

Abstract
Expandable (CTG)n repeats in the 3′ UTR of the DMPK gene are a cause of myotonic dystrophy type 1 (DM1), which leads to a toxic RNA gain-of-function disease. Mutant RNAs with expanded CUG repeats are retained in the nucleus and aggregate in discrete inclusions. These foci sequester splicing factors of the MBNL family and trigger upregulation of the CUGBP family of proteins resulting in the mis-splicing of their target transcripts. To date, many efforts to develop novel therapeutic strategies have been focused on disrupting the toxic nuclear foci and correcting aberrant alternative splicing via targeting mutant CUG repeats RNA; however, no effective treatment for DM1 is currently available. Herein, we present results of culturing of human DM1 myoblasts and fibroblasts with two small-molecule ATP-binding site-specific kinase inhibitors, C16 and C51, which resulted in the alleviation of the dominant-negative effects of CUG repeat expansion. Reversal of the DM1 molecular phenotype includes a reduction of the size and number of foci containing expanded CUG repeat transcripts, decreased steady-state levels of CUGBP1 protein, and consequent improvement of the aberrant alternative splicing of several pre-mRNAs misregulated in DM1.
Keywords: CUG Foci, CUGBP1 protein, MBNL1 protein, RNA splicing, RNA-binding proteins, myotonic dystrophy, protein kinases, protein phosphorylation
Introduction
Myotonic dystrophy type 1 (DM1) is the most common muscular dystrophy in adults1 and at present has no effective therapy for its treatment. This autosomal dominant disorder results from the expansion of CTG repeats in the 3′UTR of the DMPK gene, and its pathogenesis is mediated by the mutant transcript. DMPK transcripts containing expanded CUG repeats (CUGexp) become arrested in the nucleus and form multiple discrete inclusions, and their toxic effects are mediated through at least two RNA binding proteins: muscleblind-like 1 (MBNL1) and CUG repeat binding protein 1 (CUGBP1). Altered activity of these two antagonistic regulators of alternative splicing results from the titration of MBNL1 by the expanded CUG repeat foci and hyperphosphorylation of CUGBP1, which leads to its increased steady-state levels as shown in DM1 myoblasts, skeletal muscle, and heart tissues.2–5 Loss of MBNL1 and a gain of CUGBP1 function result in a misregulated splicing pattern of several pre-mRNAs, including chloride channel (CLCN), insulin 66 receptor (INSR), cardiac troponin T (TNNT2), and sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 1 (SERCA1),6–9 and the expression of embryonic alternative splicing isoforms predominates in adult DM1 tissues.
CUGBP1 and MBNL1 are multifunctional proteins that regulate alternative splicing and also contribute to mRNA translation and RNA stability,10–12 miRNA biogenesis,13,14 protein secretion, and localization of alternative 3′UTR isoforms.12 Their altered activity in DM1 cells is correlated with different kinase signaling pathways. Experimental evidences have shown that CUGBP1 can be phosphorylated at different sites by various protein kinases, such as cyclin-dependent kinases (CDKs), glycogen synthase kinase 3β (GSK3β), protein kinase AKT, and protein kinase C (PKC),15–18 and their unspecific activation has been reported in DM1 cells.16–18 Interestingly, as PKC activation results in CUGBP1 hyperphosphorylation in DM1, utilization of PKC-specific inhibitors Bis-1, Bis-IX, and Ro-31-8220 has been shown to block CUGBP1 upregulation in cellular models18 and the heart-specific mouse model of the disease.19 This underscores the notion that targeting protein kinases may be beneficial for alleviating molecular hallmarks of DM1 because expression of mutant DMPK transcripts can disrupt normal signaling pathways, leading to unspecific activation of protein kinases. Additional evidence of altered kinase signaling pathways in DM1 cells came from the most recent report by Botta and colleagues,20 which highlighted the unspecific activation of Src family kinases (SFK) by overexpression and nuclear localization of the protein products of MBNL1 transcripts containing exon 5 (MBNL142-43). In vitro assays showed that MBNL142-43 binds the Src-homology 3 domain of Src family kinases via proline-rich motifs, enhancing the SFK activity. Importantly, MBNL142-43 downregulation by specific short interfering RNA (siRNA) resulted in decreased levels of tyrosine-phosphorylated proteins and an improved splicing pattern of MBNL1 exon 5. This suggests an additional pathomechanism in DM1 based on an altered phosphotyrosine signaling pathway, which may be a novel therapeutic target.
Thus far, efforts to develop DM1 therapeutics have focused on drugs targeting RNA by destroying toxic CUGexp RNA and/or inhibiting its pathogenic interactions with nuclear proteins (reviewed in ref. 21). The antisense technology that utilizes morpholino CAG-25 oligonucleotides,22,23 other chemically modified CAG repeat antisense oligonucleotides,24,25 and synthetic siRNAs to target CUG repeats26 appears to be effective in DM1 cells and mouse models of the disease. Additionally, viral vector-mediated expression of hU7-snRNA-(CAG) has shown to be beneficial in DM1 myoblasts.27 Also, several bioactive small molecules that are CUG repeat binders have been reported as potential therapeutic agents for DM1 and are able to inhibit the interactions between expanded CUG RNA and MBNL1 protein.28–33 Ongoing efforts to develop novel therapeutic small-molecule candidates are critical in the search for an effective treatment for DM1. Such molecules may, in addition to CUGexp RNA, target other yet-unidentified cellular components critical for DM1 pathogenesis. Interestingly, the most recent report from the Brook laboratory indicates that targeting protein kinases with small molecules results in alleviation of molecular hallmarks of DM1.34 This was correlated with the disappearance of nuclear CUGexp RNA foci without degradation of the mutant transcripts or their translocation to the cytoplasm.
Herein, we describe the use of two small molecule ATP site-directed kinase inhibitors: the imidazolo-oxindole inhibitor C16 (6,8-Dihydro-8-[1H-imidazol-5-ylmethylene]-7H-pyrrolo [2,3-g]benzothiazol-7-one)35 and the pyrimidine-based inhibitor C51 (N-[2-{1H-indol-3-yl}ethyl]-4-[2-methyl-1H-indol-3-yl] pyrimidin-2-amine).36 Previous studies have identified these two compounds as protein kinase R (PKR) inhibitors; however, these chemicals can also exert activity against other targets because ATP-binding sites are abundant in the kinome. C16 activity against kinases other than PKR has been reported,37 yet C51 has not been characterized in this manner. C16 exhibits neuroprotective properties in various systems,37–41 including cultured mouse neurons lacking PKR, indicating that the kinase may not be its only target. The neuroprotection provided by C16 has been shown to result from inhibiting certain CDKs, including cyclin-dependent kinase 1 (CDK1), 2 (CDK2), and 5 (CDK5) as well as glycogen synthase kinases GSK3α and GSK3β. In contrast, C16 has no major in vitro inhibitory effect on pro-apoptotic kinases, including c-Jun N-terminal kinases, stress-activated protein kinases (SAPKs or p38 MAP kinases), and the death-associated protein kinases (DAPKs), or other kinases, such as mitogen-activated protein kinases 1 (MKK1), 6 (MKK6), and 7 (MKK7) and c-Raf.37
In this study, we cultured human myoblasts and fibroblasts treated with either C16 or C51 and demonstrated the mitigation of major molecular hallmarks of DM1, which included downregulation of CUGBP1 steady-state levels, a reduction of the size and number of foci containing expanded CUG repeat transcripts, and consequent improvement of the aberrant splicing of several pre-mRNAs dysregulated in DM1. Earlier studies indicated that C16 and C51 act as ATP-site specific PKR inhibitors, which suggests PKR as a therapeutic target for DM1. However, the results of prior genetic studies of the cross between the expanded CUG repeat expressing HSALR DM1 mouse model with PKR kinase domain mutant mice showed no improvement in DM1 pathogenesis, implying that this kinase is not required for development of the disease symptoms in the transgenic DM1 mouse model.5 Thus, it is likely that C16 and C51 act via other kinases to mitigate the molecular phenotype of DM1 and the possible molecular mechanism of their action is discussed.
Results
Treatment with C16 and C51 diminishes steady-state levels of CUGBP1 and corrects aberrant splicing of its pre-mRNA targets
To determine if C16 and C51 ATP site-directed kinase inhibitors (Fig. 1) can affect the steady-state levels of CUGBP1, which is known to be upregulated by the DM1 mutation,3,18,42 human myoblasts derived from DM1 patients were grown in the presence of either the compounds or vehicle alone (DMSO), and CUGBP1 expression levels were analyzed by western blot. As shown in Figure 2A and B, DM1 cells responded to treatment with either C16 or C51 by downregulating CUGBP1 expression, which was dose-dependent. C16 had a stronger impact on reducing the protein levels than C51, and this effect was observed at lower doses of the compound. In DM1 human myoblasts, the steady-state expression levels of CUGBP1 after the treatment with DMSO solvent were similar to those of untreated cells (Fig. S1A). C16 and C51 are likely to act on a post-transcriptional level to cause CUGBP1 protein downregulation because treatment with the compounds did not affect CUGBP1 transcript levels (Fig. 2C).
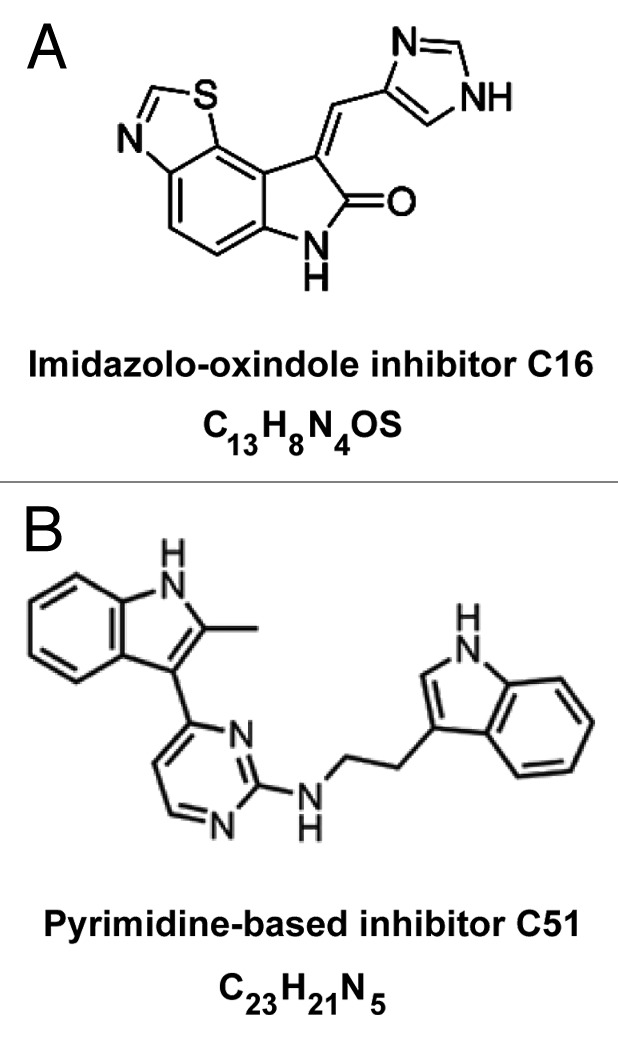
Figure 1. Structures of C16 and C51 small molecule compounds.

Figure 2. Treatment with C16 and C51 affects steady-state levels of CUGBP1 protein. (A and B) western blot analyses of CUGBP1 protein in lysates from two different human DM1 myoblasts (Line 1 and 2) grown either with or without C16 and C51 at the indicated concentrations. Treatment with C16 and C51 led to a dose-dependent downregulation of CUGBP1 steady-state levels in DM1 cells. Equal protein loading was determined by immunoblotting for two reference controls, GAPDH and α-Tubulin, to assure that the drugs affected CUGBP1 rather than any reference protein. The bar graph shows the levels of CUGBP1 as determined in independent experiments relative to α-Tubulin. The data are expressed as the mean (± SEM) of triplicate experiments. For more data on the CUGBP1 levels, please refer to Figure S1. (C) Semiquantitative RT-PCR analyses of CUGBP1 mRNA levels in human DM1 myoblasts grown either with or without C16 and C51 at the indicated concentrations. These treatments had no effect on CUGBP1 transcript levels. *, P < 0.01; **, P < 0.001 for treated vs. untreated samples.
We aimed to determine if the treatment that affected CUGBP1 protein expression caused any changes in the splicing pattern of CUGBP1-regulated pre-mRNAs.43 For this purpose we pre-selected a subset of genes (CAPZB, SORBS1, ATP2B1, ABLIM1, MTMR3, ERBB2IP, SGT2, KIDINS220, R3HDM2, CTAGE5, and ITGA6) that are mis-spliced in DM1 cells.43,44 As the majority of them expressed only one alternative splicing product in human myoblasts, measuring any changes with these genes was not feasible. Because of this, we chose MTMR3 and ITGA6, which express two alternative splicing isoforms (both alternative exon inclusion and exclusion). As shown in Figure 3A, the splicing pattern of the two pre-mRNAs was not improved in DM1 cells upon treatment with C16 and C51. This may indicate that downregulation of CUGBP1 by the two compounds was insufficient to trigger a splicing transition of the MTMR3 and ITGA6 pre-mRNAs. Thus, DM1 myoblasts were transfected with siRNA against mRNA of CUGBP1 (siCUGBP1) followed by treatment with C16 and C51. As depicted in Figure 3B, downregulation of CUGBP1 mRNA to approximately 25% after siRNA treatment alone did not affect the alternative-splicing pattern of the two pre-mRNAs compared with untreated cells. Interestingly, when siRNA-treated DM1 cells were then treated with C16 and C51, we observed a dose-dependent correction of aberrant splicing of ITGA6 and MTMR3 pre-mRNAs. No changes in the splicing pattern were observed in cells treated with control siRNA (Block-it). Since these results indicated that the ability of C16 and C51 to correct aberrant splicing might be sensitive to the expression levels of CUGBP1 protein we tested the effectiveness of the compounds in HepG2 and HEK293 non-DM1 cells that vary in CUGBP1 protein levels (Fig. S1B).45 In HepG2 cells, CUGBP1 is expressed at significantly lower levels than in HEK293 and in DM1 myoblasts. As shown in Figure 3C, HepG2 and HEK293 cells express the DM1-specific splicing pattern of several pre-mRNAs including CAPZB, SORBS1, MTMR3, and KIDINS220. Importantly, treatment with C16 or C51 resulted in correction of some of the mis-splicing events and HepG2 cells were more susceptible for changes than HEK293. Of note, is the fact that C16, which we found more effective in diminishing CUGBP1 protein levels in DM1 myoblasts, was a stronger splicing modifier in HepG2 cells and its effect was dose dependent (Fig. 3C). These results indicate that treatment with the compounds may be efficacious when tested in vivo in skeletal muscles that express CUGBP1 protein at levels that resemble the expression profile of HepG2 cells rather than that of cultured myoblasts or fibroblasts.

Figure 3. Correction of aberrant CUGBP1-dependent alternative splicing after C16 and C51 treatment is sensitive to CUGBP1 steady-state levels. (A and B) Splicing analyses of ITGA6 and MTMR3 pre-mRNAs in human DM1 myoblasts; no changes in the splicing pattern were observed after treatment with C16 and C51 alone (A); siRNA downregulation of CUGBP1 mRNA prior to compounds treatment led to an improvement of the ITGA6 and MTMR3 pre-mRNAs splicing patterns as indicated by increased expression of the normal splicing isoforms (B). (C) Representative agarose gel analysis of RT-PCR products of endogenous MTMR3, SORBS1, KIDINS220, and CAPZB pre-mRNA splicing patterns in HepG2 and HEK293 cells. DM1-specific splicing aberrations were more susceptible for changes after the drugs treatment in HepG2 cells in which CUGBP1 expression levels are significantly lower than that of HEK293. *, P < 0.01; **, P < 0.001 for treated vs. untreated samples.
In summary, these results demonstrate that C16 and C51 correct a molecular defect in DM1 cells by decreasing CUGBP1 steady-state levels and indicate that their ability to improve aberrant alternative splicing of CUGBP1-regulated targets is sensitive to the intracellular expression levels of CUGBP1 protein.
Treatment with C16 and C51 diminishes nuclear CUGRNA inclusions
To further define if C16 and C51 affect nuclear CUGexp RNA foci (another molecular hallmark of DM1 cells),5,46 we analyzed the foci number and their size in human myoblasts and fibroblasts derived from DM1 patients. RNA fluorescence in situ hybridization (FISH) was employed to visualize the CUGexp inclusions. As shown in Figure 4A–D, C16 and C51 affect the CUGexp RNA foci to a varying degree by diminishing their abundance compared with untreated and DMSO-treated cells. However, the inhibitors did not eliminate the inclusions. Similar results were obtained by analyzing DM1 fibroblasts that underwent the same inhibitor treatments (Fig. S2A and B).

Figure 4. C16 and C51 affect nuclear CUGexp RNA foci in DM1 myoblasts. (A and C) Representative RNA-FISH confocal images of cultured human DM1 myoblasts: untreated or treated with DMSO (0.2%), C16 (1.0 μM), and C51 (30 μM). Treatment with either C16 or C51 resulted in a significant reduction of the size and number of nuclear CUGexp RNA foci, whereas DMSO had no effect. Nuclei were stained with DAPI (blue); CUG RNA foci are indicated by red fluorescence. Scale bar, 2 μm (A) and 10 μm (C). For more images of nuclear CUGexp RNA foci in human DM1 cells, please refer to Figure S2A and B. (B and D) Quantitative analyses of the area and number of CUGexp RNA foci in DM1 cells that were either untreated or incubated with the indicated compounds. (E) qRT-PCR for DMPK transcripts in DM1 myoblasts treated with the indicated concentrations of C16 and C51. Expression was normalized to GAPDH mRNA. DMPK was downregulated upon treatment with C51, whereas no significant changes were detected in C16- treated cells. The bar graphs represent the mean ± SEM of different DM1 cell lines (three pulled for C16 and two for C51). For the results of the DMPK mRNA in DM1 myoblasts treated with DMSO, please refer to Figure S2C. *, P < 0.01; **, P < 0.001 for treated vs. untreated samples.
The decreased size and number of CUGexp RNA foci suggested that C16 and C51 affect the expression of DMPK transcripts. Thus, we used quantitative real-time RT-PCR (qRT-PCR) to analyze the steady-state levels of DMPK mRNA in DM1 myoblasts that were treated with the compounds. As depicted in Figure 4E, only C51 affected DMPK expression. After treatment with 30 μM C51, the total DMPK mRNA levels were downregulated by 37% in DM1 myoblasts compared with untreated cells (P < 0.001). Neither lower doses of C51 (2.0 μM) nor C16 (0.1 μM and 1.0 μM) caused any changes in the DMPK expression levels. The cells treated with DMSO solvent alone showed no changes in the transcript expression levels (Fig. S2C).
Taken together, these results demonstrate that both C16 and C51 affect the abundance of ribonuclear CUGexp aggregates; however, only C51 was able to downregulate the number of DMPK transcripts.
Treatment with C16 and C51 affects colocalization of MBNL1 with CUGexp RNA Foci
Next, we analyzed if the treatment with C16 and C51 affects MBNL1, another splicing factor involved in DM1 pathogenesis. We performed a combined RNA CUG repeat-specific FISH and MBNL1-specific immunofluorescence analysis on human DM1 myoblasts and fibroblasts that were either untreated or treated with the compounds to determine the intracellular localization of MBNL1 known to be immobilized in nuclear CUGexp RNA inclusions.5,47 As shown in Figure 5 and Figure S3A and B in untreated and DMSO-treated cells, MBNL1 co-localizes with nuclear RNA CUGexp foci and is detected predominantly in the inclusions. After treatment with either C16 or C51, we observed a displacement of MBNL1 from faint ribonuclear inclusions and its redistribution within the cell. In some of the larger foci, we detected immunofluorescence signals of MBNL1, indicating that this protein was not entirely dislodged from the RNA nuclear inclusions after treatment with either compound.
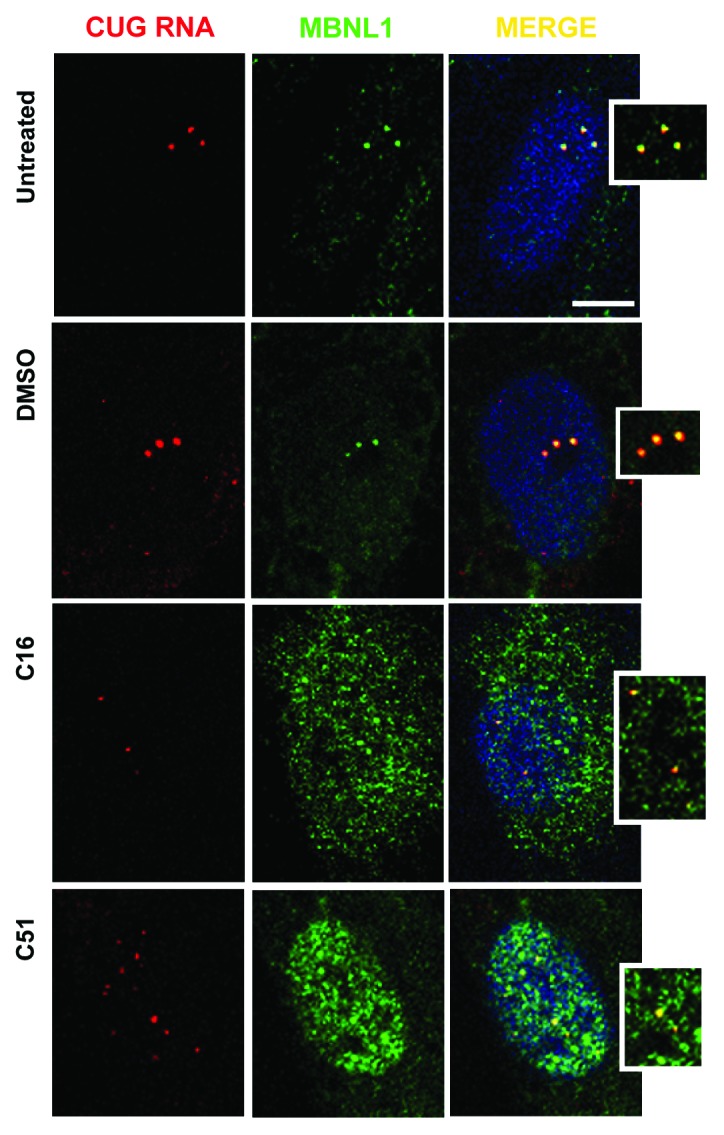
Figure 5. Treatment with C16 and C51 affects colocalization of MBNL1 with CUG RNA foci. Representative confocal images of RNA-FISH/IF of cultured human DM1 myoblasts that were either untreated or treated with DMSO (0.01%), C16 (0.5 μM), and C51 (30 μM). Treatment with C16 and C51 resulted in the dislodging of MBNL1 from faint nuclear CUGexp RNA foci and redistribution within the cells; DMSO had no such effect. Nuclei were stained with DAPI (blue); the CUGexp RNA foci are indicated by red fluorescence and MBNL1 by green fluorescence. Scale bar, 10 μm. For more images of RNA-FISH and MBNL1 immunofluorescence in DM1 fibroblasts, please refer to Figure S3A and B.
Based on this result, we sought to determine if disruption of the MBNL1/CUG repeat RNA complexes results from the binding of C16 and C51 to the repeats. Thus, we performed a filter binding assay (FBA) to measure the binding affinity of recombinant MBNL1 to synthetic (CUG)35 oligonucleotides in the presence of C16 and C51. The assay showed a lack of inhibitory potential of the compounds, even at high concentrations, on the in vitro interaction between the (CUG)35 hairpin RNA and MBNL1 (Fig. S3D). This result suggests that freeing MBNL1 from CUGexp RNA inclusions observed in situ after C16 and C51 treatment may be caused by a mechanism independent of the direct interaction between the compounds and either CUGexp RNA or MBNL1.
To conclude, colocalization of MBNL1 with mutant DMPK RNA is diminished after treatment with C16 and C51; however, this effect is not caused by the inhibitory binding of the compounds to either CUG repeat RNA or MBNL1.
Correction of MBNL1 regulated alternative splicing in DM1 cells upon C16 and C51 treatment
We sought to determine if the treatment that abolished CUGexp foci formation and sequestration of MBNL1 affects the alternative splicing of MBNL1-regulated pre-mRNAs that have been reported to be aberrantly spliced in human DM1 cells.9,27,48 For this, we tested the splicing pattern of endogenous SERCA1 exon 22 (SERCA1+E22), LDB3 exon 7 (LDB3+E7), MBNL1 exon 7 (MBNL1+E7), and DMD exon 78 (DMD+E78) by RT-PCR in proliferating human myoblasts. The splicing analyses were performed at different C16 and C51 concentrations to determine their dose-dependent effect on the aberrant splicing of tested pre-mRNAs (i.e., if they can increase the fraction of adult splicing isoforms and decrease the fetal isoforms). DM1 cells treated with either of the compounds but not DMSO alone showed a significant correction of aberrant splicing of all tested pre-mRNAs (Fig. 6A and B; Fig. S4A and B). The magnitude of this correction varied among the pre-mRNAs and was dose-dependent. In C16-treated cells, a reversal of the mis-splicing was detectable at 0.1 μM and increased with the compound concentration, with the most pronounced correction observed at the highest dose of C16 used (1.0 μM). In C51-treated cells, splicing alterations were not observed with lower doses of the compound (< 30 μM), but significant improvements in SERCA1+E22 and LDB3+E7 pre-mRNAs splicing patterns were detected at concentrations of C51 ≥ 30 μM, and no changes in DMD+E78 alternative splicing were observed with this treatment (Fig. 6A and B). Cells treated with equivalent concentrations of DMSO showed no effect on the splicing pattern (Fig. S4B).

Figure 6. Correction of aberrant MBNL1-dependent alternative splicing in DM1 cells after C16 and C51 treatment. (A and B) Representative agarose gel analysis of RT-PCR products of different splicing isoforms of several pre-mRNAs of DMD, SERCA1, MBNL1, and LDB3 in DM1 cells. The levels of the splicing variants of the pre-mRNAs were assessed by RT-PCR. DM1-specific splicing aberrations were corrected after treatment with either C16 or C51, the effects of which were dose-dependent. The experiments were performed in triplicate and quantitative results are shown as bar diagrams. The fraction of exon inclusion or exclusion (± SEM) was calculated by dividing the quantified signal of the PCR product band corresponding to the DM1-specific splice product by the total intensity of both splice products. Exon inclusion (+E); exon exclusion (-E). *, P < 0.01; **, P < 0.001. For more data on mis-splicing correction in DM1 cells, please refer to Figure S4A and B.
We also tested if C16 and C51 can rescue aberrant DM1-specific splicing in differentiated DM1 myoblasts and DM1 fibroblasts. As shown in Figure S4A, differentiated DM1 myoblasts with aberrant splicing patterns of SERCA1+E22, DMD+E78, LDB3+E7, and MBNL1+E7 responded to C16 and C51 by increasing the percentage of the adult isoforms with a corresponding decrease of the fetal isoforms. In C16-treated cells, the splicing correction was observed at both concentrations used, but the effect was more pronounced at 0.5 μM. Similarly, a correction of aberrant splicing was detected in C16-treated DM1 fibroblasts, in which three transcripts that were tested showed a dose-dependent improvement exhibited by the significant increase of adult isoforms of the SERCA1+E22, MBNL1+E7, and DMD+E78 splicing variants (Fig. S4A). In control fibroblasts and myoblasts, C16 and C51 caused no splicing alterations, and the different human cell lines expressing the normal length of CUG repeat in the DMPK gene had no splicing changes of SERCA1+E22, LDB3+E7, and DMD+E78 after treatment with the compounds (Fig. S4C).
We also determined whether C16 and C51 affected the splicing pattern of genes that are MBNL1-and CUGBP1-independent. We tested several pre-mRNAs regulated by NOVA1 (MAP4K4 and APLP2 mRNAs), PTBP1 (HMGCS1), PTBP2 (PPP3CB and CAPN3), and FOX2 (ECT2 and TSC2),49,50 in treated DM1 myoblasts. As shown in Figure S4D, no significant changes in the expression of alternative splicing patterns of the splice isoforms were observed after treatment with either compound compared with untreated cells, with the exception of the PPP3CB pre-mRNA. The splicing pattern of PPP3CB exon 13 showed the compounds dose-dependent changes detected by a decrease of expression of the isoform with the exon and this effect was more pronounced after the treatment with C16 than with C51.
Taken together, these results suggest that treatment with C16 and C51 could correct the aberrant splicing of MBNL1-dependent pre-mRNAs in DM1 cells without affecting the splicing pattern in normal non-DM1 cells. Importantly, these treatments had no effect on a subset of pre-mRNAs that are not regulated by CUGBP1 and MBNL1 splicing factors.
Discussion
Herein, we show evidence on the alleviation of DM1 molecular phenotypes associated with RNA containing expanded CUG repeats by the treatment of human DM1 myoblasts and fibroblasts with two small molecule compounds, the imidazolo-oxindole inhibitor C16 and the pyrimidine-based inhibitor C51. These compounds were previously identified as ATP-binding site-specific inhibitors and shown to block phosphorylation of PKR and other kinases.35–37 Our results indicate that C16 and C51 (or their derivatives) may have therapeutic potential in patients with DM1. Treatment with the compounds resulted in a redistribution of MBNL1 protein sequestered in CUGexp RNA foci and in a decrease of the steady-state levels of CUGBP1. These actions were correlated with correction of aberrant alternative splicing of MBNL1-dependent (SERCA1, DMD, MBNL1, LDB3) and CUGBP1-dependent (ITGA6, MTMR3, and SORBS1) pre-mRNA targets, shifting the patterns typical for DM1 cells to be more like those observed in non-DM cells. However, while the compounds caused the nuclear CUGexp RNA foci to become less abundant, as determined by in situ RNA hybridization and immunocytochemistry for MBNL1 protein, they did not prevent the formation of complexes of recombinant MBNL1 and synthetic (CUG)35 oligonucleotides as determined in vitro by the filter binding assay. Importantly, C16 and C51 did not affect DMPK mRNA levels, except for the highest concentrations of C51 used, which indicates that the mutant DMPK transcripts were rather dispersed within the nucleus after the compounds’ treatment without being degraded or released to the cytoplasm.
Thus far, several approaches have been used to develop DM1 therapeutics. The vast majority of them have focused on molecules that are CUG repeat binders and are able to inhibit the interactions between expanded CUG RNA and MBNL1 protein (reviewed in ref. 21). Given that the mechanism of RNA gain-of-function in DM1 remains elusive, and the complete list of factors affected by the toxic RNA needs to be determined, it is reasonable to examine novel candidate therapeutics that, in addition to CUGexp RNAs, could target other cellular components. Identification of such molecules has lately been described by Ketley A, et al.,34 who screened several libraries of small molecule compounds, including phosphatase and kinase inhibitors, using a medium-throughput phenotypic assay. Based on the identification of nuclear foci in DM1 cells using in situ hybridization and high-content imaging, the authors identified two compounds, chromomycin A3 and Ro 31-8220, of potential therapeutic benefit in DM1. Both compounds eliminate nuclear CUGexp RNA foci, reduce MBNL1 protein in the nucleus, affect ATP2B1 alternative splicing, and decrease steady-state levels of CELF1 protein. Interestingly, Ro 31-8220 has previously been identified as a PKC inhibitor and shown to affect the hyperphosphorylation of CELF1 and ameliorate the cardiac phenotype in a DM1 mouse model.19 However, studies by Ketley A, et al. demonstrate that Ro 31-8220 acts independently of PKC on DM1 pathomechanism, suggesting involvement of other kinases. Although the mechanism of the inhibitor action requires further investigation, the compound is likely to work independently of CUG repeat RNA binding.
Nuclear-retained DMPK CUGexp RNAs are complexed with MBNL-like proteins and other nuclear proteins and form multiprotein–RNA inclusions (Fig. 7). C16 and C51 kinase inhibitors cause diminishing of such ribonucleoprotein complexes. The mechanism of action of these two small molecular compounds in DM1 cells requires further investigation. Based on our results, their action did not involve binding to RNA containing CUG repeats. Binding to MBNL1 protein also appears unlikely because we did not detect any changes in the splicing pattern in normal fibroblasts and myoblasts after treatment with either compound. It appears possible that C16 and C51 act on the protein phosphorylation level by affecting the activity of various kinases (and thus, their downstream effectors) in the alleviation of DM1 molecular hallmarks. In this scenario, non-specific activation of CUGBP1 kinases reported in DM1 could be inhibited, resulting in reduced stability of CUGBP1 and a decrease in its steady-state levels. Moreover, C16 and C51 could affect activity of Src family kinases, which have recently been shown to bind to MBNL142-43. In this case, lower levels of tyrosine phosphorylated nuclear proteins, including splicing factors, could correct aberrant pre-mRNA splicing. Additionally, modification of the activity of various kinases that affect other proteins could trigger rearrangements within proteins of the CUGexp RNA foci, including their displacement and relocalization within the cell. In such a scenario, MBNL1 would be dislodged from diminishing ribonuclear inclusions as their constituents dissolve. Although the two kinase inhibitors do not seem to bind to CUG repeats or MBNL1, we cannot exclude the possibility that the small molecules load into proteins other than MBNL1, which are bound initially into foci, and the compound–protein complexes affect the loading of MBNL1 and other proteins into the foci. The C51 inhibitor may also work, at least partially, by downregulating mutant DMPK expression.
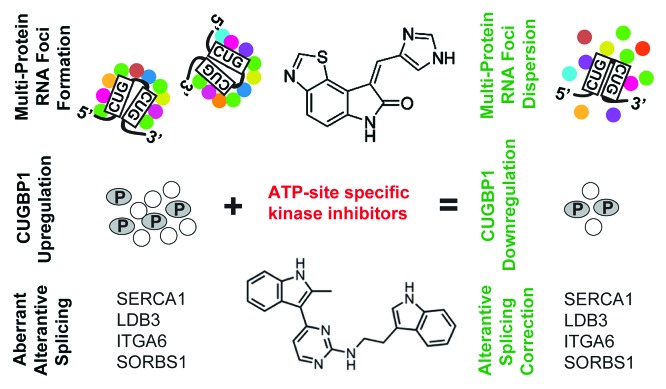
Figure 7. Kinase inhibitors as potential therapeutics for DM1. DM1 molecular phenotype is characterized by formation of nuclear multi-protein CUGexp RNA foci, CUGBP1 protein hyperphosphorylation, and upregulation, as well as aberrant alternative splicing of several pre-mRNAs. These features result, among others, from unspecific activation of various protein kinases in the presence of mutant DMPK transcripts. Utilization of small-molecule kinase inhibitors, C16 and C51, causes significant improvements of DM1 phenotype and their mechanism of action do not involve targeting of mutant CUG repeat transcripts.
The latest results by Botta, et al.20 suggest an additional pathomechanism in DM1 based on unspecific activation of Src family kinases. Altered activity of other kinases correlated with DM1 has previously been described by others.15–18 As a support of this notion, a recent report from the Brook laboratory34 indicated that small molecule kinase inhibitor Ro 31-8220 alleviates DM1 molecular hallmarks by eliminating nuclear CUGexp RNA foci and affecting two splicing factors involved in DM1 pathogenesis, MBNL1, and CELF1 proteins. Importantly, similar observations come from our study in which two different kinase inhibitors were tested. Although the specificity of inhibitors used by Brook and ourselves need further investigation, these results underscore the concept that targeting protein kinases may be beneficial in DM1.
Materials and Methods
Cell culture
Primary human myoblast and fibroblast cultures of unaffected and DM1 patients were used. Normal non-DM1 fibroblast cell lines (GM07492, GM07525, and GM08399) and DM1 patient-derived fibroblasts (GM03987, GM03989, and GM04033) were purchased from Coriell Cell Repositories. These cells, along with human embryonic kidney 293 (HEK293) cells and human hepatoma liver (HepG2) cells, were cultured in MEM medium (Lonza) supplemented with 10% FBS (Sigma), 1% Glutamax (Cellgro, Mediatech Inc.), 1x antibiotic/antimycotic solution (Sigma), and 1x MEM non-essential amino acid solution (Sigma). DM1 myoblast cell lines (9886, > 200 CTG repeats; 10010, > 200 CTGs; and 10011, > 350 CTGs) were used along with sex- and age-matched control non-DM1 myoblasts (10104, 9648, 10701). All muscle cells were cultured in low D-glucose (1.0 g/L) DMEM (Cellgro, Mediatech Inc.) supplemented with 15% FBS, 1x antibiotic/antimycotic solution, 5 μg/ml insulin (Sigma), 0.5 mg/ml BSA (Sigma), 10 ng/ml hEGF (Sigma), and 0.39 μg/ml dexamethasone (Sigma). To trigger myoblast differentiation, growth medium was removed from subconfluent cultures and replaced by DMEM medium supplemented with 0.5% horse serum (Sigma), 1x antibiotic/antimycotic solution, 10 μg/ml insulin, and 10 μg/ml apo-transferrin (Sigma).51
In vitro treatment with C16 and C51 and siRNA transfection
C16 was purchased from Sigma-Aldrich and C51 was synthesized at Weill Cornell’s Chemistry Core, and kindly provided by John Griffin and Ruslana Bryk. Both compounds were prepared as 10 mM stock solutions in anhydrous DMSO and stored at 20 °C. Further dilutions were made in sterile 1x PBS immediately prior to cell treatments. For the in vitro experiments, C16 was used at different concentrations based on previously published data (i.e., 0.05, 0.1, 0.2, 0.5, and 1.0 μM),39-41 whereas C51 was used at 2.0, 10, and 30 μM.36 In DMSO-treated cells, the solvent percentage corresponded to its concentration in the diluted active compounds (0.0005, 0.001, 0.002, 0.005, 0.01, and 0.3%). Human cells were counted and seeded at the same density in 6-well plates in complete medium (as described above) with addition of C16, C51, or DSMO. The treatments were conducted for 7 d with media changes occurring every 24 h.
For siRNA transfection, 50 nM of siCUGBP1 duplex (Futuresynthesis) (Table S3) was diluted in serum and antibiotic-free Opti-MEM (Invitrogen) and mixed with Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol. The mixture was added to the cells grown in a 6-well plate and incubated at 37 °C for 4 h. Fluorescent control siRNAs (Block-it Fluorescent Oligo, Invitrogen) were used to monitor transfection efficiency. At 72 h post-transfection, either C16 or C51 was added to the cell media, and growth was monitored for an additional 72 h.
RNA FISH and immunostaining
RNA FISH/IF was performed in DM1 cultured human myoblasts and fibroblasts as previously described.52 For details, please see Supplementary Material. To characterize the nuclear RNA foci, approximately 100 cells were selected at random from each treatment group. The same exposure was set up for all groups of images from a single experiment, and a z-stack sufficient to cover all of the foci in the nucleus was acquired for each cell with a slice thickness of 1 μm using either PL-Apo 63X or 100X/1.4 oil objective in conjunction with a cooled AxioCam HRc camera; images in one stack were overlaid and saved as TIFF files. Determination of the CUG RNA nuclear foci number was performed manually whereas quantitative analysis of the foci area (measured as squared pixels) was performed with ImageJ software (NIH) and analyzed using Microsoft Excel. All images were taken at the Electron and Confocal Microscopy Core at Adam Mickiewicz University (Poznan).
Western blotting
Cell protein extracts were prepared in 1xPB homogenate buffer (60 mM Tris-HCl, 2% SDS, 10% sucrose, 2 mM PMSF) and the protein concentration was measured by a NanoDrop spectrophotometer. Approximately 10–20 μg of protein lysates were separated on 12% SDS-PAGE and transferred onto nitrocellulose membranes (Sigma). Non-specific binding was blocked with 5% non-fat milk in TBS containing 0.1% Tween-20 (TBST) for 1 h at room temperature, and blots were incubated overnight at 4 °C with the following primary antibodies: anti-CUGBP1 (3B1) (Santa Cruz Biotechnology), anti-GAPDH (Millipore), and anti-α-Tubulin (Covance). After the overnight treatment, the membranes were washed in TBST and then incubated with secondary HRP-conjugated antibody. After several washes, immunoreactive bands were visualized using the WesternBright Quantum detection kit (Advansta Corp.).
RT-PCR analysis of aberrant alternative splicing pattern and quantitative real-time RT-PCR
Total RNA was extracted from cells with TRIzol reagent (Sigma) according to the manufacturer’s protocol. RNA concentration was measured with a NanoDrop spectrophotometer, and approximately 1 μg of RNA was reverse-transcribed with random primers (Promega) using SuperScript III reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. All cDNAs were diluted 3-fold with water prior to PCR. GoTaq Flexi DNA Polymerase (Promega) and 0.1 mM dNTP mix, 1.5 mM MgCl2, and 0.8 μM of each primer were used for the RT-PCR assays. For specifics on the primers and PCR conditions used to determine the splicing products, please refer to Table S1. The PCR amplification products were separated on a 1.8% agarose gel in 0.5xTBE buffer, stained with 0.5 μg/ml ethidium bromide, and quantified using Gel-Pro 3.1 software. Each cDNA sample was assayed in at least three independent RT-PCR reactions. The fractions of the alternative splicing isoforms were calculated by dividing the intensity of the PCR product band corresponding to the DM1-specific splicing variant by the total intensity of both splicing forms, i.e., normal and DM1-specific.
SYBR Green RT-PCR analysis of DMPK expression was performed according to the manufacturer’s recommendations (Applied Biosystems) using a LightCycler®480 SW1.5.1 real-time PCR system (Roche). RT-PCR was performed at an annealing temperature of 60 °C for 45 cycles with the primers shown in Table S2. Each sample was assayed in three independent RT-PCR reactions, and the data were normalized to the level of GAPDH mRNA.
Statistical analysis
All of the experiments were performed at least three times, and the representative results are shown. The data are presented as the mean ± SEM for experiments involving untreated and C16- and C51-treated cells. Statistical significance was determined by two-tailed the Student t test, and P values of < 0.01 or < 0.001 (depicted in figures as * or **, respectively) were considered to be statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Anti-MBNL1 (A2764) was a gift from Dr Charles Thornton. We would like to thank Jacquelynn E Larson for her editorial comments. Human DM1 and control non-DM1 myoblasts were obtained from the Telethon Biobank Network and EuroBioBank.
Funding
This work was supported by a grant from the Polish Ministry of Science and Higher Education (N N401 572140 to Wojciechowska M and 2011/01/B/NZ1/01603 to Sobczak K) and by the TEAM program grant to Sobczak K from the Foundation for Polish Science (co-financed by the European Union within the European Regional Development Fund) and by European Regional Development Fund within Innovative Economy Program (POIG.01.03.01-30-098/08) to Krzyzosiak WJ.
Glossary
Abbreviations:
- DM1
myotonic dystrophy type 1
- DMPK
dystrophia myotonica protein kinase
- FISH
fluorescence in situ hybridization
- IF
immunofluorescence
- CUGBP1
CUG repeat-binding protein
- ATP
Adenosine-5′-triphosphate
- PKR
protein kinase R
- DMSO
dimethyl sulfoxide
- SERCA1 (ATP2A1)
sarcoplasmic/endoplasmic reticulum calcium ATPase 1
- MBNL1
muscleblind-like 1 protein
- CUGexp RNA
mutant CUG repeat transcript
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- qRT-PCR
quantitative real time RT-PCR
- LNA
locked nucleic acids
- LDB3
LIM domain-binding protein 3
- DMD
dystrophin
- ITGA6
Integrin alpha-6
- MTMR3
myotubularin-related protein 3
- CAPZB
capping protein (actin filament) muscle Z-Line, beta
- SORBS1
sorbin and SH3 domain containing 1
- ATP2B1
ATPase, Ca++ Transporting, Plasma Membrane 1
- ABLIM1
actin binding LIM protein 1
- ERBB2IP
erbb2 interacting protein
- SGT2
small glutamine-rich tetratricopeptide repeat (TPR)-containing, beta
- KIDINS220
kinase D-interacting substrate, 220 kDa
- R3HDM2
R3H domain containing 2
- CTAGE5
CTAGE family, member 5
- MAP4K4
mitogen-activated protein kinase kinase kinase kinase 4
- APLP2
amyloid beta (A4) precursor-like protein 2
- HMGCS1
3-hydroxy-3-methylglutaryl-CoA synthase 1
- 38 PPP3CB
protein phosphatase 3, catalytic subunit, beta isozyme
- CAPN3
calpain 3, (p94)
- ECT2
epithelial cell transforming sequence 2 oncogene
- TSC2
tuberous sclerosis 2
References
- 1.Harper PS. Myotonic Dystrophy 3rd edn., (W.B. Saunders, London), 2001. [Google Scholar]
- 2.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001;276:7820–6. doi: 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- 3.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–11. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA. Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res. 2005;97:1152–5. doi: 10.1161/01.RES.0000193598.89753.e3. [DOI] [PubMed] [Google Scholar]
- 5.Mankodi A, Teng-Umnuay P, Krym M, Henderson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54:760–8. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- 6.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 7.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–41. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–88. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 9.Hino S, Kondo S, Sekiya H, Saito A, Kanemoto S, Murakami T, Chihara K, Aoki Y, Nakamori M, Takahashi MP, et al. Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1. Hum Mol Genet. 2007;16:2834–43. doi: 10.1093/hmg/ddm239. [DOI] [PubMed] [Google Scholar]
- 10.Timchenko NA, Wang GL, Timchenko LT. RNA CUG-binding protein 1 increases translation of 20-kDa isoform of CCAAT/enhancer-binding protein beta by interacting with the alpha and beta subunits of eukaryotic initiation translation factor 2. J Biol Chem. 2005;280:20549–57. doi: 10.1074/jbc.M409563200. [DOI] [PubMed] [Google Scholar]
- 11.Timchenko LT, Salisbury E, Wang GL, Nguyen H, Albrecht JH, Hershey JW, Timchenko NA. Age-specific CUGBP1-eIF2 complex increases translation of CCAAT/enhancer-binding protein beta in old liver. J Biol Chem. 2006;281:32806–19. doi: 10.1074/jbc.M605701200. [DOI] [PubMed] [Google Scholar]
- 12.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–24. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18:840–5. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Costa JM, Garcia-Lopez A, Zuñiga S, Fernandez-Pedrosa V, Felipo-Benavent A, Mata M, Jaka O, Aiastui A, Hernandez-Torres F, Aguado B, et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum Mol Genet. 2013;22:704–16. doi: 10.1093/hmg/dds478. [DOI] [PubMed] [Google Scholar]
- 15.Jin J, Wang GL, Salisbury E, Timchenko L, Timchenko NA. GSK3beta-cyclin D3-CUGBP1-eIF2 pathway in aging and in myotonic dystrophy. Cell Cycle. 2009;8:2356–9. doi: 10.4161/cc.8.15.9248. [DOI] [PubMed] [Google Scholar]
- 16.Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, Woodgett J, Killian J, Timchenko NA, Timchenko LT. GSK3β mediates muscle pathology in myotonic dystrophy. J Clin Invest. 2012;122:4461–72. doi: 10.1172/JCI64081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, Wang GL, Albrecht JH, Timchenko LT. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res. 2008;314:2266–78. doi: 10.1016/j.yexcr.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang GS, Kuyumcu-Martinez MN, Sarma S, Mathur N, Wehrens XH, Cooper TA. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J Clin Invest. 2009;119:3797–806. doi: 10.1172/JCI37976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Botta A, Malena A, Tibaldi E, Rocchi L, Loro E, Pena E, Cenci L, Ambrosi E, Bellocchi MC, Pagano MA, et al. MBNL142 and MBNL143 gene isoforms, overexpressed in DM1-patient muscle, encode for nuclear proteins interacting with Src family kinases. Cell Death Dis. 2013;4:e770. doi: 10.1038/cddis.2013.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–9. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–5. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, Furling D, Platenburg GJ, Gourdon G, Thornton CA, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci U S A. 2009;106:13915–20. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.González-Barriga A, Mulders SA, van de Giessen J, Hooijer JD, Bijl S, van Kessel ID, van Beers J, van Deutekom JC, Fransen JA, Wieringa B, et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol Ther Nucleic Acids. 2013;2:e81. doi: 10.1038/mtna.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol Ther. 2013;21:380–7. doi: 10.1038/mt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.François V, Klein AF, Beley C, Jollet A, Lemercier C, Garcia L, Furling D. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat Struct Mol Biol. 2011;18:85–7. doi: 10.1038/nsmb.1958. [DOI] [PubMed] [Google Scholar]
- 28.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc. 2009;131:9767–79. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc Natl Acad Sci U S A. 2009;106:16068–73. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol. 2012;7:856–62. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jahromi AH, Nguyen L, Fu Y, Miller KA, Baranger AM, Zimmerman SC. A novel CUG(exp)·MBNL1 inhibitor with therapeutic potential for myotonic dystrophy type 1. ACS Chem Biol. 2013;8:1037–43. doi: 10.1021/cb400046u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jahromi AH, Honda M, Zimmerman SC, Spies M. Single-molecule study of the CUG repeat-MBNL1 interaction and its inhibition by small molecules. Nucleic Acids Res. 2013;41:6687–97. doi: 10.1093/nar/gkt330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Natl Acad Sci U S A. 2009;106:18551–6. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ketley A, Chen CZ, Li X, Arya S, Robinson TE, Granados-Riveron J, Udosen I, Morris GE, Holt I, Furling D, et al. High content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC independent pathway in Myotonic Dystrophy cell lines. Hum Mol Genet. 2013;23:1551–62. doi: 10.1093/hmg/ddt542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jammi NV, Whitby LR, Beal PA. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem Biophys Res Commun. 2003;308:50–7. doi: 10.1016/S0006-291X(03)01318-4. [DOI] [PubMed] [Google Scholar]
- 36.Bryk R, Wu K, Raimundo BC, Boardman PE, Chao P, Conn GL, Anderson E, Cole JL, Duffy NP, Nathan C, et al. Identification of new inhibitors of protein kinase R guided by statistical modeling. Bioorg Med Chem Lett. 2011;21:4108–14. doi: 10.1016/j.bmcl.2011.04.149. [DOI] [PubMed] [Google Scholar]
- 37.Chen HM, Wang L, D’Mello SR. A chemical compound commonly used to inhibit PKR, 8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one, protects neurons by inhibiting cyclin-dependent kinase. Eur J Neurosci. 2008;28:2003–16. doi: 10.1111/j.1460-9568.2008.06491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjević K, et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell. 2011;147:1384–96. doi: 10.1016/j.cell.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimazawa M, Hara H. Inhibitor of double stranded RNA-dependent protein kinase protects against cell damage induced by ER stress. Neurosci Lett. 2006;409:192–5. doi: 10.1016/j.neulet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- 40.Shimazawa M, Ito Y, Inokuchi Y, Hara H. Involvement of double-stranded RNA-dependent protein kinase in ER stress-induced retinal neuron damage. Invest Ophthalmol Vis Sci. 2007;48:3729–36. doi: 10.1167/iovs.06-1122. [DOI] [PubMed] [Google Scholar]
- 41.Page G, Rioux Bilan A, Ingrand S, Lafay-Chebassier C, Pain S, Perault Pochat MC, Bouras C, Bayer T, Hugon J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience. 2006;139:1343–54. doi: 10.1016/j.neuroscience.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 42.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–47. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 43.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105:20333–8. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen S, Talukdar I, Webster NJ. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol Cell Biol. 2009;29:871–80. doi: 10.1128/MCB.01709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wojciechowska M, Krzyzosiak WJ. Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum Mol Genet. 2011;20:3811–21. doi: 10.1093/hmg/ddr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–70. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 48.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–97. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 49.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–7. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 2012;26:1626–42. doi: 10.1101/gad.191338.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, Schoser B, Puymirat J. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. 2009;36:181–90. doi: 10.1016/j.nbd.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 52.de Mezer M, Wojciechowska M, Napierala M, Sobczak K, Krzyzosiak WJ. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011;39:3852–63. doi: 10.1093/nar/gkq1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.