
Figure 1. A high-fructose diet increases hepatic de-novo lipogenesis and a high-fat diet increases hepatic chylomicron remnant uptake.
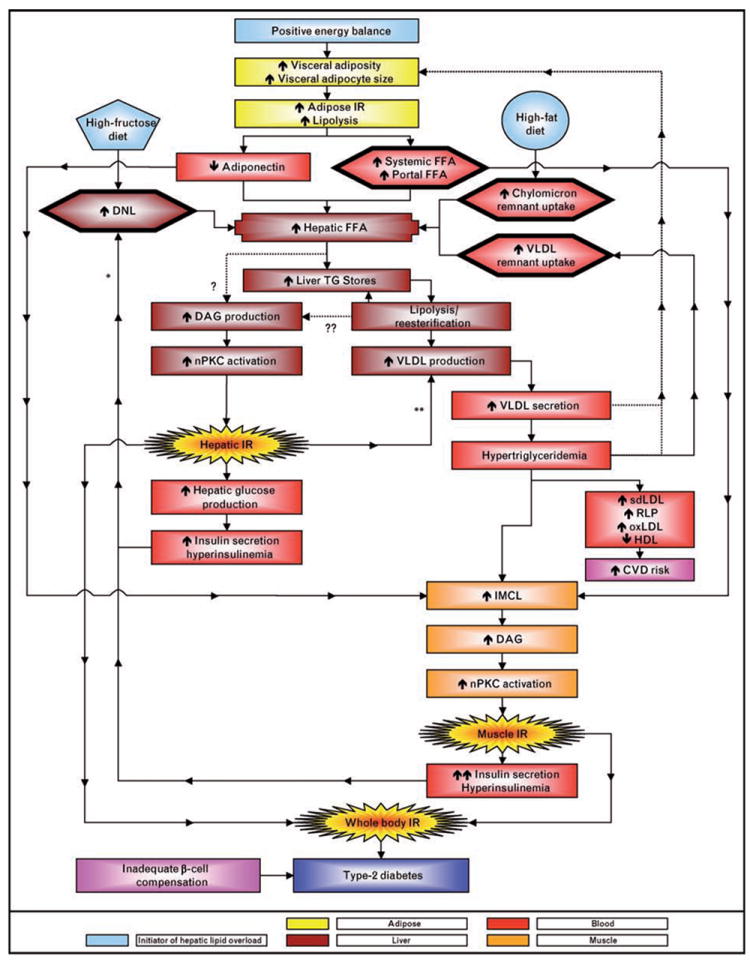
Either diet can produce a hepatic lipid overload along with, or independently of, visceral adiposity and increased portal free fatty acid (FFA) delivery. Visceral adiposity with adipocyte hypertrophy has been hypothesized to reduce adiponectin production and delivery to the liver which would be expected to promote hepatic lipid accumulation. The esterification of hepatic FFA to triglycerides (TG) stored in hepatocytes can increase diacylglycerol (DAG) levels (?); as can the lipolysis, re-esterification, recycling of cytosolic TG that is associated with VLDL assembly (??). Either or both of these sources of DAG may lead to activation of nPKC and impaired hepatic insulin signaling/insulin resistance. VLDL production is regulated by the hepatic lipid supply and is further upregulated by insulin resistance. Hyperinsulinemia may increase DNL because of insulin’s ability to activate SREBP1-c (*). Insulin promotes ApoB degradation and inhibits MTP(**), and both of these processes are likely to be downregulated in the insulin-resistant liver. Increased VLDL production and secretion lead to hypertriglyceridemia. Whether increased VLDL secretion and elevated triglyceride levels directly promote visceral adiposity warrants further investigation (???). Postprandial hypertriglyceridemia increases cardiovascular disease (CVD) risk by promoting lipid/lipoprotein remodeling leading to increased circulating concentrations of small dense LDL (sdLDL), remnant lipoproteins (RLP), and oxidized LDL (oxLDL) and decreased concentrations of HDL. Hypertriglyceridemia and increased levels of circulating FFA can promote accumulation of IMCL, DAG production, and nPKC activation and impaired insulin signaling in skeletal muscle. The end result is whole body insulin resistance, which, when accompanied by inadequate pancreatic beta cell compensation, leads to type 2 diabetes.