
Figure 1. Likely phases of RA development, emphasizing CXCR3+ effector T cells.
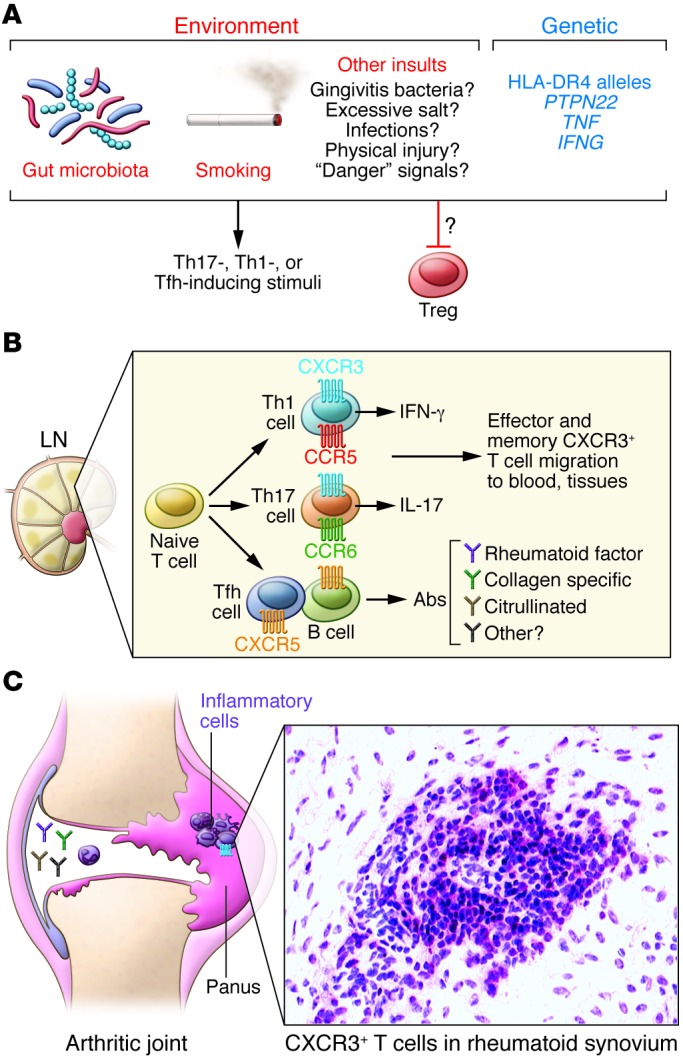
(A) Environmental and genetic triggers, including gut microbiota, smoking, infection, and PTPN22, TNF, and IFNG polymorphisms may promote Th17 and Th1 differentiation, and limit Treg development. Numerous environmental stimuli likely trigger disease through poorly understood processes. RA-associated genetic predispositions, including HLA-DR4 alleles, indicate that T cells drive RA; however, the pathogenic T cell subset is unknown. Dysfunctional MHCII and/or T cell responses could also alter gut microbiota, subsequently influencing disease. (B) During the lymphoid phase, naive T cells differentiate into disease-associated effector and memory T cells. IFN-γ–producing Th1 cells generally express CCR5 and CXCR3, whereas Th17 cells express CCR6 and CXCR3 to some extent. Tfh cells, which facilitate B cell antibody production, express CXCR5. Following generation in the lymph nodes (LN), CXCR3+ effector/memory T cells enter the blood and are a minority of T cells. During this stage, development and distribution of autoreactive effector cells and autoantibody-producing cells promote loss of immune tolerance. (C) The articular phase involves numerous cell types and inflammatory mediators, although the sequence of events remains uncertain. CXCR3+ effector/memory T cells are enriched in RA synovium, where their cytokine products, such as IFN-γ or IL-17, may drive inflammation and leukocyte recruitment. Additionally, deposition of immune complexes and complement activation in joints may initiate inflammation and leukocyte recruitment. Leukocyte products may damage joint tissue or promote synovial fibroblast proliferation. It is not clear whether CXCR3+ Th1 or Th17 cells underlie rheumatoid joint pathogenesis. See ref. 12 for more detailed information.