyze the data. Intensity of fluorescence was measured, and the percent maximum was presented in the overlaid histograms.
Cholera Toxin Glycosylation Assay
Expression, purification, and generation of cholera toxin conjugated with the glycosylation motif (GGG-K(biotin)-NNSTG) was done as described previously (21). For the glycosylation assay, a similar number (about 20 × 106) of KBM7WT, GPR107GT, and GPR107GT + GPR107 cells were washed once in PBS and resuspended in 300 μl of Opti-MEM containing a final concentration of 20 nm of CTx-GGGK(biotin)-NNSTG. Cells were incubated for 30 min at room temperature, and the medium containing the toxin was removed after a gentle centrifugation. The cells were resuspended in 3 ml of Iscove's modified Dulbecco's medium supplemented with 10% heat-inactivated fetal serum and chased for different time points at 37 °C and 5% CO2. At the indicated time points during the chase, cells were collected and lysed in Tris buffer (150 mm NaCl, 5 mm MgCl2, 25 mm Tris-HCl, pH 7.4) containing 0.5% Nonidet P-40. Because the CTx also carries biotin probe, we recovered it from the lysate using NeutrAvidin beads. The toxin was eluted from the beads by boiling for 5 min in 1% SDS sample buffer. The glycosylation of CTx was detected by Western blotting using streptavidin-HRP and ECL substrate. The film was scanned, and the bands were quantified using ImageQuant software (GE Healthcare).
RESULTS
Haploid Genetic Screen Identifies Host Factors Required for PE Intoxication
To identify host proteins important for PE intoxication, we performed a genetic screen that exploits KBM7 cells, haploid except for chromosome 8 (23, 29). We mutagenized KBM7 cells using a gene-trap vector to obtain a collection of ∼100 × 106 mutants. We then intoxicated these mutant cells with 50 ng/ml PE, a concentration that kills the majority of wild type KBM7 cells. To identify genes that render cells resistant to PE, massively parallel sequence analysis was performed on genomic DNA isolated from the pooled clones. Gene-trap insertions in genes identified in the selected cell population were compared with gene-trap insertion events in a nonselected control population, and genes significantly enriched for mutations were thus identified. Among these, nine genes had already been implicated in PE intoxication, whereas the other eight genes were not previously known to be involved in the PE pathway (Fig. 1, A and B).
FIGURE 1.

Loss-of-function genome-wide genetic screen in KBM7 cells identifies host factors used by PE, as confirmed by CRISPR/Cas9-gene editing system in HeLa cells. A, bubble plot showing the genes identified in the haploid genetic screen using PE. The size of the bubble is correlated with the number of independent insertions (shown in brackets). On the x axis, genes are ranked based on their alphabetic order. The y axis shows the −log of the p value of the enrichment of gene-trap insertions in the PE-selected cell population as compared with the mutagenized control cells. B, host factors identified in the screen are presented according to their possible function in the PE pathway. Novel genes identified in our screen are marked in red. C, schematic representation of the DNA amplicon used for genotyping of mutants generated using CRISPR/Cas9-gene editing strategy. D, surveyor assay for Cas9-mediated cleavage of the indicated genes in HeLa cells. Accordingly, genomic DNA was extracted, and the region that flanks the sequence targeted by the sgRNA was amplified by PCR. The amplicons were then subjected to endonuclease digestion that specifically cleaves at the site of the newly generated substitution and monitored by agarose electrophoresis. Note that unique cleavage products were observed in the mutant cell lines but not in the control. E, PE intoxication assay of mutant HeLa cells generated using CRISPR/Cas9-gene editing system. Cell viability was determined and shown as percent relative to nonintoxicated cells. Error bars represent S.D. of three independent experiments performed in duplicate.
Validation of the Haploid Genetic Screen Using CRISPR/Cas9-gene Editing System
Because the screen was performed in the haploid KBM7 cell line, we extended these observations to another cell line to exclude cell type-specific effects. We performed PE intoxication assays on mutant HeLa cell populations, generated using CRISPR/Cas9 genome editing (25), for eight hits identified in the KBM7 screen. The introduction of mutations in the gene of interest was assessed by Surveyor assay that relies on digestion of amplified genomic DNA by an endonuclease that specifically cleaves at the site of the newly generated substitution. Accordingly, the region that flanks the sequence targeted by the sgRNA was amplified by PCR. The amplicons were then subjected to digestion and monitored by agarose electrophoresis. Unique cleavage products were observed in the mutant cell lines but not in the wild type control (Fig. 1D) showing the enrichment of gene knock-outs in these cell lines. As expected, these mutant cell populations showed increased resistance to PE as compared with the wild type control (Fig. 1E). Genes identified in the KBM7 screen as hits were thus confirmed by an independent genome editing approach in a different cell type.
In the following sections we describe the categories of mutations observed in our haploid genetic screen performed in KBM7 cells.
Mutations in the Diphthamide Biosynthetic Pathway Leads to PE Resistance
PE transfers an ADP-ribose group from NAD+ to diphthamide, a conserved, post-translationally modified histidine residue unique to eEF2 resulting in inhibition of protein synthesis (30). In eukaryotic cells, several genes are known to be involved in the biosynthesis of diphthamide (31). We observed a significant enrichment of mutations in DPH1, DPH2, DPH4, and DPH7 (WDR85) (Fig. 1A), indicating good coverage of the screen.
PE Trafficking in the Endocytic Pathway
PE enters cells via receptor-mediated endocytosis and reaches its final destination, the cytosol, via vesicular transport and translocation across an intracellular membrane (Fig. 8D). Prior to cytosolic delivery of its catalytic domain, PE undergoes proteolytic cleavage by furin, a protease localized in the TGN and in late endocytic compartments (32). The identification of furin in our screen is therefore consistent with the known requirement of PE for a proteolytic conversion. Moreover, LRP1, one of the known receptors for PE, undergoes a furin-mediated cleavage to generate the active form of the receptor (33). As we will show below, furin acts on yet other components essential for PE intoxication. The closely related proteins LRP1 and LRP1B are the known receptor(s) for PE (7, 8). However, these proteins were not found in our screen. This might be because of the redundancy of their role as a receptor of PE or these proteins are essential for the survival of KBM7 cells.
FIGURE 8.

GPR107 is essential for retrograde trafficking of cholera toxin. A, equal numbers of KBM7WT, GPR107GT, and reconstituted GPR107GT cells were intoxicated with CTx-GGGK(biotin)-NNSTG for 30 min and chased for the indicated time points. Cells were lysed, and the glycosylation of CTx was analyzed by Western blotting using streptavidin-HRP. B, quantification of experiments performed as described in A. C, schematic representation of the intracellular pathway of PE based on the results of the genetic screen. Novel genes identified in the screen are shown in red.
A heterotetrameric tethering factor named the Golgi-associated retrograde protein (GARP) complex that comprises vacuolar protein sorting 51 (VPS51), VPS52, VPS53, and VPS54, promotes fusion of endosome-derived retrograde transport carriers with the TGN (34). In our screen, we identified VPS52, VPS53, and VPS54 of the GARP complex. HeLa cells lacking VPS53, resulting from CRISPR/Cas9-mediated inactivation of the corresponding gene, acquire resistance to PE (Fig. 1E), demonstrating that the GARP complex is essential for PE transport from endosomes to the TGN.
Trafficking of membrane proteins is also involves adaptor protein complexes (35). AP-1 mediates protein sorting between the TGN and early endosomes and is composed of four subunits, including AP1M1 (36). We identified AP1M1 as a novel host factor essential for PE intoxication (Fig. 1A); HeLa cells with mutations in AP1M1 become resistant to PE (Fig. 1E).
Retrograde Transport of PE from Golgi-to-ER
To modify eEF2, PE must cross a membrane barrier(s), but it is unclear whether PE travels through the Golgi to reach the ER as a point of escape. To address this, we performed microscopy studies in HeLa cells using PE conjugated with Atto647 (Fig. 2, A and B). We found that PE partially co-localizes with GPR107 (Fig. 2D), a protein that is localized at the TGN (see below). In our screen, we also implicated proteins involved in retrograde Golgi-to-ER transport, including the KDEL receptor 1 (KDELR1), which retrieves proteins from the Golgi for delivery to the ER through recognition of a C-terminal KDEL motif (Fig. 1A). To test whether PE reaches the ER, we installed a 3×Gly-Lys-(biotin or Atto647) extended with the RDEL sequence or 3×Gly-Lys-biotin without the RDEL sequence at the C terminus of PE via a sortase-mediated transacylation reaction (Fig. 2, A and B) (19, 20). Both biotinylated and fluorescently labeled PE that carry the RDEL motif are toxic, whereas PE lacking the RDEL motif is not (Fig. 2C). The RDEL sequence immediately adjacent to the C terminus is thus required for PE intoxication. This is consistent with a previous report suggesting that the KDEL receptor plays a role in the retrieval of PE to the ER (37).
FIGURE 2.

PE is transported through the Golgi to reach the ER. A, crystal structure of PE depicting its three domains and the C-terminal site where biotin or fluorophore is conjugated using sortase-mediated labeling strategy. B, sortase-catalyzed attachment of biotin or Atto647 dye to PE. Reactions were analyzed by SDS-PAGE with visualization by Coomassie gel, streptavidin-HRP blot, and typhoon image showing PE was successfully labeled with biotin or Atto647 bearing RDEL at the C terminus. See “Experimental Procedures” for details. C, HeLa cells were intoxicated with different concentrations of PE-WT, PE-without RDEL motif (PE-LPETG or PE-LPETG-K-biotin) or labeled PE carrying RDEL motif at the C-terminal (PE-(biotin)-RDEL or PE-(Atto647)-RDEL), and cell viability was determined and shown as percent relative to nonintoxicated cells. Error bars represent S.D. of three experiments performed in duplicate. D, confocal images of HeLa cells expressing C-terminally GFP-tagged GPR107 that were intoxicated with PE-(Atto647)-RDEL. Note that PE partially colocalizes with GPR107. E, similar number (1 × 106) of GPR107 null and wild type KBM7 cells were intoxicated with 50 ng/ml PE for 30 min, and the cell lysates (∼20 μg) were analyzed by immunoblots using PE and GAPDH antibodies.
In addition to KDELR1, we also identified Sec1 family domain-containing protein 1 (SCFD1) and an oligosaccharyltransferase complex subunit (OSTC) as host factors used by PE, and neither of these proteins had previously been implicated in PE intoxication. Consistent with our KBM7 screen, pools of HeLa cells mutated for either SCFD1 or OSTC show increased resistance to intoxication (Fig. 1E). SCFD1 (also called rsly1) regulates Golgi-to-ER retrograde protein transport, possibly through its association with syntaxin 5 (38, 39). OSTC is predicted to form stable complexes with the Sec61 complex, a protein-conducting channel that translocates nascent polypeptides across the ER membrane (40).
GPR107 Is Required for PE and Campylobacter jejuni CDT Intoxication
We identified GPR107 (synonyms are KIAA1624 and LUSTR1) as one of the hits with the highest enrichment of gene-trap insertions in the surviving cells (Fig. 3A). GPR107 was likewise identified in a screen performed with cytolethal distending toxin (cjCDT) secreted by C. jejuni (23). GPR107 is also required for intoxication of mouse cells by ricin, as identified in a genetic screen performed in haploid mouse embryonic stem cells (41). Beyond its assignment to the family of GPCRs, nothing is known about GPR107 (42, 43). CDTs produced by Escherichia coli, Aggregatibacter actinomycetemcomitans, and Haemophilus ducreyi retain the capacity to kill GPR107 null cells, whereas cjCDT no longer does so (23). CDTs are believed to exert their toxicity in the nucleus by triggering the cell cycle checkpoint, causing G2 arrest, which eventually leads to cell death (44). Even though their mechanisms of intoxication are different, identification of GPR107 in the PE and cjCDT screens suggests that they share a common host factor at some stage en route to their final destination. Because GPR107-deficient and wild type KBM7 cells bind PE equally well (Fig. 2E), we can reasonably exclude a surface receptor function. The sensitivity of intra-Golgi trafficking to pertussis toxin (17) indicates involvement of a G-protein, and presumably this implies the existence of coupled GPCR(s) and effector(s). GPR107 may be a candidate for such a GPCR.
FIGURE 3.

GPR107-deficient KBM7 cells are resistant to PE and C. jejuni CDT but sensitive to the other CDTs. A, map of unique insertion sites in the GPR107 gene in the surviving KBM7 cell population. Boxes denote transmembrane domains. B, RT-PCR analysis showing the absence of GPR107 transcript in the clonal cell line carrying gene-trap insertions in the GPR107 gene. C, KBM7WT, GPR107GT, and GPR107GT cells reconstituted with GPR107 cDNA were intoxicated with PE; cell viability was determined and shown as percent relative to nonintoxicated cells. Error bars represent S.D. of three experiments performed in duplicate. D, cells were intoxicated for 18 h with CDTs of different bacterial origin, stained with propidium iodide, and subjected to FACS analysis to assess the G2/M cell cycle arrest. Intensity of fluorescence was measured, and the percent maximum was presented in the overlaid histograms.
Ectopic Expression of GPR107 Restores Sensitivity to PE and cjCDT in GPR107-null Cells
To validate the conclusion that GPR107 is necessary for PE and cjCDT intoxication, a PE-resistant mutant clone (GPR107GT) that carried a defined gene-trap insertion was isolated (Fig. 3B). Treatment of these cells with cjCDT showed resistance of GPR107GT cells to intoxication (Fig. 3D). Introduction of HA-tagged GPR107 into GPR107-deficient cells fully restored sensitivity to both PE and cjCDT, confirming that GPR107 is indeed required for intoxication (Fig. 3, C and D). As expected, GPR107 null cells are sensitive to CDTs from other species (Fig. 3D) (23). A pool of HeLa cells exposed to the appropriate CRISPR/Cas9 construct to target the GPR107 gene shows increased resistance to PE (Fig. 1E).
GPR107 Is Localized in the Trans-Golgi Network
To determine the subcellular localization of GPR107, we established a HeLa cell line that stably expresses GPR107 with an HA tag at its C terminus. Confocal microscopy showed that GPR107 colocalizes with TGN46, a marker for the TGN, but not with the cis/medial Golgi marker, Giantin (Fig. 4, A and C). Upon addition of BFA, GPR107 redistributed over small vesicles as did TGN46, but GPR107 and TGN46 did not completely colocalize, placing GPR107 in a distinct Golgi sub-compartment (Fig. 4B). GPR107 also partially colocalizes with the endoprotease furin (Fig. 4D). We were unable to detect GPR107 at the cell surface (Fig. 4E), where we employed the palmitoylated/myristoylated-tagged red fluorescent protein as a plasma membrane marker(45).
FIGURE 4.
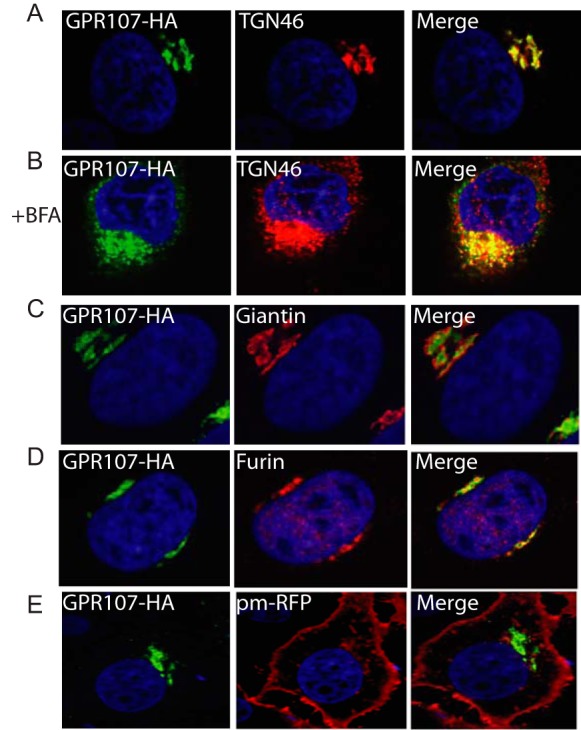
GPR107 is localized in the trans-Golgi network. A, confocal images of HeLa cells expressing C-terminally HA-tagged GPR107. Cells were co-stained for HA and for the trans-Golgi network marker TGN46. B, cells were treated with BFA for 3 h and co-stained as described in A. C–E, cells were costained with HA and the cis/medial-Golgi marker Giantin, furin, or cell surface marker palmitoylated/myristoylated-tagged red fluorescent protein (pm-RFP). In all images the nuclei were stained with DAPI (blue). Note that GPR107 colocalizes with TGN46 but not with Giantin, and the presence of both red and green vesicular structures in the BFA-treated cells is indicative of at least partial segregation of GPR107 and TGN46.
GPR107 Undergoes Proteolytic Cleavage
GPR107 contains seven transmembrane segments with a combined molecular mass of ∼52 kDa for the protein backbone, which carries three predicted N-linked glycosylation sites (Fig. 5A). To assess maturation and turnover of GPR107, we performed pulse-chase analysis in HeLa cells that stably express C-terminally HA-tagged GPR107. We observed that GPR107 undergoes a proteolytic cleavage to yield two fragments of 17 and 35 kDa (molecular mass estimates made after complete deglycosylation of immunoprecipitated material with PNGase F) (Fig. 5C). A single cleavage is therefore likely responsible for the generation of the two fragments. The larger fragment bears the HA epitope and therefore corresponds to the C terminus of GPR107 (Fig. 5C).
FIGURE 5.

GPR107 is cleaved by furin and the disulfide bond that associates the cleaved fragments is essential for its activity. A, predicted topological structure of GPR107. N-Glycosylation sites, disulfide bond linkage, and the furin cleavage site are indicated. B, sequence alignment of the furin-like cleavage site present at the N-terminal region of GPR107 from different eukaryotes. C, HeLa cells expressing C-terminally HA-tagged GPR107WT and GPR107R182A were labeled with [35S]methionine/cysteine for 30 min and chased for the indicated time points. Cells were lysed and immunoprecipitated with anti-HA coupled beads, subjected to PNGase F treatment, analyzed by SDS-PAGE, and autoradiography. D, BFA untreated and treated C-terminally HA-tagged GPR107WT-expressing HeLa cells were labeled with [35S]methionine/cysteine for 30 min and chased for different time points. Cells were lysed and immunoprecipitated with anti-HA coupled beads, subjected to Endo H treatment, and analyzed as described in C. E, HeLa cells expressing C-terminally HA-tagged GPR107WT and GPR107C109/228A were labeled with [35S]methionine/cysteine for 30 min, chased, and immunoprecipitated as described in C. Unless otherwise indicated, immunoprecipitates were eluted and analyzed in reducing conditions. F, HeLa cells expressing C-terminally HA-tagged GPR107WT were labeled with [35S]methionine/cysteine for 30 min and chased in the presence or absence of the lysosomal inhibitor concanamycin A. Cells were lysed and immunoprecipitated with anti-HA coupled beads, and samples were processed as described in C (left panel). Alternatively, immunoprecipitate at the 0-h chase time point was subjected to furin digestion and analyzed by SDS-PAGE and autoradiography (right panel). G, KBM7WT, GPR107GT, and GPR107GT cells reconstituted with GPR107WT, GPR107R182A, or GPR107C109A/C228A were intoxicated with PE; cell viability was determined and presented as percent relative to nonintoxicated cells. Error bars represent S.D. of three experiments performed in duplicate.
GPR107 Is Cleaved by Furin and the Cleaved Fragment Remains Associated with Disulfide Bond
Where in the cell does cleavage occur? What is the enzyme responsible for cleavage?
GPR107 cleavage is blocked by exposure of cells to BFA (Fig. 5D), whereas agents that compromise lysosomal function such as concanamycin A have little effect on cleavage (Fig. 5F, left panel). Because cleavage occurs relatively soon after synthesis and GPR107 is localized predominantly in the TGN, we explored the role of furin, the major processing protease of the secretory pathway, residing also in the TGN (46). GPR107 contains an extended furin recognition site that includes KSKR, a variant of the classical furin cleavage motif (Arg-Xaa-(Lys/Arg)-Arg) (Fig. 5, A and B). Although not common among furin substrates, in GPR107 the Lys residue replaces the first conserved Arg, similar to the furin cleavage site found in the Ebola virus glycoprotein precursor (47). To assess its possible role as a cleavage site, we generated several mutants of GPR107 centered on the KSKR sequence and tested their sensitivity to cleavage in HeLa cell transfectants. A single mutation, R182A, abolished cleavage of GPR107 (Fig. 5C).
In an alternative approach, we pulse-labeled HeLa cells that express HA-tagged GPR107 with [35S]methionine/cysteine for 30 min, immunoprecipitated GPR107, and then subjected the immunoprecipitate to digestion with furin. Under these conditions, immunoprecipitated GPR107 was cleaved by furin and yielded a C-terminal fragment similar in mobility to that produced in living cells (Fig. 5F, right panel). GPR107 co-localized with furin at the TGN, consistent with furin's proposed role in the proteolytic conversion of GPR107 (Fig. 4D).
To determine whether the furin-processed fragments of GPR107 remain associated after cleavage, we examined the behavior of GPR107 by SDS-PAGE under nonreducing conditions. The predicted furin cleavage site is straddled by two cysteine residues, which could form the only possible disulfide bond in the predicted extracellular portion of GPR107. When analyzed under nonreducing conditions, the cleavage fragments indeed remained associated. The C109A/C228A mutation eliminates the possibility of forming this single disulfide bond and abolished the covalent association of the two cleavage fragments (Fig. 5E), further demonstrating that the two cleavage fragments are indeed disulfide-linked.
Is proteolytic cleavage of GPR107 important for PE and cjCDT intoxication? We introduced the cleavage-resistant form of GPR107 (GPR107R182A-HA) into GPR107-deficient cells and examined its ability to restore sensitivity to PE and cjCDT. We found that GPR107R182A-HA restored sensitivity to both PE and cjCDT to a level similar to that of wild type GPR107 (Fig. 5G). Furin cleavage of GPR107 is thus dispensable for PE intoxication. However, expression of the C109A/C228A mutant in GPR107-deficient cells only partially restored sensitivity to PE (Fig. 5G), indicating that the disulfide bond that preserves the interaction between the two cleavage products contributes to GPR107 function.
N-terminal Region of GPR107 Is Required for Its Function
We generated a truncation mutant (GPR107Δ40–182) that lacks the N-terminal fragment of GPR107 to determine the role of this fragment in PE intoxication. This construct carries the N-terminal GPR107 signal peptide (amino acids 1–39) to allow proper ER insertion of the mutant GPR107 (Fig. 6A). GPR107Δ40–182 localized to the TGN, similar to wild type GPR107 (Fig. 6B), establishing that the requisite TGN targeting signals are contained elsewhere in the GPR107 sequence. However, expression of GPR107Δ40–182 in GPR107GT cells failed to restore sensitivity to PE intoxication (Fig. 6C). The N-terminal extracellular region of GPR107 is therefore critical for its function in PE transport.
FIGURE 6.

N-terminal region of GPR107 is essential for its activity. A, topological structure of GPR107Δ40–182. B, confocal images of HeLa cells expressing C-terminally HA-tagged GPR107Δ40–182. Cells were costained for HA and for the trans-Golgi network marker TGN46. The nuclei were stained with DAPI (blue). C, KBM7WT, GPR107GT, and GPR107GT cells reconstituted with GPR107Δ40–182 were intoxicated with PE, and cell viability was determined and shown as percent relative to nonintoxicated cells. Error bars represent S.D. of three independent experiments performed in duplicate.
GPR107 Contributes to Retrograde Transport of Cholera Toxin
To gain more insight into the biological function of GPR107, we set out to test its involvement in both anterograde and retrograde trafficking. First, we examined by pulse-chase analysis whether or not the absence of GPR107 affects protein secretion. Media were collected at the different chase time points and analyzed by SDS-PAGE and autoradiography. No obvious differences were observed between the products released by GPR107-deficient and wild type KBM7 cells (Fig. 7A). The absence of GPR107 was also without effect on the maturation of class I MHC products, type I membrane glycoproteins that traffic via the constitutive secretory pathway to the cell surface. GPR107-deficient cells show maturation of class I MHC products at a rate similar to that seen in wild type cells (Fig. 7B), as assessed by rate and extent of acquisition of Endo H resistance. In our haploid genetic screen, we identified furin as one of the hits required for intoxication by PE. Furin is a protease that cleaves not only PE (5) but also its receptor (LRP1; Gu et al. (33)) and GPR107 (this study). Consequently, we examined the maturation/trafficking of furin in GPR107-deficient cells. Furin not only recycles within the secretory pathway but it is also secretes (46). No difference was found between GPR107-deficient and wild type KBM7 cells in maturation or secretion of furin (Fig. 7C). Confocal microscopy on GPR107-depleted HeLa cells showed that the distribution of furin was similar to that of the control cells (Fig. 7D, left panel). GPR107 is thus dispensable for anterograde transport/secretion of furin.
FIGURE 7.

GPR107 is dispensable for anterograde protein transport and for maintaining the morphological structure of the trans-Golgi network. A, equal number of wild type and GPR107-deficient KBM7 cells were labeled with [35S]methionine/cysteine for 10 min and chased at 37 °C or kept on ice. Supernatants were collected at the indicated time points and analyzed by SDS-PAGE and autoradiography. B, wild type and cells lacking GPR107 were pulse-labeled with [35S]methionine/cysteine as in A and chased for the indicated time points. Cells were lysed, and class I MHC molecules were recovered using W6/32 antibody, treated with Endo H, and analyzed by SDS-PAGE and autoradiography. C, KBM7WT and GPR107GT expressing C-terminally HA-tagged furin were labeled with [35S]methionine/cysteine for 20 min and chased for different time points. Both the cells and the supernatants were collected in parallel, lysed, and immunoprecipitated with anti-HA-coupled beads and where indicated subjected to PNGase F treatment and then analyzed by SDS-PAGE and autoradiography. furini is intracellular furin; furins is secreted furin. D, HeLa cells expressing HA-tagged GPR107 were transduced with the control shGFP or two sets of shRNA that targets GPR107. Cell lysates were prepared from these samples, and knockdown efficiency was examined by Western blotting using anti-HA HRP. E, confocal images of control and GPR107-depleted HeLa cells before or at different time points after BFA treatment. Cells were stained for the trans-Golgi network marker TGN46 or furin (left panel). In all images, the nuclei were stained with DAPI (blue).
Given its localization to the trans-Golgi network, does the lack of GPR107 affect the structure of the Golgi? Analysis by confocal microscopy on wild type and GPR107-depleted HeLa cells for the trans-Golgi marker TGN46 before and after BFA treatment (Fig. 7D) showed no difference in overall structure or in the recovery from Golgi fragmentation post-BFA treatment (Fig. 7D). GPR107 is therefore not obviously involved in maintaining the structure of this organelle.
To examine the role of GPR107 in retrograde transport, we used a modified version of cholera toxin (CTx), equipped with a glycosylation motif and biotin at the C terminus of the A1 subunit (Guimaraes et al. (21)). A similar number of KBM7WT, GPR107GT, and GPR107GT cells reconstituted with GPR107 cDNA were intoxicated with CTx-GGGK(biotin)-NNSTG for 30 min and then chased for 1 and 3 h. Glycosylation of CTx indicates arrival in the ER and is accompanied by an increase in apparent molecular weight, as detected by Western blotting using streptavidin-HRP. Cells that lack GPR107 showed a reduced level of CTx glycosylation compared with control cells (Fig. 8, A and B). GPR107GT cells reconstituted with GPR107 cDNA (overexpressors) showed an increased level of CTx glycosylation (Fig. 8, A and B), suggesting that GPR107 contributes to retrograde transport of CTx and possibly of PE as well.
We generated a similarly engineered version of PE, equipped with a glycosylation motif extended with an ER retrieval signal sequence and modified with a biotin residue (PE-GGGK(biotin)-NNSTGKDEL), to probe the role of GPR107 in retrograde transport of PE. However, we were unable to detect any glycosylated PE even in the wild type or GPR107-null cells reconstituted with GPR107 cDNA overexpressors (data not shown). This is consistent with our microscopy results, where we were likewise unable to detect PE in compartments other than the endocytic pathway (data not shown). This is entirely consistent with the notion that only very few molecules of PE need to make it to the final destination, the cytosol, to achieve intoxication.
DISCUSSION
Bacterial toxins are valuable tools to dissect the physiology of mammalian cells. The coevolution of pathogens and their hosts results in strategies in which host factors are exploited to the advantage of the invader (48, 49). We set out to expand our knowledge of how a bacterial toxin takes advantage of host cell machinery by performing a genome-wide genetic screen and molecular characterization of the hits.
Loss-of-function haploid genetic screens using human KBM7 cells have led to the identification of host factors essential for viral (29, 50, 51) and bacterial (52) pathogenesis as well as for other bacterial toxins (21, 29, 53). We used CRISPR/Cas9-mediated gene editing in HeLa cells to support the results obtained in the KBM7 screen. GPR107 is one of the novel genes identified in our genetic screen in KBM7 cells as a host factor used by PE. GPR107 is ubiquitously expressed and localizes to the TGN. It is conserved in higher eukaryotes, including Caenorhabditis elegans, fruit fly, zebra fish, and Arabidopsis thaliana. GPR107 has the hallmarks of a GPCR, but if an endogenous ligand exists it remains to be identified, because GPR107 is unlikely to have evolved to enable intoxication by ricin or by bacterial toxin.
The GPR107 null cells obtained in our genetic screens are resistant to CDT from C. jejuni (cjCDT), but remain sensitive to other CDTs (E. coli and A. actinomycetemcomitans). How does GPR107 participate in intoxication by PE and cjCDT? Our inability to detect GPR107 at the cell surface makes it unlikely that GPR107 serves as the receptor, although the presence of a small number of surface-disposed GPR107 molecules would have escaped detection by the approaches used here. We consider a role for GPR107 as a shared receptor for PE and cjCDT less plausible also in view of the widely divergent molecular structures of PE and cjCDT. Finally, our finding that GPR107 null cells bind PE in quantities similar to the parental KBM7 cell line also argues against a surface receptor role for GPR107. There is a consensus that the closely related proteins LRP1 and LRP1B are receptors for PE (7, 8). Although GPCR signaling has mostly been considered as confined to the cell surface, recent evidence points to the possibility of intracellular signaling as well (54).
Proteins involved in retrograde trafficking identified in the PE screen (GARP complex, KDELR1, AP1M1, SCFD1, and OSTC) did not appear as hits in the cjCDT screen (23), demonstrating that the pathways of intoxication by PE and cjCDT overlap only in part. Interestingly, such a high degree of specificity shown by individual toxins in co-opting trafficking pathways may represent an evolutionary adaptation of these bacterial products toward optimizing their ability to reach specific cellular targets in a fully active form.
Membrane vesiculation at the TGN requires the trimeric G-protein Gβγ subunits. Addition of Gβγ subunits stimulates Golgi vesiculation in vitro through activation of PKD (13, 55, 56). The heterotrimeric G-proteins Gαi3 and Gαs have been localized to the Golgi apparatus (57). Overexpression of Gαi3 and the use of reagents that perturb the function of G-proteins demonstrate the involvement of heterotrimeric G-proteins in vesicular transport (17). PE and cjCDT might both require G-proteins for retrograde trafficking through the Golgi. To the best of our knowledge, the GPCR(s) associated with G-proteins at the TGN remain(s) unknown. GPR107 is therefore a candidate for a receptor that controls certain aspects of intra-Golgi trafficking.
GPR107 is cleaved by furin, and the cleaved fragments remain associated, suggesting a conceptual analogy with protease-activated GPCRs such as the thrombin receptor (58, 59). In its role as a pro-protein convertase, cleavage by furin may activate the substrate, but proteolysis can also inactivate or modify the activity of the furin substrate (60). If GPR107 were a furin-activated GPCR that undergoes cleavage in the Golgi, then this cleavage might be required for GPR107 function. Our finding that expression of a cleavage-resistant form of GPR107 in GPR107-null cells restores sensitivity to both PE and cjCDT shows that cleavage is dispensable for intoxication. However, this does not rule out the possibility that cleavage is important for other cellular processes that might involve GPR107. Furthermore, the cleavage-resistant version of GPR107 may still allow a segment of its N-terminal domain to engage the ligand-binding pocket of GPR107 and activate it. Our attempts to recapitulate the GPR107 null phenotype through shRNA-mediated knockdown have failed, perhaps because only a few GPR107 molecules may suffice to exert its normal function. Consequently, if the cleavage-resistant version of GPR107 would show much reduced activity compared with its cleavable counterpart, it is uncertain whether we would have been able to accurately score such a difference.
Despite its presence in the TGN and seemingly normal localization pattern compared with intact GPR107, the mutant lacking the N-terminal region of GPR107 fails to restore sensitivity to PE when expressed in GPR107-deficient cells. Furthermore, compromising the disulfide bridge that links the two fragments together affects GPR107 function. This may be due to the fact that any modification at the N terminus affects its ability to bind its ligand and/or interactors that are necessary for its function. The disruption of the disulfide bond may lead to the assembly of an unstable or improperly folded form of GPR107 with a reduced ability to interact with its protein cargo.
Our biochemical data exclude an obvious role for GPR107 in anterograde trafficking. However, cells that lack GPR107 showed a reduced level of glycosylation of the engineered cholera toxin reporter. Reconstitution of GPR107-null cells with GPR107 cDNA, which presumably causes overexpression of GPR107 to levels that exceed wild type levels, resulted in an increased level of cholera toxin glycosylation. Combined, these data are consistent with the involvement of GPR107 in retrograde trafficking, supported also by the fact that GPR107 is localized at the Golgi and that PE uses the retrograde trafficking pathway to reach its final destination, the cytosol.
In conclusion, we identify several novel cellular components used by PE and thus provide a far more detailed map of the PE intoxication pathway than what was known until now (Fig. 8C). Bacterial toxins can thus help identify novel host components involved in key intracellular trafficking steps. The exact contribution of GPR107 to normal host physiology deserves exploration in depth. Given its ubiquitous patterns of expression and conservation among higher eukaryotes (61), GPR107 might belong to the GPCRs that likely orchestrate G-protein-dependent events at the Golgi apparatus (14, 17).
Acknowledgments
We thank Irene Wuethrich, Malini Varadarajan, Sharvan Sehrawat, Eduardo Guillen, and Paul Koenig for technical support and valuable discussions. We also thank Tom DiCesare for illustrations and Wendy Salmon of the Keck facility for imaging. We thank Robin Ross of New England Regional Center of Excellence bimolecule production core facility for large scale production of P. aeruginosa exotoxin A.
This work was supported, in whole or in part, by National Institutes of Health Grant AI087879 (to H. L. P.). This work was also supported by the Netherlands Organization for Scientific Research (to F. G. T.) and New England Regional Center of Excellence Harvard Medical School (to C. P. G.).
This article was selected as a Paper of the Week.
- PE
- P. aeruginosa exotoxin A
- GPCR
- G-protein-coupled receptor
- GARP
- Golgi-associated retrograde protein
- PNGase
- peptide:N-glycosidase
- Endo H
- endoglycosidase H
- CDT
- cytolethal distending toxin
- TGN
- trans-Golgi network
- ER
- endoplasmic reticulum
- BFA
- brefeldin A
- CTx
- cholera toxin
- OSTC
- oligosaccharyltransferase complex.
REFERENCES
- 1. Bonifacino J. S., Glick B. S. (2004) The mechanisms of vesicle budding and fusion. Cell 116, 153–166 [DOI] [PubMed] [Google Scholar]
- 2. De Matteis M. A., Luini A. (2008) Exiting the Golgi complex. Nat. Rev. Mol. Cell Biol. 9, 273–284 [DOI] [PubMed] [Google Scholar]
- 3. Sandvig K., Skotland T., van Deurs B., Klokk T. I. (2013) Retrograde transport of protein toxins through the Golgi apparatus. Histochem. Cell Biol. 140, 317–326 [DOI] [PubMed] [Google Scholar]
- 4. Allured V. S., Collier R. J., Carroll S. F., McKay D. B. (1986) Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. U.S.A. 83, 1320–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wedekind J. E., Trame C. B., Dorywalska M., Koehl P., Raschke T. M., McKee M., FitzGerald D., Collier R. J., McKay D. B. (2001) Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity. J. Mol. Biol. 314, 823–837 [DOI] [PubMed] [Google Scholar]
- 6. Weldon J. E., Pastan I. (2011) A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 278, 4683–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pastrana D. V., Hanson A. J., Knisely J., Bu G., Fitzgerald D. J. (2005) LRP 1 B functions as a receptor for Pseudomonas exotoxin. Biochim. Biophys. Acta 1741, 234–239 [DOI] [PubMed] [Google Scholar]
- 8. Kounnas M. Z., Morris R. E., Thompson M. R., FitzGerald D. J., Strickland D. K., Saelinger C. B. (1992) The α2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 267, 12420–12423 [PubMed] [Google Scholar]
- 9. Cai H., Reinisch K., Ferro-Novick S. (2007) Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev. Cell 12, 671–682 [DOI] [PubMed] [Google Scholar]
- 10. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 11. Fukuda M. (2008) Regulation of secretory vesicle traffic by Rab small GTPases. Cell. Mol. Life Sci. 65, 2801–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bomsel M., Mostov K. (1992) Role of heterotrimeric G proteins in membrane traffic. Mol. Biol. Cell 3, 1317–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takizawa P. A., Yucel J. K., Veit B., Faulkner D. J., Deerinck T., Soto G., Ellisman M., Malhotra V. (1993) Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell 73, 1079–1090 [DOI] [PubMed] [Google Scholar]
- 14. Jamora C., Takizawa P. A., Zaarour R. F., Denesvre C., Faulkner D. J., Malhotra V. (1997) Regulation of Golgi structure through heterotrimeric G proteins. Cell 91, 617–626 [DOI] [PubMed] [Google Scholar]
- 15. Irannejad R., Wedegaertner P. B. (2010) Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein βγ subunits. J. Biol. Chem. 285, 32393–32404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jamora C., Yamanouye N., Van Lint J., Laudenslager J., Vandenheede J. R., Faulkner D. J., Malhotra V. (1999) Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell 98, 59–68 [DOI] [PubMed] [Google Scholar]
- 17. Stow J. L., de Almeida J. B., Narula N., Holtzman E. J., Ercolani L., Ausiello D. A. (1991) A heterotrimeric G protein, Gαi-3, on Golgi membranes regulates the secretion of a heparan sulfate proteoglycan in LLC-PK1 epithelial cells. J. Cell Biol. 114, 1113–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gray G. L., Smith D. H., Baldridge J. S., Harkins R. N., Vasil M. L., Chen E. Y., Heyneker H. L. (1984) Cloning, nucleotide sequence, and expression in Escherichia coli of the exotoxin A structural gene of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 81, 2645–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Popp M. W., Antos J. M., Ploegh H. L. (2009) Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci. Chapter 15, Unit 15.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guimaraes C. P., Witte M. D., Theile C. S., Bozkurt G., Kundrat L., Blom A. E., Ploegh H. L. (2013) Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1787–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guimaraes C. P., Carette J. E., Varadarajan M., Antos J., Popp M. W., Spooner E., Brummelkamp T. R., Ploegh H. L. (2011) Identification of host cell factors required for intoxication through use of modified cholera toxin. J. Cell Biol. 195, 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mungall A. J., Palmer S. A., Sims S. K., Edwards C. A., Ashurst J. L., Wilming L., Jones M. C., Horton R., Hunt S. E., Scott C. E., Gilbert J. G., Clamp M. E., Bethel G., Milne S., Ainscough R., Almeida J. P., Ambrose K. D., Andrews T. D., Ashwell R. I., Babbage A. K., Bagguley C. L., Bailey J., Banerjee R., Barker D. J., Barlow K. F., Bates K., Beare D. M., Beasley H., Beasley O., Bird C. P., Blakey S., Bray-Allen S., Brook J., Brown A. J., Brown J. Y., Burford D. C., Burrill W., Burton J., Carder C., Carter N. P., Chapman J. C., Clark S. Y., Clark G., Clee C. M., Clegg S., Cobley V., Collier R. E., Collins J. E., Colman L. K., Corby N. R., Coville G. J., Culley K. M., Dhami P., Davies J., Dunn M., Earthrowl M. E., Ellington A. E., Evans K. A., Faulkner L., Francis M. D., Frankish A., Frankland J., French L., Garner P., Garnett J., Ghori M. J., Gilby L. M., Gillson C. J., Glithero R. J., Grafham D. V., Grant M., Gribble S., Griffiths C., Griffiths M., Hall R., Halls K. S., Hammond S., Harley J. L., Hart E. A., Heath P. D., Heathcott R., Holmes S. J., Howden P. J., Howe K. L., Howell G. R., Huckle E., Humphray S. J., Humphries M. D., Hunt A. R., Johnson C. M., Joy A. A., Kay M., Keenan S. J., Kimberley A. M., King A., Laird G. K., Langford C., Lawlor S., Leongamornlert D. A., Leversha M., Lloyd C. R., Lloyd D. M., Loveland J. E., Lovell J., Martin S., Mashreghi-Mohammadi M., Maslen G. L., Matthews L., McCann O. T., McLaren S. J., McLay K., McMurray A., Moore M. J., Mullikin J. C., Niblett D., Nickerson T., Novik K. L., Oliver K., Overton-Larty E. K., Parker A., Patel R., Pearce A. V., Peck A. I., Phillimore B., Phillips S., Plumb R. W., Porter K. M., Ramsey Y., Ranby S. A., Rice C. M., Ross M. T., Searle S. M., Sehra H. K., Sheridan E., Skuce C. D., Smith S., Smith M., Spraggon L., Squares S. L., Steward C. A., Sycamore N., Tamlyn-Hall G., Tester J., Theaker A. J., Thomas D. W., Thorpe A., Tracey A., Tromans A., Tubby B., Wall M., Wallis J. M., West A. P., White S. S., Whitehead S. L., Whittaker H., Wild A., Willey D. J., Wilmer T. E., Wood J. M., Wray P. W., Wyatt J. C., Young L., Younger R. M., Bentley D. R., Coulson A., Durbin R., Hubbard T., Sulston J. E., Dunham I., Rogers J., Beck S. (2003) The DNA sequence and analysis of human chromosome 6. Nature 425, 805–811 [DOI] [PubMed] [Google Scholar]
- 23. Carette J. E., Guimaraes C. P., Wuethrich I., Blomen V. A., Varadarajan M., Sun C., Bell G., Yuan B., Muellner M. K., Nijman S. M., Ploegh H. L., Brummelkamp T. R. (2011) Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat. Biotechnol. 29, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Humphray S. J., Oliver K., Hunt A. R., Plumb R. W., Loveland J. E., Howe K. L., Andrews T. D., Searle S., Hunt S. E., Scott C. E., Jones M. C., Ainscough R., Almeida J. P., Ambrose K. D., Ashwell R. I., Babbage A. K., Babbage S., Bagguley C. L., Bailey J., Banerjee R., Barker D. J., Barlow K. F., Bates K., Beasley H., Beasley O., Bird C. P., Bray-Allen S., Brown A. J., Brown J. Y., Burford D., Burrill W., Burton J., Carder C., Carter N. P., Chapman J. C., Chen Y., Clarke G., Clark S. Y., Clee C. M., Clegg S., Collier R. E., Corby N., Crosier M., Cummings A. T., Davies J., Dhami P., Dunn M., Dutta I., Dyer L. W., Earthrowl M. E., Faulkner L., Fleming C. J., Frankish A., Frankland J. A., French L., Fricker D. G., Garner P., Garnett J., Ghori J., Gilbert J. G., Glison C., Grafham D. V., Gribble S., Griffiths C., Griffiths-Jones S., Grocock R., Guy J., Hall R. E., Hammond S., Harley J. L., Harrison E. S., Hart E. A., Heath P. D., Henderson C. D., Hopkins B. L., Howard P. J., Howden P. J., Huckle E., Johnson C., Johnson D., Joy A. A., Kay M., Keenan S., Kershaw J. K., Kimberley A. M., King A., Knights A., Laird G. K., Langford C., Lawlor S., Leongamornlert D. A., Leversha M., Lloyd C., Lloyd D. M., Lovell J., Martin S., Mashreghi-Mohammadi M., Matthews L., McLaren S., McLay K. E., McMurray A., Milne S., Nickerson T., Nisbett J., Nordsiek G., Pearce A. V., Peck A. I., Porter K. M., Pandian R., Pelan S., Phillimore B., Povey S., Ramsey Y., Rand V., Scharfe M., Sehra H. K., Shownkeen R., Sims S. K., Skuce C. D., Smith M., Steward C. A., Swarbreck D., Sycamore N., Tester J., Thorpe A., Tracey A., Tromans A., Thomas D. W., Wall M., Wallis J. M., West A. P., Whitehead S. L., Willey D. L., Williams S. A., Wilming L., Wray P. W., Young L., Ashurst J. L., Coulson A., Blöcker H., Durbin R., Sulston J. E., Hubbard T., Jackson M. J., Bentley D. R., Beck S., Rogers J., Dunham I. (2004) DNA sequence and analysis of human chromosome 9. Nature 429, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guschin D. Y., Waite A. J., Katibah G. E., Miller J. C., Holmes M. C., Rebar E. J. (2010) A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 [DOI] [PubMed] [Google Scholar]
- 27. Wang H., Yang H., Shivalila C. S., Dawlaty M. M., Cheng A. W., Zhang F., Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tafesse F. G., Sanyal S., Ashour J., Guimaraes C. P., Hermansson M., Somerharju P., Ploegh H. L. (2013) Intact sphingomyelin biosynthetic pathway is essential for intracellular transport of influenza virus glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 110, 6406–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carette J. E., Guimaraes C. P., Varadarajan M., Park A. S., Wuethrich I., Godarova A., Kotecki M., Cochran B. H., Spooner E., Ploegh H. L., Brummelkamp T. R. (2009) Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235 [DOI] [PubMed] [Google Scholar]
- 30. Jørgensen R., Merrill A. R., Yates S. P., Marquez V. E., Schwan A. L., Boesen T., Andersen G. R. (2005) Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature 436, 979–984 [DOI] [PubMed] [Google Scholar]
- 31. Su X., Lin Z., Lin H. (2013) The biosynthesis and biological function of diphthamide. Crit. Rev. Biochem. Mol. Biol. 48, 515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas G. (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gu M., Gordon V. M., Fitzgerald D. J., Leppla S. H. (1996) Furin regulates both the activation of Pseudomonas exotoxin A and the quantity of the toxin receptor expressed on target cells. Infect. Immun. 64, 524–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonifacino J. S., Hierro A. (2011) Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 21, 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonifacino J. S., Lippincott-Schwartz J. (2003) Coat proteins: shaping membrane transport. Nat. Rev. Mol. Cell Biol. 4, 409–414 [DOI] [PubMed] [Google Scholar]
- 36. Mills I. G., Praefcke G. J., Vallis Y., Peter B. J., Olesen L. E., Gallop J. L., Butler P. J., Evans P. R., McMahon H. T. (2003) EpsinR: an AP1/clathrin interacting protein involved in vesicle trafficking. J. Cell Biol. 160, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jackson M. E., Simpson J. C., Girod A., Pepperkok R., Roberts L. M., Lord J. M. (1999) The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 112, 467–475 [DOI] [PubMed] [Google Scholar]
- 38. Araç D., Dulubova I., Pei J., Huryeva I., Grishin N. V., Rizo J. (2005) Three-dimensional structure of the rSly1 N-terminal domain reveals a conformational change induced by binding to syntaxin 5. J. Mol. Biol. 346, 589–601 [DOI] [PubMed] [Google Scholar]
- 39. Williams A. L., Ehm S., Jacobson N. C., Xu D., Hay J. C. (2004) rsly1 binding to syntaxin 5 is required for endoplasmic reticulum-to-Golgi transport but does not promote SNARE motif accessibility. Mol. Biol. Cell 15, 162–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shibatani T., David L. L., McCormack A. L., Frueh K., Skach W. R. (2005) Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain Sec61, TRAP, and two potential new subunits. Biochemistry 44, 5982–5992 [DOI] [PubMed] [Google Scholar]
- 41. Elling U., Taubenschmid J., Wirnsberger G., O'Malley R., Demers S. P., Vanhaelen Q., Shukalyuk A. I., Schmauss G., Schramek D., Schnuetgen F., von Melchner H., Ecker J. R., Stanford W. L., Zuber J., Stark A., Penninger J. M. (2011) Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell 9, 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yosten G. L., Redlinger L. J., Samson W. K. (2012) Evidence for an interaction of neuronostatin with the orphan G protein-coupled receptor, GPR107. Am. J. Physiol. Regul. Integr. Comp. Physiol. 303, R941–R949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edgar A. J. (2007) Human GPR107 and murine Gpr108 are members of the LUSTR family of proteins found in both plants and animals, having similar topology to G-protein coupled receptors. DNA Seq. 18, 235–241 [DOI] [PubMed] [Google Scholar]
- 44. Lara-Tejero M., Galán J. E. (2001) CdtA, CdtB, and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect. Immun. 69, 4358–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fairn G. D., Ogata K., Botelho R. J., Stahl P. D., Anderson R. A., De Camilli P., Meyer T., Wodak S., Grinstein S. (2009) An electrostatic switch displaces phosphatidylinositol phosphate kinases from the membrane during phagocytosis. J. Cell Biol. 187, 701–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vey M., Schäfer W., Berghöfer S., Klenk H. D., Garten W. (1994) Maturation of the trans-Golgi network protease furin: compartmentalization of propeptide removal, substrate cleavage, and COOH-terminal truncation. J. Cell Biol. 127, 1829–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Volchkov V. E., Feldmann H., Volchkova V. A., Klenk H. D. (1998) Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U.S.A. 95, 5762–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wiertz E. J., Jones T. R., Sun L., Bogyo M., Geuze H. J., Ploegh H. L. (1996) The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779 [DOI] [PubMed] [Google Scholar]
- 49. Ahn K., Gruhler A., Galocha B., Jones T. R., Wiertz E. J., Ploegh H. L., Peterson P. A., Yang Y., Früh K. (1997) The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6, 613–621 [DOI] [PubMed] [Google Scholar]
- 50. Carette J. E., Raaben M., Wong A. C., Herbert A. S., Obernosterer G., Mulherkar N., Kuehne A. I., Kranzusch P. J., Griffin A. M., Ruthel G., Dal Cin P., Dye J. M., Whelan S. P., Chandran K., Brummelkamp T. R. (2011) Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jae L. T., Raaben M., Riemersma M., van Beusekom E., Blomen V. A., Velds A., Kerkhoven R. M., Carette J. E., Topaloglu H., Meinecke P., Wessels M. W., Lefeber D. J., Whelan S. P., van Bokhoven H., Brummelkamp T. R. (2013) Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340, 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rosmarin D. M., Carette J. E., Olive A. J., Starnbach M. N., Brummelkamp T. R., Ploegh H. L. (2012) Attachment of Chlamydia trachomatis L2 to host cells requires sulfation. Proc. Natl. Acad. Sci. U.S.A. 109, 10059–10064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Papatheodorou P., Carette J. E., Bell G. W., Schwan C., Guttenberg G., Brummelkamp T. R., Aktories K. (2011) Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. U.S.A. 108, 16422–16427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Irannejad R., Tomshine J. C., Tomshine J. R., Chevalier M., Mahoney J. P., Steyaert J., Rasmussen S. G., Sunahara R. K., El-Samad H., Huang B., von Zastrow M. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495, 534–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baron C. L., Malhotra V. (2002) Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295, 325–328 [DOI] [PubMed] [Google Scholar]
- 56. Campelo F., Malhotra V. (2012) Membrane fission: the biogenesis of transport carriers. Annu. Rev. Biochem. 81, 407–427 [DOI] [PubMed] [Google Scholar]
- 57. de Almeida J. B., Doherty J., Ausiello D. A., Stow J. L. (1993) Binding of the cytosolic p200 protein to Golgi membranes is regulated by heterotrimeric G proteins. J. Cell Sci. 106, 1239–1248 [DOI] [PubMed] [Google Scholar]
- 58. Coughlin S. R. (2000) Thrombin signalling and protease-activated receptors. Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 59. Vu T. K., Hung D. T., Wheaton V. I., Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 60. Molloy S. S., Anderson E. D., Jean F., Thomas G. (1999) Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 [DOI] [PubMed] [Google Scholar]
- 61. Gorbushin A. M., Klimovich A. V., Iakovleva N. V. (2009) Himasthla elongata: effect of infection on expression of the LUSTR-like receptor mRNA in common periwinkle haemocytes. Exp. Parasitol. 123, 24–30 [DOI] [PubMed] [Google Scholar]
- 62. Abi-Habib R. J., Liu S., Bugge T. H., Leppla S. H., Frankel A. E. (2004) A urokinase-activated recombinant diphtheria toxin targeting the granulocyte-macrophage colony-stimulating factor receptor is selectively cytotoxic to human acute myeloid leukemia blasts. Blood 104, 2143–2148 [DOI] [PubMed] [Google Scholar]