
Figure 2. Proteomic workflows for identification and quantitation of phosphorylated proteins and peptides.
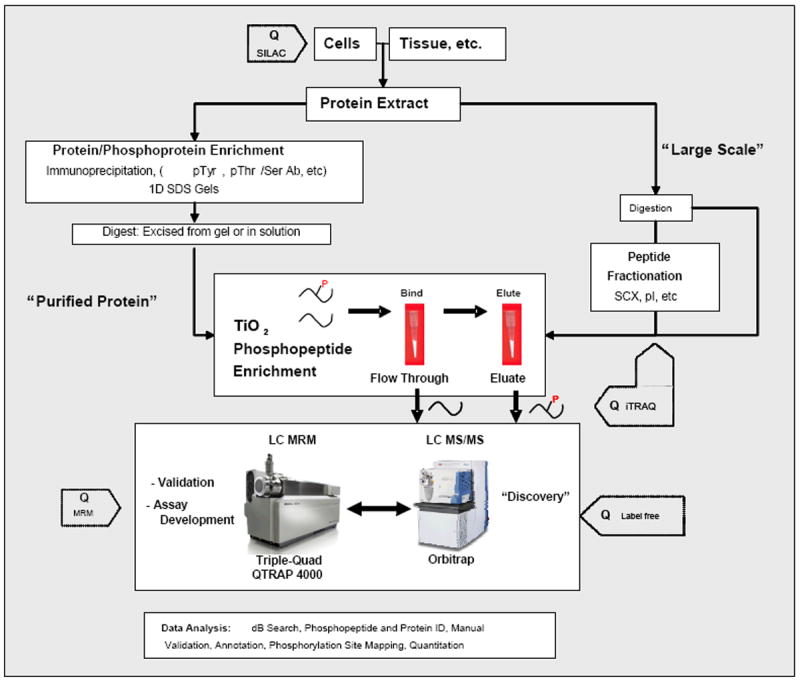
Workflow is divided into two options: one for TiO2 enrichment of phosphopeptides after the protein(s) of interest are purified or immunoprecipitated and another for more complex phosphopeptide mixtures directly extracted from tissue or whole cell extracts. The former is a more targeted approach to discover numerous sites of phosphorylation on proteins of interest, whereas the latter is directed towards more global discovery based approaches. For most of these approaches, three different quantitative methods (SILAC, iTRAQ, and label-free quantitation) can be introduced at steps indicated by “Q” to determine changes of phosphorylation between two or more samples.