

Abstract
Oxidative stress has long been implicated in the pathogenesis of various neurodegenerative disorders such as Alzheimer’s disease and stroke. While high levels of oxidative stress are generally associated with cell death, a slight rise of reactive oxygen species (ROS) levels can be protective by “preconditioning” cells to develop a resistance against subsequent challenges. However, the mechanisms underlying such preconditioning (PC)-induced protection are still poorly understood. Previous studies have supported a role of ERK5 (mitogen-activated protein [MAP] kinase 5) in neuroprotection and ischemic tolerance in the hippocampus. In agreement with these findings, our data suggest that ERK5 mediates both hydrogen peroxide (H2O2)-induced PC as well as nerve growth factor (NGF)-induced neuroprotection. Activation of ERK5 partially rescued pheochromocytoma PC12 cells as well as primary hippocampal neurons from H2O2-caused death, while inhibition of ERK5 abolished NGF or PC-induced protection. These results implicate ERK5 signaling as a common downstream pathway for NGF and PC. Furthermore, both NGF and PC increased the expression of the transcription factor, KLF4, which can initiate an anti-apoptotic response in various cell types. Induction of KLF4 by NGF or PC was blocked by siERK5, suggesting that ERK5 is required in this process. siKLF4 can also attenuate NGF- or PC-induced neuroprotection. Overexpression of active MEK5 or KLF4 in H2O2-stressed cells increased Bcl-2/Bax ratio and the expression of NAIP (neuronal apoptosis inhibitory protein). Taken together, our data suggest that ERK5/KLF4 cascade is a common signaling pathway shared by at least two important mechanisms by which neurons can be protected from cell death.
Keywords: ERK5, KLF4, Oxidative stress, Preconditioning, NGF, Neuroprotection
Introduction
Oxidative stress-induced cell damage has been implicated in the physiological process of aging as well as the pathology of a variety of disorders associated with aging, such as Alzheimer’s disease (AD) and stroke. Studies in AD models support that early molecular alterations in degenerating neurons include increased generation of reactive oxygen species (ROS) to activate apoptotic pathways. However, while high levels of oxidative stress is generally associated with neuronal degeneration and death, a slight rise of ROS level can be substantially protective by “preconditioning” (PC) cells to develop a resistance against subsequent challenges (Wiese et al. 1995; Lee and Um 1999; Kim et al. 2001; Dirnagl et al. 2009). For example, hydrogen peroxide (H2O2)-induced PC is known to confer adaptive cytoprotection against subsequent oxidative stress-related injury in various cell types (Angeloni et al. 2011; Mo et al. 2012). Nevertheless, the precise mechanism by which PC protects against oxidative injury, especially in the brain, remains unknown.
The ERK/mitogen-activated protein kinase (MAPK) pathway is not only involved in the perception of responses to oxidative stress, but also a key mediator of neuroprotection. For example, estrogen activated ERK1/2 signaling pathway in global ischemia-induced hypoxia, and the MEK (upstream activator kinase for ERK) inhibitor PD98059 blocked the neuroprotection by estrogen (Lebesgue et al. 2009). However, such studies used pharmacological inhibitors for MEK, including PD98059 or U0126, which inhibit both MEK1 and MEK5, the upstream activator of ERK1/2 and ERK5, respectively (Davies et al. 2000). Therefore, it is plausible that the effects previously assigned to ERK1/2 are actually a function of ERK5. Importantly, Wang et al. reported that cerebral ischemic PC activated ERK5 in the hippocampal CA1 region of rats, and that ERK5 signaling contributes to ischemic tolerance (Wang et al. 2006a). These data and ongoing work in our laboratory led to our hypothesis that ERK5 is a key component of the protection associated with hypoxic PC.
Known activators for ERK5 include growth factors such as nerve growth factor (NGF) (Fukuhara et al. 2000) and oxidative stress (Fukuhara et al. 2000; Scapoli et al. 2004; Cavanaugh et al. 2006). While there is considerable information regarding ERK5’s upstream activators, little is known regarding the downstream effectors of ERK5, especially in the CNS. In a recent report by Ohnesorge et al., transcription factor Krüppel-like factor 4 (KLF4) was identified as a novel executioner of MEK5/ERK5 pathway to elicit an anti-apoptotic and anti-inflammatory phenotype in human endothelial cells (Ohnesorge et al. 2010). KLF4 is well known for its role in stem cell biology (Takahashi and Yamanaka 2006), cardiovascular disease (Dong and Huang 2009) and cancer (Cho et al. 2007; Akaogi et al. 2009). And though KLF4 has been implicated in neuron regeneration (Moore et al. 2009), brain tumor formation (Nakahara et al. 2010) and neuronal apoptosis (Zhu et al. 2009), a mechanistic understanding of its role in the CNS is extremely limited.
During a recent large-scale screening, Dijkmans et al. identified KLF4 as a major immediate-early gene induced by NGF in the rat pheochromocytoma PC12 cells (Dijkmans et al. 2009). PC12 cells have been widely used as a model to elucidate the mechanisms by which NGF exerts its anti-apoptotic effects (Greene 1978). In Dijkmans’ report, the authors found that the NGF-induced increase in KLF4 expression was partially abolished by U0126 (inhibitor against MEK1/2 and MEK5), suggesting an involvement of the ERKs (Dijkmans et al. 2009). Given that ERK5 signaling induces KLF4 expression in peripheral cells to counteract apoptosis (Ohnesorge et al. 2010), and that NGF activates ERK5 in PC12 cells (Obara et al. 2009), we hypothesized that NGF and/or PC protects neuronal cells from apoptosis via activation of an ERK5/KLF4 pathway. By using both PC12 cells and mouse primary hippocampal neurons as the complimentary models, we examined (1) the effect of blocking ERK signaling on KLF4 induction by NGF and PC, and (2) whether depletion of ERK5 or KLF4 by RNAi can attenuate the neuroprotection elicited by NGF and PC against an H2O2 insult. Potential downstream gene targets of KLF4 to exert its protective functions were examined as well.
Methods
Cell culture and treatment
Rat pheochromocytoma PC12 cells were grown in RPMI 1640 media containing 5 % fetal bovine serum and 10 % heat-inactivated horse serum (HIHS) and maintained at 37 °C in 5 % CO2 humidified incubator. NGF (Millipore, Billerica, MA, USA) was added at 100 ng/ml. For cell viability experiments, cells were exposed to NGF or PC (tert-butyl-hydrogen peroxide; 50 μM) for 24 h followed by a 24-h exposure to 250 μM H2O2tert-butyl-hydrogen peroxide (Sigma, St. Louis, MO, USA).
Full details of the mouse study were approved by the Institutional Animal Care and Use Committee at the University of North Texas Health Science Center. All mice were handled according to the Guide for the Care and Use of Laboratory Animals. Primary cultures of hippocampal neurons were prepared from neonatal murine pups (C57BL/6NHSd mice, Harlan) as described by Sarkar et al. (2008) with modifications. Briefly, hippocampal tissue isolated from newborn mice (postnatal days 2–4) was dissociated with trypsin and DNase I for 10 min at 37 °C. The tissue was then washed twice with Neurobasal-A medium containing B-27 and further dissociated by gentle titration using a graded series of fine polished Pasteur pipettes. After centrifugation at 200 × g for 3 min at 4 °C, hippocampal neurons were resuspended in Neurobasal-A/B-27 medium, passed through a cell strainer with 40 μm mesh, and plated at 1.0 × 105 cells/cm2 on culture dishes precoated with poly-d-lysine. The culture dishes were kept at 37 °C in humidified 95 % air and 5 % CO2. The initial culture medium was replaced after 5 h; subsequently, half of the medium was changed every 3 days. At day in vitro (DIV) 2, 1-β-arabinofuranosylcytosine (AraC) was added to a final concentration of 5 μM to prevent glial proliferation. Treatments of the primary cultures started at DIV 7.
Specific inhibition of MEK5 was achieved using the pharmacological inhibitors, BIX01288 or BIX01289 (Selleckchem, Houston, TX, USA) at 30 μM, which was applied 30 min before NGF or PC administration. For transfection experiments, siRNA duplexes of ERK5 or KLF4 were transfected with Gene Silencer reagent (Genlantis, San Diego, CA, USA). Total RNA or protein was isolated 24 h post transfection.
Calcein AM cell viability assay
PC12 cells or primary neurons were cultured in a 96-well flat-bottomed plate. Calcein AM (Molecular Probes, Oregon, USA) staining was used to determine the number of viable cells in each well. The eight wells in the same column on the plate were assigned to the same treatment, and mean from these eight wells was considered as a single “n”. Each treatment was repeated in three different experiments. At the end of treatment, cells were incubated with calcein AM (2 μM) at 37 °C for 30 min and rinsed with PBS. Live cells were distinguished by hydrolysis by intracellular esterase of calcein-AM to calcein, which fluorescence when excited by 485 nm. Emission at 538 nm was measured with an Fluorescence Microplate Reader (Molecular Devices Corporation, Sunnyvale, CA, USA) and analyzed by Softmax-PRO. Data were expressed as the mean fluorescent peak height of samples normalized to percent of the control value.
Plasmids and transient transfection
Expression vectors encoding mouse wild-type ERK5 and constitutively active MEK5 (CA-MEK5) were generous gifts from Dr. Bradford Berk (University of Rochester, Rochester, NY, USA). PC12 cells were transfected with plasmids using TransIT-Neural® Transfection Reagent (Mirus Bio LLC, Madison, WI, USA) at 60–80 % confluency per manufacturer’s protocol. Total RNA or protein was isolated 24 h post transfection.
RNA isolation and cDNA synthesis
Total RNA was extracted from cultures and DNase-treated using the RNeasy Lipid Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Concentrations of extracted RNA were calculated from the absorbance at 260 nm. The quality of RNA was assessed by absorption at 260 and 280 nm (A260/A280 ratios of 1.9–2.0 were considered acceptable). Total RNA (2 μg) was reverse transcribed into cDNA in a total volume of 20 μl using the High-Capacity DNA Archive Kit (Roche Applied Science, Indianapolis, IN, USA), according to the manufacturer's instructions.
Primers and probes for quantitative real-time RT-PCR
PCR primers and probes for the target genes and the endogenous control, GAPDH, were purchased as Assay-On-Demand (Applied Biosystems Inc, Foster City, CA). The assays were supplied as 20× mix of PCR primers (900 nM) and TaqMan probes (200 nM). The KLF4 assay (Rn00821506_g1) contained FAM (6-carboxy-fluorescein phosphoramidite) dye label at the 5′ end of the probes and minor groove binder and nonfluorescent quencher at the 3′ end of the probes. The GAPDH assay (Rn9999999_s1) contained VIC-labeled probes. The assays were optimized for use on an ABI Prism Sequence Detection System at the default machine settings.
Quantitative real-time RT-PCR
The reaction mixture contains water, 2× qPCR Master Mix (Eurogentec), and 20× Assay-On-Demand for BDNF. A separate reaction mixture was prepared for the endogenous control, GAPDH. The reaction mixture was aliquoted in a 96-well plate and cDNA (100 ng of RNA converted to cDNA) was added to bring the final volume to 30 μl. Each sample was analyzed in triplicate. Amplification and detection were performed using the ABI 7300 Sequence Detection System (Applied Biosystems) with the following profile: 2 min hold at 50 °C (UNG activation), 10 min hold at 95 °C, followed by 40 cycles of 15 s at 95 °C (denaturation) and 1 min at 60 °C (annealing and extension). Sequence Detection Software 1.3 (Applied Biosystems) was used for data analysis. The comparative CT method (2−ΔΔCt) was used to calculate the relative changes in target gene expression. In the comparative CT method, the amount of target, normalized to an endogenous control (GAPDH) and relative to a calibrator (untreated control), is given by the (2−ΔΔCt) equation. Quantity is expressed relative to a calibrator sample that is used as the basis for comparative results. Therefore, the calibrator was the baseline (vehicle-treated control) sample and all other treatment groups were expressed as an n-fold (or percentage) difference relative to the control (Livak and Schmittgen 2001). The average and standard deviation of (2−ΔΔCt) were calculated for the values from five independent experiments, and the relative amount of target gene expression for each sample was plotted using the GraphPad Prism 4 software (San Diego, CA).
Western blotting
After treatment with NGF or PC, cells were harvested with lysis buffer containing protease and phosphatase inhibitors, as described previously (Singh et al. 1999). After homogenization, samples were centrifuged at 99,000 × g for 15 min at 4 °C, and the resulting supernatants were evaluated for total protein concentrations using the Bio-Rad DC (Bio-Rad Laboratories, Inc.) protein assay kit. Sample lysates were loaded onto a sodium dodecyl sulfate/10 % polyacrylamide gel, subjected to electrophoresis, and subsequently transferred onto a polyvinylidene difluoride membrane (0.22 μm pore size; Bio-Rad Laboratories, Inc.). The membrane was blocked for 1 h with 5 % non-fat milk in 0.2 % Tween-containing Tris-buffered saline solution before application of the primary antibody. The following primary antibodies were used: The antibodies against phospho-ERK1/2 (#9101, Thr202/Tyr204, 1:1,000), phospho-ERK5 (#3371, Thr218/Tyr220, 1:500), ERK5 (#3372, 1:1,000), GAPDH (14C10, 1:1,000) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies to ERK1 (C-16, sc-93, ERK2 (c-14, sc-154), KLF4 (F-8, sc-166238, 1:500), NAIP2 (A-17, sc-11068, 1:500), Bax (6A7, sc-23959, 1:500), Bcl-2 (C-2, sc-7382, 1:500) were from Santa Cruz Technology. β-Actin antibody (A3854, clone AC-15) was from Sigma.
Antibody binding to the membrane was detected using a secondary antibody (either goat anti-rabbit or rabbit anti-goat) conjugated to horseradish peroxidase (1:20,000; Pierce Chemical Co., Rockford, IL, USA) and visualized using enzyme-linked chemiluminescence (Pierce ECL Western Blotting Substrate; Thermo Scientific, Illinois, USA) with the aid of the AlphaInnotech imaging system.
siRNA and transfection
AllStars negative siRNA control, pre-designed siRNA duplexes against ERK5 (SI05429620 for rat, SI01300663 for mouse), KLF4 (SI01529150 for rat, SI01083579 for mouse), ERK1 (SI01906163 for rat, SI01300569 for mouse) and ERK2 (SI01906037 for rat, SI02672117) were purchased from Qiagen. Briefly, PC12 cells or mouse primary neuronal cultures were transfected with the siRNA duplexes using Gene Silencer reagent (Genlantis) per manufacturer protocol. The siRNA (μg)/Gene Silencer (μl) ratio is 1:5. Total RNA was isolated 24 h post-transfection. Silencing of ERK5 and KLF4 was assessed by real-time PCR.
Statistical analysis
At least three independent experiments were conducted for Western blotting, Calcein assay and qRT-PCR. Densitometric analysis of the Western blots was performed using Alpha Innotech Image Analysis software (Cell Biosciences, Santa Clara, CA, USA). Data were subjected to ANOVA, followed by Tukey’s analysis for the assessment of group differences, and presented as a bar graph depicting the mean ± SEM, using GraphPad Prism software (San Diego, CA, USA). A p value <0.05 was considered to indicate statistical significance.
Results
Both NGF and low dose H2O2 preconditioning protect PC12 cells against high dose H2O2-induced oxidative stress
Since a low level of H2O2 is considered to be a “preconditioning” signal to protect cells while a high level of H2O2 triggers cell death, it is critical to determine the appropriate doses of H2O2 used under the two different circumstances. The rat pheochromocytoma neuron-like PC12 cells have been widely used as a model to study H2O2-induced oxidative stress (Li et al. 2008; Nair and Olanow 2008; Aliaghaei et al. 2014), therefore, we started our investigation of PC neuroprotection with this model. To examine the effects of different concentrations of H2O2 on cell viability, PC12 cells were exposed to various concentrations of H2O2 (50 to 1000 μM) for 24 h, and cell viability was determined by CalceinAM assay. In agreement with previous reports (Guyton et al. 1996), 0–100 μM H2O2 did not affect PC12 cell viability significantly, while concentrations higher than 250 μM induced a significant cell death (Fig. 1a). Therefore, we used 50 μM H2O2 as the PC concentration and 250 μM H2O2 as the oxidative stress concentration in the subsequent experiments with PC12 cells.
Fig. 1.

NGF and PC (low-dose H2O2 preconditioning) protect PC12 cells against H2O2-induced oxidative stress. a PC12 cells were treated with increasing concentrations of H2O2 for 24 h before cell viability was assessed by CalceinAM assay (n = 4). H2O2 (50–100 μM) did not affect the cell viability (n.s. no statistic significance compared to control), while doses higher than 250 μM significantly decreased the cell viability (**p < 0.01 compared to control). H2O2 (50 μM) was chosen as the preconditioning dose, and 250 μM H2O2 was used as the oxidative stress concentration in the following experiments. b PC12 cells were pre-treated with PC or NGF for 24 h prior to treatment with a cell death-inducing concentration of H2O2 (250 μM) for another 24 h, then cell viability was assessed (n = 3. **p < 0.01, ***p < 0.001 compared to control; ### p < 0.001 compared to H2O2 group)
Because NGF and PC can protect various types of neurons against oxidative stress (Kim et al. 2001; Cao et al. 2007; Fuenzalida et al. 2007), we first tried to determine whether both NGF and PC protect PC12 cells from oxidative stress in this study. We pretreated PC12 cells with NGF (100 ng/ml) or PC (50 μM H2O2) for 24 h and then stressed the cells with 250 μM H2O2 for another 24 h. Cell viability was measured by Calcein AM assay. We found that NGF or PC pretreatment significantly rescued PC12 cells from H2O2-induced death (Fig. 1b). Interestingly, the combination of NGF and PC pretreatment did not further increase cell viability compared to NGF or PC pretreatment alone.
NGF and PC activated ERK1/2 and ERK5 signaling
Since both the ERK1/2 and ERK5 pathways have been implicated as mediators of neuroprotection, we sought to determine whether ERK1/2 and/or ERK5 are activated by NGF or PC in our experimental system. Consistent with the literature, NGF resulted in a significant increase in ERK1/2 phosphorylation (indicating activation) (Fig. 2). In addition, ERK5 phosphorylation was also elicited. Consistent with the protective effects of PC, we found that PC was also effective at eliciting ERK1/2 an ERK5 phosphorylation. Notably, NGF- and PC- induced ERK activation had very different kinetics: NGF-induced phosphorylation of ERK5 was evident within the first hour of treatment and gradually declined back to the basal level at 4–6 h. PC induced a delayed ERK5 phosphorylation with a maximal effect at 4 h. In contrast, the dynamic patterns of ERK1/2 activation by NGF and PC are similar to each other, both of which reached peak within 1 h and then continuously declined to the baseline. The different kinetics indicate an intriguing difference between the ERK1/2 versus ERK5 signaling.
Fig. 2.
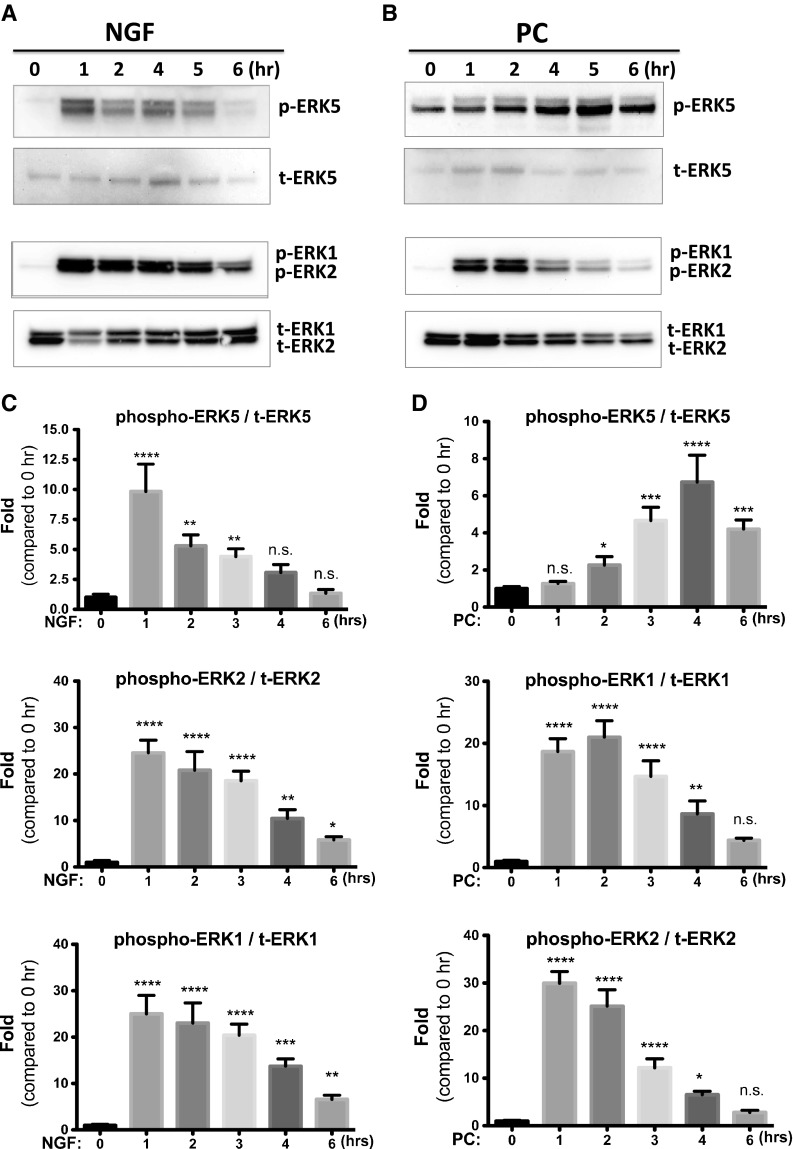
NGF and PC activate the ERK pathways. PC12 cells were treated with NGF (a, c) or PC (b, d) for indicated times, and phosphorylation of ERKs was examined by Western blotting (n = 3). Both NGF and PC activated ERK5 and ERK1/2. A representative blot is shown in a and b. Densitometric quantification of phosphor-ERK versus total ERK is shown in c and d
NGF- and PC-induced neuroprotection requires ERK5 signaling
Next, we tested the hypothesis that blocking ERK signaling by chemical inhibitors affected the effect of NGF and PC on cell viability. To dissect the potential role of ERK5 versus ERK1/2 in meditating NGF or PC’s effect, we used PD184352 as the selective inhibitor against MEK1/2 to block activation of ERK1/2 (Su et al. 2011). PC12 cells were pre-incubated with for 30 min before NGF or PC administration, then exposed to H2O2 for another 24 h before cell viability was assessed by Calcein AM assay. Interestingly, PD184352 had no effect on NGF- and PC-induced cytoprotection (data not shown), suggesting that ERK1/2 pathway did not mediate this effect. Next, we used BIX02188 or BIX02189, two recently identified specific inhibitors against MEK5, to investigate the potential involvement of ERK5 pathway (Tatake et al. 2008). Both BIX02188 and BIX02189 completely blocked the protection against H2O2 by NGF (Fig. 3a) or PC (Fig. 3b), supporting that the protection is mediated via ERK5. These two inhibitors by themselves did not block H2O2-decreased cell viability (Fig. 3a and b). Further, NGF, PC or the inhibitors alone did not change cell viability compared to control (Fig. 3c), indicating that change in the basal activity of ERK5 does not affect PC12 neuronal survival.
Fig. 3.

ERK 5 signaling mediates NGF- and PC-induced neuroprotection against H2O2. PC12 cells were pre-treated with BIX01288 or BIX01289 (specifically inhibiting ERK5 activation) for 30 min before NGF or PC was applied for 24 h. Then cells were stressed with H2O2 for another 24 h before cell viability was assessed (n = 3). The ERK5 inhibitors blocked the protective effect of NGF (A) and PC (B). BIX01288 or BIX01289 alone did not rescue the cells from H2O2 (a, b). NGF, PC or the inhibitors alone did not affect cell viability (c) (**, @@: p < 0.01; ***, ###, @@@: p < 0.001; ns no significance)
Overexpression of active MEK5 protects PC12 cells from H2O2
The pharmacological effects of BIX02188 or BIX02189 supported that ERK5 plays an important role in mediating NGF- and PC-induced protection. To test this hypothesis further, we used a constitutively active MEK5 construct, (CA-MEK5) (Pi et al. 2004), to specifically activate ERK5. We have previously reported that expression of this construct resulted in a significant increase of ERK5 phosphorylation without affecting ERK1/2 phosphorylation (Su et al. 2011), demonstrating the specificity of the MEK5 mutant. Consistent with our hypothesis of a protective role of ERK5 on neuronal survival against oxidative stress, we found that transfection with CA-MEK5 led to an increase in cell survival in the face of the H2O2 insult (Fig. 4). This data, when combined with Figs. 2 and 3, strongly support that NGF and PC signal through the ERK5 pathway to inhibit oxidative stress-induced cell death.
Fig. 4.

Overexpressing active MEK5 rescued cells from H2O2. PC12 Cells were transfected with control vector or constitutively active MEK5 (CA-MEK5). Twenty-four hours post-transfection, cells were stressed by H2O2 before cell viability was assessed (n = 3). Activation of MEK5 partially inhibited H2O2-induced cell death. # p < 0.05; **p < 0.01
NGF and PC up-regulated the expression of KLF4
Knowledge regarding the downstream effectors of ERK5 signaling in neurons is extremely limited, with a small body of research focused on the transcription factor MEF2 (Liu et al. 2003; Shalizi et al. 2003). Since ERK5 has been a focus of interests in cardiovascular diseases, we searched the literature in the vascular system and found that KLF4 has been reported to be an important mediator for ERK5 (Ohnesorge et al. 2010; Clark et al. 2011; Sunadome et al. 2011). Importantly, NGF has been reported to transiently up-regulate KLF4 mRNA level in PC12 cells (Dijkmans et al. 2009). These data support the likelihood that KLF4 is a plausible target of the neuroprotective/ERK5 pathway.
To test the hypothesis that KLF4 is a downstream target of NGF and PC in PC12 cells, we measured KLF4 mRNA and protein levels in response to NGF or PC stimulation. Given that KLF4 has generally been considered as an early response gene in various tissues (Dijkmans et al. 2009; Godmann et al. 2010), coupled with our observation that its potential upstream activator, ERK5, was activated by NGF or PC within an hour of stimulation (Fig. 2), we used the time frame of 1 to 6 h to assess the effect of NGF and PC on KLF4 expression. In line with published results, KLF4 mRNA was rapidly increased by NGF within 1 h, and then declined to basal levels (Fig. 5a). Interestingly, PC also induced a significant KLF4 mRNA increase, but with different kinetics: the time for KLF4 mRNA to reach peak levels was delayed to 3 h after treatment, and the overall effect was weaker than the effect induced by NGF. We further tested if NGF or PC changed the levels of KLF4 protein. To this end, Western blotting data confirmed the findings obtained through qPCR (Fig. 5b), supporting our hypothesis that KLF4 is a target of both NGF and PC. There was a slight delay of KLF4 protein level change compared to the mRNA change, which may reflect the time required for KLF4 translation.
Fig. 5.

KLF4 is a downstream target of NGF and PC. PC12 Cells were treated with NGF or PC for indicated times (n = 3), then mRNA or protein level of KLF4 was measured by qPCR (a) or Western blotting (b), respectively. GAPDH was used as loading control. Both NGF and PC induced an increase in KLF4 mRNA and protein levels. Densitometric quantification KLF4 protein versus GAPDH was shown in c (****p < 0.0001 compared to 0 h)
ERK5 mediates NGF- and PC-induced KLF4 expression
Since our data indicated that the ERK5 signaling cascade is activated by NGF as well as PC in PC12 cells (Fig. 2), and ERK5 activation is associated with the induction of KLF4 in other cell types (Ohnesorge et al. 2010), we asked if ERK5 mediated NGF- and PC-increased KLF4 in PC12 cells. Given that chemical inhibitors may suffer from specificity issues and further, and comparing different pharmacological agents may be confounded by their relative efficacy against their presumptive targets (i.e., efficacy of U0126 against ERK1/2 vs. ERK5), we elected to employ an RNAi approach to selectively knock down the expression of ERK5, ERK1 or ERK2 in PC12 cells. We previously reported a successful depletion of ERK5 expression in rat primary astrocytes using this method (Su et al. 2011). Indeed, transfection of siERK5 significantly inhibited NGF- or PC-induced KLF4 up-regulation, while siERK1/2 had no effect on KLF4 mRNA levels (Fig. 6), suggesting that ERK5 is the mediator for NGF and PC to induce KLF4 expression in PC12 cells.
Fig. 6.

ERK5 mediates NGF- and PC-induced KLF4 expression. PC12 Cells were transfected with siRNA scramble control, siERK5, siERK1 or siERK2 for 24 h before NGF or PC was applied for another 1 or 3 h, respectively. KLF4 mRNA was measured by qPCR (n = 3). siERK5 abolished NGF- or PC-induced KLF4 expression (**p < 0.01 compared to siControl; ## p < 0.01 compared to siControl + NGF or PC). siERK5, siERK1 or siERK2 by themselves had no effect compared to siControl. siERK1 + NGF (or PC) or siERK2 + NGF (or PC) showed no statistical difference compared to siControl + NGF (or PC)
Both ERK5 and KLF4 are required for NGF- and PC-induced protection
Since ERK5, as well as KLF4, are known to play a role in the regulation of apoptosis (Shalizi et al. 2003; Ohnesorge et al. 2010), we hypothesized that both ERK5 and KLF4 play a role in mediating NGF- or PC-increased cell viability against H2O2 stress. Again, to avoid the non-specific effects by chemical inhibitors, we employed RNAi-mediated knocking-down of ERK5 or KLF4 in PC12 cells. Because NGF and PC also activated the ERK1/2 pathways (Fig. 2), siRNA against ERK1 or ERK2 was tested in parallel. Cells transfected with a scrambled siRNA control, siRNA specifically against ERK5 or KLF4 were pre-incubated with NGF for 24 h, and then treated with H2O2 (Fig. 7a). While the scramble siRNA control did not alter the protective effects of NGF, either siEKR5 or siKLF4 significantly attenuated the protective effect, suggesting that both ERK5 and KLF4 are required for NGF to exert its effect on cell viability. A second experiment with PC showed a similar inhibition of the protective effect by either siERK5 or siKLF4 (Fig. 7b). These data support our hypothesis that ERK5/KLF4 is a common downstream signaling pathway shared by NGF and PC. In agreement with our previous finding that PD184352 (specific inhibitor against ERK1/2) had no effect on NGF- or PC-enhanced cell viability, siERK1 or siERK2 did not affect protection conferred by NGF (Fig. 7c) or PC (Fig. 7d), suggesting that ERK1/2 signaling does not mediate cytoprotection downstream of these two neuroprotectants.
Fig. 7.

Both ERK5 and KLF4 are required for NGF- and PC-induced protection. PC12 Cells were transfected with siControl, siERK5, or siKLF4 for 24 h before NGF or PC was applied for another 24 h. Cells were then stressed with H2O2, and assessed for cell viability (n = 4). Both siERK5 and siKLF4 attenuated NGF-induced (a) or PC-induced (b) cell survival. siERK1 or siERK2 had no effect on NGF-induced (c) or PC-induced (d) protection (@: p < 0.05; **, ##, @@: p < 0.01; ***, ###: p < 0.001; ****, ####: p < 0.000)
Activation of the ERK5/KLF4 pathway up-regulates the expression of antiapoptotic genes
The published literature suggests that one major mechanism underlying the cytoprotective effects of KLF4 is through the inhibition of apoptosis. To test the hypothesis that the ERK5/KLF4 pathway affects apoptosis, we used an apoptotic PCR array (SABiosciences, rat apoptotic PCR array PARN-012Z) to screen potential candidate apoptotic genes in KLF4-overexpressed PC12 cells. Cells transfected with empty vector or KLF4 encoding plasmid for 24 h were processed for PCR array analysis. Comparison of the mRNA profile of KLF4-transfected PC12 cells to the vector-transfected cells revealed several genes whose expression levels were changed more than 2-fold. Genes that were up-regulated include well-known protective factors such as Bcl-2, NAIP2 (neuronal apoptosis inhibitory protein-2), Birc5, FAIM (Fas apoptosis inhibitory molecule), and genes that were down-regulated include Bax, Bid, Caspase1, and C-IAP1 (Cellular Inhibitors of Apoptosis 1) (data not shown). As a follow-up, we investigated whether activating the ERK5/KLF4 pathway under conditions of oxidative stress affected the expression of these genes identified by PCR array. PC12 cells were transfected with empty vector, a KLF4 encoding plasmid, or a plasmid encoding constitutively active MEK5 (CA-MEK5) for 24 h, then exposed to 250 μM H2O2 for another 24 h. Cell lysates were subjected to Western blot to determine the expression of NAIP2, Bcl-2 and Bax (Fig. 8). In both scenarios, NAIP2 protein level and the ratio of Bcl-2 : Bax were significantly elevated, supporting our hypothesis that up-regulated expression of anti-apoptotic proteins and down-regulation of pro-apoptotic proteins contribute to the cell survival induced by ERK5/KLF4 signaling.
Fig. 8.

Activation of ERK5/KLF4 signaling regulates apoptotic genes. PC12 Cells were transfected with control vector, encoding vector for KLF4 or constitutively active MEK5 for 24 h. Total cellular protein was isolated and analyzed for expression of NAIP2, Bax and Bcl-2 by Western blotting (n = 3). β-Actin was used as loading control. A representative blot is shown in a. b Densitometry of Bcl-2/Bax ratio and NAIP versus β-Actin (*p < 0.05, **p < 0.01 vs. control)
RNAi-mediated knocking-down of ERK5 or KLF4 attenuated NGF- and PC-induced protection in primary hippocampal neurons
Low level of H2O2 PC has also been reported to protect primary neurons from subsequent oxidative stress induced by higher concentration of H2O2 or oxygen-glucose deprivation (OGD) (Furuichi et al. 2005) (Chang et al. 2008). To determine if ERK5/KLF4 signaling pathway plays an important role in protecting primary neurons against oxidative stress, we first examined the cell viability of mouse primary hippocampal neurons exposed to increasing H2O2 concentrations for 24 h. In general, primary neurons were more vulnerable to H2O2 treatment compared to PC12 cells, while 0–25 μM H2O2 did not produce significant cell toxicity (as measured by CalceinAM assay), 50 μM H2O2 killed 45–60 % neurons (Fig. 9). Therefore, we used 25 μM H2O2 as the PC protectant concentration and 50 μM H2O2 as the oxidative stressor in our following experiments with primary cultures. We found that similar to our results from PC12 cells, 24-h pretreatment of NGF and PC significantly protected neurons against another 24-h exposure to H2O2 (Fig. 9b). Importantly, transfecting these neurons with siRNA against either ERK5 or KLF4 abolished NGF- and PC-induced protection (Fig. 9c and d). Efficient knocking-down of target gene levels with the siRNA was confirmed with qPCR (data not shown).
Fig. 9.

ERK5/KLF4 pathway is required for NGF- and PC-induced protection against oxidative stress in mouse primary hippocampal neurons. a Primary hippocampal neurons were treated with increasing concentrations of H2O2 for 24 h before cell viability was assessed by CalceinAM assay (n = 4). 25 μM H2O2 was chosen as the preconditioning concentration, while 50 μM H2O2 was used as the oxidative stressor in the following experiments. b Primary neurons were pre-treated with PC or NGF for 24 h prior to treatment with H2O2 (50 μM) for another 24 h, then cell viability was assessed (n = 3). Both PC and NGF rescued neurons from H2O2 (***p < 0.001, ****p < 0.0001 vs. control, respectively). # p < 0.05, ### p < 0.001 versus H2O2. c, d Primary neurons were transfected with siControl, siERK5 or siKLF4 for 24 h before NGF or PC was applied for another 24 h. Cells were then stressed with H2O2, and assessed for cell viability (n = 3). While NGF and PC both increased cell viability against H2O2 (# p < 0.05, ## p < 0.01), siERK5 or siKLF4 abolished this effect (@: p < 0.05 compared to siControl + H2O2 + NGF [or PC]). NGF or PC alone did not affect cell viability (ns between siControl + NGF [or PC] vs. siControl). Without H2O2 stress, siERK5 or siKLF4 did not affect the protective effect by NGF or PC (ns between siERK5 + NGF [or PC], siKLF4 + NGF [or PC] vs. siControl + NGF [or PC])
Discussion
Numerous epidemiological and experimental studies support a unifying role for oxidative stress in the pathogenesis of many cardiovascular and neurodegenerative disorders, including AD and stroke (Briones and Touyz 2010; Miller et al. 2010). In particular, the increased ROS, including H2O2, could kill neurons directly or indirectly by activating apoptotic pathways. However, while high levels of oxidative stress is generally associated with neuronal degeneration and death, a slight rise of reactive ROS level can be protective by “preconditioning” cells to develop a resistance against subsequent challenges (Wiese et al. 1995; Lee and Um 1999; Kim et al. 2001; Dirnagl et al. 2009). It is believed that de novo synthesis of H2O2-responsive proteins accumulated during PC accounts for H2O2 adaptation (Wiese et al. 1995). Therefore, revealing the downstream transcription factors responsible for the PC-elicited protection will help elucidate the underlying molecular mechanism.
One clue stems from the observation that the MAP kinase ERK5 is recruited both under conditions of H2O2-induced neuronal PC as well as following application of NGF (Suzaki et al. 2002; Jou 2008). In the present study, we investigated the neurprotective role of ERK5 against oxidative stress in PC12 cells. These cells are widely used as a model to study PC protection, and signaling cascades known to mediate PC effect in these cells include SAPK/JNK (Kim et al. 2001), PI3K/Akt (Mo et al. 2012) and ERK5 (Suzaki et al. 2002). Here, we provide evidence, for the first time, for a key mechanism whereby the ERK5/KLF4 signaling pathway functions as a convergent point of various upstream signals (such as NGF and PC) to protect neurons from oxidative stress. Our conclusion is strongly supported by the findings that (1) NGF and PC rapidly activated ERK5 and subsequently increased the expression of KLF4; (2) Activating ERK5 signaling by transfecting the active form of its upstream kinase MEK5 protected cells from H2O2; (3) inhibiting ERK5/KLF4 by chemical inhibitors or RNAi-mediated depletion abolished PC- or NGF neuroprotection; and (4) activating ERK5/KLF4 pathway regulated pro- and anti-apoptotic genes in a manner that favors survival.
ERKs play key roles in many physiological processes, including cellular proliferation, differentiation, and gene expression (Roux and Blenis 2004). The best-studied members of this family, ERK1/2, have been considered critical in promoting cell survival against oxidative stress in cultured primary neurons (Hota, Hota et al. 2012; Sanchez et al. 2012) as well as PC12 cells (Wruck et al. 2007; Xiao et al. 2011). However, these studies utilized the pharmacological inhibitors U0126 and PD98059, which have been previously used as selective inhibitors of the ERK1/2 signaling pathway but recently reported to effectively inhibit ERK5 signaling pathway as well (Suzaki et al. 2004; Su et al. 2011). In fact, ERK1/2 and ERK5 pathways can play either similar or opposite roles within the same system. For example, Cavanaugh et al. (2006) reported that in a dopaminergic cell line, both ERK1/2 and ERK5 pathways promote neuronal survival under basal conditions, while only ERK1/2 pathway was neuroprotective in response to oxidative stress. We also recently described distinct roles for ERK1/2 and ERK5 activities on BDNF expression such that ERK5 exerts tonic inhibitory control over BDNF expression, while the ERK1/2 pathway is recruited to preferentially mediate stimulus-driven increases in BDNF expression (Su et al. 2011). Therefore, it is critical to delineate the roles of ERK5 versus ERK1/2 when studying a physiological effect.
ERK5 is well known to be sensitive to oxidative stress, and regulate apoptosis and inflammation in oxidatively stressed tissues, especially within the cardiovascular system (for review, see Nigro et al. 2011). Nevertheless, the physiological functions of ERK5 in the CNS and peripheral nervous system (PNS) has been poorly studied, presumably due to the lack of selective pharmacological inhibitor(s), until the recently reported BIX02188 and BIX02189 compounds (Tatake et al. 2008). Further, the technical challenge of delivering interfering molecules (i.e., siRNAs) efficiently into neurons has also been only more recently addressed. Existing literature suggests a role of ERK5 in neuronal development (Cundiff et al. 2009), adult neurogenesis in hippocampus and the olfactory bulb (Pan et al. 2013; Pan et al. 2013), neuronal survival in both CNS and PNS (Watson et al. 2001; Pazyra-Murphy et al. 2009), and neuropathic pain in the spine (Obata et al. 2007). A few studies have identified neurotrophins, including NGF, BDNF and NT-3 as major physiological activators of ERK5 in neurons (Watson et al. 2001; Wang et al. 2006b; Ohtsuka et al. 2009; Yu et al. 2012). Furthermore, Suzaki et al. (2002) reported that in PC12 cells, ERK5 was rapidly (within 5 min) activated by 30–300 μM H2O2, which resulted in increased DNA binding of transcription factor MEF2 to promote cell survival. Because both NGF and PC are known neuroprotectants, we asked whether or not NGF and PC can activate ERK5 in PC12 cells. Indeed, both NGF and PC (50 μM H2O2) induced significant ERK5 phosphorylation. Importantly, inhibiting ERK5 activation by the selective MEK5 inhibitors, BIX02188 and BIX02189, abolished the neuroprotection by NGF or PC, suggesting an indispensible role of ERK5 in mediating this effect (Fig. 2). Interestingly, NGF- and PC-induced ERK5 activation showed different kinetics: NGF triggered a rapid but transient increase in ERK5 phosphorylation, with a maximal effect at 1 h. In comparison, PC induced a gradual and sustained ERK5 phosphorylation that reached the plateau at 3–6 h after treatment. The same pattern was also observed with NGF- and PC-induced ERK1/2 phosphorylation (Fig. 2, lower panels). Since the differential activation kinetics of the ERK1/2 pathway can exert distinct biological effects (Seidman et al. 2001; Murphy and Blenis 2006), it will be interesting to determine if this also holds true for ERK5. However, for the effects examined by the current study (i.e., cell survival), both NGF- and PC-induced ERK5 signaling resulted in cytoprotection against oxidative stress. It is worthwhile to point out that combination of NGF and PC pretreatment did not further increase cell viability compared to NGF or PC pretreatment alone, which argues that NGF and PC have no addictive/synergistic effects, and support a common downstream route for cytoprotection.
The effectors of ERK5 signaling in neurons are poorly studied. In vascular endothelium, transcription factor KLF4 has been reported as an ERK5 downstream target, and activating the ERK5/KLF4 pathway affects the expression of more than 200 genes, resulting in an overall anti-inflammatory and anti-apoptotic phenotype (Ohnesorge et al. 2010). Our data support that the transcription factor KLF4 is an important downstream molecule for ERK5 to exert its function. We observed similar kinetics for KLF4 induction as seen in ERK5 activation by NGF and PC (Fig. 5). Depleting ERK5 by siRNA abolished NGF- and PC-increased KLF4, supporting a requirement of ERK5 to mediate this effect. Importantly, siRNA against either ERK5 or KLF4 inhibited the protective effects of NGF and PC against oxidative stress (Fig. 6). Co-transfecting siRNA against both ERK5 and KLF4 showed no synergistic effect, suggesting that these two signaling molecules reside within the same signaling cascade. To further explore the underlying mechanism for KLF4 to promote survival, we screened ~90 genes well known for their roles in apoptosis and identified several as KLF4 targets, including NIAP, Bcl-2 and Bax (Fig. 8). Our data suggest that inhibiting apoptosis contributes to, at least partially, the overall protective effect of KLF4 in neurons.
Results are equivocal in current literature regarding KLF4’s role in cellular survival, and comparisons are complicated by differences in the utilized species, age of animals, and experimental procedures. Depending on the cell and tissue types used in specific studies, KLF4 has been shown to either inhibit, promote, or have no effect on apoptosis. For example, somatically knocking out KLF4 confirmed a role in promoting survival of natural killers cells in the spleen (Park et al. 2012). In breast cancer cells, KLF4 was induced during histone deacetylase inhibitor treatment, and regulated the extrinsic apoptosis pathway by inhibiting caspase cleavage (Ky et al. 2009). To the contrary, Li et al. 2010 reported that KLF4 promotes H2O2-induced apoptosis via down-regulating Bcl-2 and up-regulating Bax in leukemia. Zhu et al. (2009) showed that in primary cortical neuron cultures, KLF4 overexpression induced the activation of caspase-3 in response to glutamate excitotoxicity. In a recent Science paper, Moore et al. detected no differences in survival between KLF4- and control-transfected hippocampal neurons, although KLF4 suppresses axon and dendrite initiation and elongation in this system (Clark et al. 2013). Our data presented herein support the conclusion that KLF4 is an anti-apoptotic factor. Our recent study in a N27 rat dopaminergic neuronal cell line also suggested that siRNA depletion of KLF4 leads to an increase of caspase-1 activation (data not shown). Therefore, it is plausible, based on our data, that KLF4 inhibits apoptosis in various neuronal cell types.
To this end, we sought to investigate whether ERK5/KLF4 signaling also mediates neuroprotection by NGF and PC in primary hippocampal neurons. A concentration response to H2O2 showed a different kinetic in primary neurons compared to PC12 cells: while 50 μM H2O2 for 24 h does not change cell viability in PC12 cells (Fig. 1a), the same concentration caused a significant cellular death in primary neurons (Fig. 9a). In comparison, much higher concentrations of H2O2 were required to achieve the same “killing” effects in PC12 cells (Fig. 1a), suggesting that H2O2-induced cell death is highly cell type-specific. We tested whether 25 μM H2O2 can be used as a PC protectant in primary neurons, and indeed this concentration of H2O2, as well as NGF, rescued cells against subsequent 50 μM H2O2-induced cell death (Fig. 9b). Literature regarding the primary neuronal survival in response to increasing concentrations of H2O2 is equivocal. Chen et al. reported that in rat embryonic hippocampal neurons, 10 μM H2O2 started to induce a significant cell death, and 30 μM H2O2 killed 30 % of cultured neurons. In contrast, Chang et al. (2008) showed 15 μM H2O2 preconditioned mouse primary cortical neurons against subsequent 50 μM H2O2-induced oxidative stress. The apparent controversy in the current literature may result from the variations in animal species (mouse vs. rat), age of pups used (embryonic vs. neonatal), and different protocols used by various laboratories to establish the primary cultures.
To examine the potential involvement of ERK5/KLF4 pathway in NGF- and PC-evoked protection, we individually knocked down each of these signaling molecules by siRNA and found that similar to our PC12 results (Fig. 7), siRNA against either ERK5 or KLF4 attenuated NGF’s or PC’s protection (Fig. 9c,d). These data further support our conclusion that the ERK5/KLF4 pathway may function as a common mechanism for NGF and PC to exert their neuroprotective effect.
In summary, our findings that the ERK5/KLF4 signaling cascade promotes neuronal survival have several important implications. Although KLF4 has been well known in stem cell biology of CNS, the current study represents one of a very few existing efforts to explore a physiological function of KLF4 in neurons and further, will advance our understanding of the molecular mechanisms by which neurons are protected against oxidative stress. Second, it is plausible that the ERK5/KLF4 cascade may function as a convergence point for various protective signals to counteract oxidative stress-induced neuronal death. Since oxidative stress is thought to be a major player in many aging-associated diseases such as stroke and AD, KLF4 may serve as a therapeutic target, that when activated, counteracts oxidative stress under such conditions.
Acknowledgements
This work was supported by funds from the American Federation of Aging Research, and American Heart Association to CS, funds from the National Institutes of Health (NIA, AG022550, AG027956), the Alzheimer's Association, and the Texas Garvey Foundation to MS, and funds from the Texas Garvey Foundation, and American Heart Association to RLC.
References
- Akaogi K, Nakajima Y, et al. KLF4 suppresses estrogen-dependent breast cancer growth by inhibiting the transcriptional activity of ERalpha. Oncogene. 2009;28(32):2894–2902. doi: 10.1038/onc.2009.151. [DOI] [PubMed] [Google Scholar]
- Aliaghaei A, Khodagholi F, et al (2014) Conditioned media of choroid plexus epithelial cells induces Nrf2-activated phase II antioxidant response proteins and suppresses oxidative stress-induced apoptosis in PC12 Cells. J Mol Neurosci [DOI] [PubMed]
- Angeloni C, Motori E, et al. H2O2 preconditioning modulates phase II enzymes through p38 MAPK and PI3K/Akt activation. Am J Physiol Heart Circ Physiol. 2011;300(6):H2196–H2205. doi: 10.1152/ajpheart.00934.2010. [DOI] [PubMed] [Google Scholar]
- Briones AM, Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep. 2010;12(2):135–142. doi: 10.1007/s11906-010-0100-z. [DOI] [PubMed] [Google Scholar]
- Cao Y, Liu JW, et al. Synergistic protective effect of picroside II and NGF on PC12 cells against oxidative stress induced by H2O2. Pharmacol Rep. 2007;59(5):573–579. [PubMed] [Google Scholar]
- Cavanaugh JE, Jaumotte JD, et al. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J Neurosci Res. 2006;84(6):1367–1375. doi: 10.1002/jnr.21024. [DOI] [PubMed] [Google Scholar]
- Chang S, Jiang X, et al. Exogenous low dose hydrogen peroxide increases hypoxia-inducible factor-1alpha protein expression and induces preconditioning protection against ischemia in primary cortical neurons. Neurosci Lett. 2008;441(1):134–138. doi: 10.1016/j.neulet.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YG, Song JH, et al. Genetic and epigenetic analysis of the KLF4 gene in gastric cancer. APMIS. 2007;115(7):802–808. doi: 10.1111/j.1600-0463.2007.apm_643.x. [DOI] [PubMed] [Google Scholar]
- Clark PR, Jensen TJ, et al. MEK5 is activated by shear stress, activates ERK5 and induces KLF4 to modulate TNF responses in human dermal microvascular endothelial cells. Microcirculation. 2011;18(2):102–117. doi: 10.1111/j.1549-8719.2010.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark VE, Erson-Omay EZ, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339(6123):1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cundiff P, Liu L, et al. ERK5 MAP kinase regulates neurogenin1 during cortical neurogenesis. PLoS One. 2009;4(4):e5204. doi: 10.1371/journal.pone.0005204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, et al. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkmans TF, van Hooijdonk LW, et al. Identification of new Nerve Growth Factor-responsive immediate-early genes. Brain Res. 2009;1249:19–33. doi: 10.1016/j.brainres.2008.10.050. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, et al. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HM, Huang L. Role of the transcription factor Kruppel-like factor 4 (KLF4) in the pathogenesis of atherosclerosis. Zhonghua Xin Xue Guan Bing Za Zhi. 2009;37(10):950–952. [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, et al. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282(51):37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Marinissen MJ, et al. Signaling from G protein-coupled receptors to ERK5/Big MAPK 1 involves Galpha q and Galpha 12/13 families of heterotrimeric G proteins. Evidence for the existence of a novel Ras AND Rho-independent pathway. J Biol Chem. 2000;275(28):21730–21736. doi: 10.1074/jbc.M002410200. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Liu W, et al. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J Neurosci Res. 2005;79(6):816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- Godmann M, Kosan C, et al. Kruppel-like factor 4 is widely expressed in the mouse male and female reproductive tract and responds as an immediate early gene to activation of the protein kinase A in TM4 Sertoli cells. Reproduction. 2010;139(4):771–782. doi: 10.1530/REP-09-0531. [DOI] [PubMed] [Google Scholar]
- Greene LA. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J Cell Biol. 1978;78(3):747–755. doi: 10.1083/jcb.78.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Liu Y, et al. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271(8):4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- Hota KB, Hota SK, et al. Acetyl-l-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus. 2012;22(4):723–736. doi: 10.1002/hipo.20934. [DOI] [PubMed] [Google Scholar]
- Jou MJ. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv Drug Deliv Rev. 2008;60(13–14):1512–1526. doi: 10.1016/j.addr.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Kim DK, Cho ES, et al. Adaptive concentrations of hydrogen peroxide suppress cell death by blocking the activation of SAPK/JNK pathway. J Cell Sci. 2001;114(Pt 23):4329–4334. doi: 10.1242/jcs.114.23.4329. [DOI] [PubMed] [Google Scholar]
- Ky N, Lim CB, et al. KLF4 suppresses HDACi induced caspase activation and the SAPK pathway by targeting p57(Kip2) Apoptosis. 2009;14(9):1095–1107. doi: 10.1007/s10495-009-0368-0. [DOI] [PubMed] [Google Scholar]
- Lebesgue D, Chevaleyre V, et al. Estradiol rescues neurons from global ischemia-induced cell death: multiple cellular pathways of neuroprotection. Steroids. 2009;74(7):555–561. doi: 10.1016/j.steroids.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Um HD. Hydrogen peroxide suppresses U937 cell death by two different mechanisms depending on its concentration. Exp Cell Res. 1999;248(2):430–438. doi: 10.1006/excr.1999.4409. [DOI] [PubMed] [Google Scholar]
- Li RC, Morris MW, et al. Neuroglobin protects PC12 cells against oxidative stress. Brain Res. 2008;1190:159–166. doi: 10.1016/j.brainres.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhao J, et al. KLF4 promotes hydrogen-peroxide-induced apoptosis of chronic myeloid leukemia cells involving the bcl-2/bax pathway. Cell Stress Chaperones. 2010;15(6):905–912. doi: 10.1007/s12192-010-0199-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cavanaugh JE, et al. ERK5 activation of MEF2-mediated gene expression plays a critical role in BDNF-promoted survival of developing but not mature cortical neurons. Proc Natl Acad Sci U S A. 2003;100(14):8532–8537. doi: 10.1073/pnas.1332804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Miller AA, Budzyn K, et al. Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin Sci (Lond) 2010;119(1):1–17. doi: 10.1042/CS20090649. [DOI] [PubMed] [Google Scholar]
- Mo L, Yang C, et al. PI3K/Akt signaling pathway-induced heme oxygenase-1 upregulation mediates the adaptive cytoprotection of hydrogen peroxide preconditioning against oxidative injury in PC12 cells. Int J Mol Med. 2012;30(2):314–320. doi: 10.3892/ijmm.2012.1002. [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326(5950):298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31(5):268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Nair VD, Olanow CW. Differential modulation of Akt/glycogen synthase kinase-3beta pathway regulates apoptotic and cytoprotective signaling responses. J Biol Chem. 2008;283(22):15469–15478. doi: 10.1074/jbc.M707238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara Y, Northcott PA, et al. Genetic and epigenetic inactivation of Kruppel-like factor 4 in medulloblastoma. Neoplasia. 2010;12(1):20–27. doi: 10.1593/neo.91122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro P, Abe J, et al. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15(5):1405–1414. doi: 10.1089/ars.2010.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara Y, Yamauchi A, et al. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J Biol Chem. 2009;284(35):23564–23573. doi: 10.1074/jbc.M109.027821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, et al. Roles of extracellular signal-regulated protein kinases 5 in spinal microglia and primary sensory neurons for neuropathic pain. J Neurochem. 2007;102(5):1569–1584. doi: 10.1111/j.1471-4159.2007.04656.x. [DOI] [PubMed] [Google Scholar]
- Ohnesorge N, Viemann D, et al. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4) J Biol Chem. 2010;285(34):26199–26210. doi: 10.1074/jbc.M110.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka M, Fukumitsu H, et al. Neurotrophin-3 stimulates neurogenetic proliferation via the extracellular signal-regulated kinase pathway. J Neurosci Res. 2009;87(2):301–306. doi: 10.1002/jnr.21855. [DOI] [PubMed] [Google Scholar]
- Pan YW, Storm DR et al. (2013) Role of adult neurogenesis in hippocampus-dependent memory, contextual fear extinction and remote contextual memory: new insights from ERK5 MAP kinase. Neurobiol Learn Mem [DOI] [PMC free article] [PubMed]
- Park CS, Lee PH, et al. Kruppel-like factor 4 (KLF4) promotes the survival of natural killer cells and maintains the number of conventional dendritic cells in the spleen. J Leukoc Biol. 2012;91(5):739–750. doi: 10.1189/jlb.0811413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazyra-Murphy MF, Hans A, et al. A retrograde neuronal survival response: target-derived neurotrophins regulate MEF2D and bcl-w. J Neurosci. 2009;29(20):6700–6709. doi: 10.1523/JNEUROSCI.0233-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi X, Yan C, et al. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94(3):362–369. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Tripathy D, et al. Pigment epithelium-derived factor (PEDF) protects cortical neurons in vitro from oxidant injury by activation of extracellular signal-regulated kinase (ERK) 1/2 and induction of Bcl-2. Neurosci Res. 2012;72(1):1–8. doi: 10.1016/j.neures.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Huang RQ, et al. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A. 2008;105(39):15148–15153. doi: 10.1073/pnas.0802379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapoli L, Ramos-Nino ME, et al. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004;23(3):805–813. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- Seidman R, Gitelman I, et al. The role of ERK 1/2 and p38 MAP-kinase pathways in taxol-induced apoptosis in human ovarian carcinoma cells. Exp Cell Res. 2001;268(1):84–92. doi: 10.1006/excr.2001.5262. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, et al. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci. 2003;23(19):7326–7336. doi: 10.1523/JNEUROSCI.23-19-07326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Setalo G, Jr, et al. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci. 1999;19(4):1179–1188. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C, Underwood W, et al. ERK1/2 and ERK5 have distinct roles in the regulation of brain-derived neurotrophic factor expression. J Neurosci Res. 2011;89(10):1542–1550. doi: 10.1002/jnr.22683. [DOI] [PubMed] [Google Scholar]
- Sunadome K, Yamamoto T, et al. ERK5 regulates muscle cell fusion through Klf transcription factors. Dev Cell. 2011;20(2):192–205. doi: 10.1016/j.devcel.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Suzaki Y, Yoshizumi M, et al. Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: potential role in cell survival following oxidative insults. J Biol Chem. 2002;277(11):9614–9621. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- Suzaki Y, Yoshizumi M, et al. BMK1 is activated in glomeruli of diabetic rats and in mesangial cells by high glucose conditions. Kidney Int. 2004;65(5):1749–1760. doi: 10.1111/j.1523-1755.2004.00576.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tatake RJ, O'Neill MM, et al. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun. 2008;377(1):120–125. doi: 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- Wang RM, Yang F, et al. Preconditioning-induced activation of ERK5 is dependent on moderate Ca2+ influx via NMDA receptors and contributes to ischemic tolerance in the hippocampal CA1 region of rats. Life Sci. 2006;79(19):1839–1846. doi: 10.1016/j.lfs.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Wang Y, Su B, et al. Brain-derived neurotrophic factor activates ERK5 in cortical neurons via a Rap1-MEKK2 signaling cascade. J Biol Chem. 2006;281(47):35965–35974. doi: 10.1074/jbc.M605503200. [DOI] [PubMed] [Google Scholar]
- Wang W, Pan YW, et al. Extracellular signal-regulated kinase 5 (ERK5) mediates prolactin-stimulated adult neurogenesis in the subventricular zone and olfactory bulb. J Biol Chem. 2013;288(4):2623–2631. doi: 10.1074/jbc.M112.401091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, et al. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci. 2001;4(10):981–988. doi: 10.1038/nn720. [DOI] [PubMed] [Google Scholar]
- Wiese AG, Pacifici RE, et al. Transient adaptation of oxidative stress in mammalian cells. Arch Biochem Biophys. 1995;318(1):231–240. doi: 10.1006/abbi.1995.1225. [DOI] [PubMed] [Google Scholar]
- Wruck CJ, Claussen M et al (2007) Luteolin protects rat PC12 and C6 cells against MPP + induced toxicity via an ERK dependent Keap1-Nrf2-ARE pathway. J Neural Transm Suppl(72):57–67 [DOI] [PubMed]
- Xiao H, Lv F, et al. Deprenyl prevents MPP(+)-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology. 2011;290(2–3):286–294. doi: 10.1016/j.tox.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Yu SJ, Grider JR, et al. Up-regulation of brain-derived neurotrophic factor is regulated by extracellular signal-regulated protein kinase 5 and by nerve growth factor retrograde signaling in colonic afferent neurons in colitis. Exp Neurol. 2012;238(2):209–217. doi: 10.1016/j.expneurol.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Tai C, et al. Glutamatergic stimulation triggers rapid Krupple-like factor 4 expression in neurons and the overexpression of KLF4 sensitizes neurons to NMDA-induced caspase-3 activity. Brain Res. 2009;1250:49–62. doi: 10.1016/j.brainres.2008.11.013. [DOI] [PubMed] [Google Scholar]