

Abstract
Human apurinic/apyrimidinic endonuclease 1 (APE1) functions mainly in DNA repair as an enzyme removing AP sites and in redox signaling as a coactivator of various transcription factors. Based on these multifunctions of APE1 within cells, numerous studies have reported that the alteration of APE1 could be a crucial factor in development of human diseases such as cancer and neurodegeneration. In fact, the study on the combination of an individual's genetic make-up with environmental factors (gene-environment interaction) is of great importance to understand the development of diseases, especially lethal diseases including cancer. Recent reports have suggested that the human carcinogenic risk following exposure to environmental toxicants is affected by APE1 alterations in terms of gene-environment interactions. In this review, we initially outline the critical APE1 functions in the various intracellular mechanisms including DNA repair and redox regulation and its roles in human diseases. Several findings demonstrate that the change in expression and activity as well as genetic variability of APE1 caused by environmental chemical (e.g., heavy metals and cigarette smoke) and physical carcinogens (ultraviolet and ionizing radiation) is likely associated with various cancers. These enable us to ultimately suggest APE1 as a vital marker for the prediction of environmental carcinogenesis risk.
1. Introduction
Apurinic/apyrimidinic endonuclease/redox effector factor 1 (APE1/Ref-1, APEX1, here referred to as APE1) has multifunctions as a base excision repair enzyme and as a redox coactivator of a number of important transcription factors. Human APE1 protein working as a monomer is comprised of 318 amino acids and is divided into two different domains [1]. The N-terminal domain, including the nuclear localization signal (NLS) region, exerts the redox regulation activity, while the C-terminal domain is employed in the endonuclease activity at abasic site [2]. It is well known that these two domains operate independently, as explained in various reports about the mutation of specific amino acids such as Cysteine 65 (at the N-terminal end) and Histidine 309 (at the C-terminal end) in each domain [3, 4]. Unlike human APE1 protein, there is only one active function in Escherichia coli Xth and Saccharomyces cerevisiae APN2 which is APE1 endonuclease activity [5]. Human APE1 homology with other organisms includes the C-terminus which is highly conserved among various classes while the N-terminal domain is mostly conserved in mammal [6, 7].
APE1 is involved not only as key element of base excision repair and as a redox factor for regulation of transcription factors, but also as an RNA modulator and transcriptional repressor. On account of the multifunction of APE1 in humans, this protein is suggested as a crucial target in the pathology of cancers, neuronal diseases, aging, among others. Along with the APE1 related studies in pathology, previous research in our group demonstrated the correlation between environmental toxicants such as heavy metals and redox factors, which are important in the suppression of carcinogenesis [8–11]. Gene-environment interaction is regarded as an essential point to investigate diseases caused by environmental toxicants.
In this review, we begin with an exploration of various intracellular functions of APE1 which include DNA repair and redox regulation and continue to summarize the human pathologies related to APE1 alterations. Notably, we emphasize APE1 as a potential marker for risk prediction of environmental diseases induced by exposure to heavy metals, cigarette smoke toxicants, and radiation in terms of gene-environment interaction.
2. Multifunctions of APE1
A transacting protein, APE1, is considered an essential protein for maintaining cellular stability through various intracellular functions including the regulation of DNA repair and redox status (Figure 1). Since numerous authors have recently reviewed the extensive studies related to APE1's multifunction [13–16], our review focuses on the APE1's distinct functions.
Figure 1.
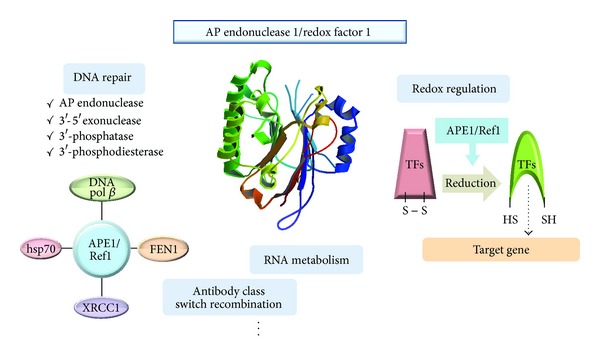
Multifunction of APE1/Ref1. APE1 has varied and independent functions and has an essential role in maintaining cellular stability. APE1 enzymatically restores damaged DNA bases and interacts with other proteins involved in DNA repair. APE1 regulates redox status of various transcription factors including AP-1 and p53 with its Cys residue. The crystal structure of APE1 (PDB ID : 1HD7) was identified by Beernink et al. [12].
2.1. APE1 in DNA Repair Mechanisms
2.1.1. DNA Repair Activity of APE1
Base excision repair (BER), one of the DNA repair mechanisms, is crucial to maintenance of genomic stability by restoring damaged DNA bases. Through the BER pathway, an apurinic/apyrimidinic (AP) site is temporarily generated by glycosylase [17, 18] and can increase cytotoxicity and mutagenicity caused by blocking DNA replication [19]. APE1 recognizes the AP site and plays a vital role in initiation of BER as cleavage 5′-phosphodiester bonds at the AP site, generating a 3′-hydroxyl group and a 5′-2′-deoxyribose phosphate (dRP) group [20]. AP endonuclease activity of APE1 was demonstrated by Marenstein et al. [17] to show that the protein acts on the AP site in double-stranded DNA as well as single-stranded DNA suggesting it may act on each substrate during different nuclear states. Also, they showed that the efficiency of its activity was comparable in the two different forms of DNA.
A ubiquitously expressed protein, APE1, has been identified as carrying not only an AP endonuclease activity, but also other catalytic activities such as showing 3′-5′ exonuclease, 3′-phosphatase, and 3′-phosphodiesterase activity [21–24]. These additional activities excise 3′-end blocking groups which might induce genomic instability like mismatched bases. Although mammalian APE1 has weak activity of 3′-damage excision relative to other phyla (E. coli or yeast) [25–27], this activity is required to remove 3′-end blocking groups produced by radiation, ROS, or mammalian glycosylases NEIL1 and NEIL2 [1, 28–30].
2.1.2. APE1 Interaction with Other Proteins Associating BER
APE1 is also involved in DNA repair processes by interacting with other components of DNA repair, such as DNA polymerase β, proliferating cell nuclear antigen (PCNA), X-ray cross-species complementing 1 (XRCC1), and others, in addition to its own independent biochemical activity. Various proteins interacting with APE1 are involved in the BER process by stimulating the protein's activity, recruiting other factors, or enhancing the AP endonuclease activity. Interactions between APE1 and DNA polymerase β or flap endonuclease 1 (FEN1) induce the removal of the remaining abasic site at the 5′-end of the cleavage site after excision of the AP site by APE1 [31, 32]. DNA polymerase β synthesizes new matching nucleotides and then removes 5′-end of APE1 products to proceed to the last step of BER, whereas FEN1 merely cleaves downstream of the DNA nick generating multiple gaps for DNA polymerases [28, 33, 34]. Additionally, interaction between FEN1 and PCNA enhances the excision reaction as well as coordinating long-patch BER [32]. Previous research in our group demonstrated that the interaction of growth arrest and DNA damage inducible, alpha (Gadd45α) with PCNA plays a critical role in modulating BER activity by affecting the involvement of APE1 [35, 36]. In addition, our group showed that the interaction between Gadd45α and both APE1 and PCNA is induced by p53-mediated BER activity with organic selenium compounds [37]. Physical association of APE1 with XRCC1 is strongly implicated in the processing and repair of ssDNA breaks in mammalian cells [38, 39], leading to reinforcing of AP endonuclease and 3′-phosphodiesterase activity [40]. Indeed, heat-shock protein 70 (HSP70) is one of the heat shock protein members involved in general stress response and can be induced by heat shock, oxidation, and other stresses [41]. Additionally, several reports demonstrated that a direct interaction between APE1 and HSP70 strongly stimulated the endonuclease activity of APE1 at AP site, as evident by APE assay with varying amounts of HSP70 [42–44]. This suggested that HSP70 might be involved in the protective mechanism (so called BER) against oxidative stress. Altogether, the multiple contributions of APE1 to DNA repair activity in processing BER are evidence that this protein is crucial for maintaining genetic integrity and cellular existence.
2.2. Redox Activity of APE1
2.2.1. Principal of Redox Activity in APE1
Along with the repair activity of APE1, this protein is a redox factor that regulates various transcription factors [17, 45]. The modulation of transcription factors activity is controlled by the redox state of reactive Cys residues both in the DNA-binding domain of some transcription factors and in the N-terminus domain of APE1. APE1 is a unique redox factor because of the lack of C-X-X-C motif which exists in most redox regulatory factors, such as thioredoxin, and because of adequate position of Cys residues allowing the formation of disulfide bonds in redox process [46, 47]. It has been reported that the Cys 65 residue in APE1 is crucial for redox-activation [6, 48], in spite of the buried location of Cys 65 making it poorly accessible by other proteins [49]. In thiol-mediated redox reactions, a Cys 65 residue of APE1 serves as the nucleophilic residue to form disulfide bonds between APE1 and the target transcription factor causing a conformational change of APE1 [46, 50]. After the mixed disulfide bond formation, another Cys residue in APE1, Cys 93, acts as a resolving residue for reduction of the target protein resulting in the oxidized redox factor [46].
2.2.2. Regulation of Transcription Factors by Redox Regulation
APE1 has been identified as a modulator of DNA-binding activity of various redox-sensitive transcription factors including both ubiquitous (AP-1, Egr-1, NF-κB, p53, CREB, and HIF-1α) and tissue-specific (PEBP-2, Pax-5 and Pax-8, and TTF-1) by regulating the redox state [46, 51–60]. Among these transcription factors affected by the redox activity of APE1, AP-1 and p53 have been reported to be involved in carcinogenesis [46]. Activator protein-1 (AP-1), which is involved in cellular proliferation, differentiation, and apoptosis, was first shown to have regulated DNA binding activity caused by APE1 because of a reducing Cys residue in the AP-1's DNA binding domain [51, 61]. APE1 enhances the DNA-binding activity of AP-1 by promoting its dimerization by regulating redox of the basic DNA binding domain of c-Jun and C-Fos [61]. APE1 also controls DNA binding activity of p53 by reducing it which is a transcriptional regulatory protein and a critically crucial tumor suppressor [55, 62]. Although p53 appears to have a redox-independent mechanism, its redox activation can be induced by a general redox factor because of low binding affinity to DNA in oxidized p53 [62, 63]. p53 affects many DNA repair pathways which depend on genotoxic stress; obviously APE1 plays a crucial role in the regulation of DNA repair [64, 65].
2.3. Other Functions of APE1
2.3.1. RNA Metabolism and APE1
The relation between APE1 and RNA metabolism has been widely investigated recently. APE1 can degrade a basic site in RNA, which suggests that APE1 plays a role in RNA quality control by removing damaged RNA templates [97, 98]. Barnes and colleague documented that human APE1 is able to cleave an RNA molecule at the coding region determinant of the c-myc mRNA [99]. This RNA modulation activity of APE1 is controlled by the N-terminal amino acids, which indicate that APE1 is involved in the posttranslational regulation, even though this mechanism is until now not sufficiently clear.
2.3.2. Immunoglobulin Modification by APE1
A newly identified role of APE1 is in the antibody class switch recombination (CSR). Earlier study reported a slight decline of CSR in an APE1-haplodeficient mouse model [100]. More recently, direct evidence shows a critical role of APE1 in CSR in mouse B cell line [101]. However, there are many discordant studies about immunoglobulin CSR, and further investigations are necessary to identify exact mechanisms.
3. APE1 in Human Pathologies
Since APE1 is well known to have AP endonuclease and redox activities [102], and more recently discovered functions, such as affecting RNA metabolism [97], the importance of APE1 in human pathologies is not unexpected. Since the role of APE1 in human pathologies including cancer has been recently reviewed in detail [1, 13, 103], we will only briefly discuss the issue.
3.1. Cancer
APE1 has received significant attention as an attractive target for pharmacological treatment in some cancer types. Alteration of APE1 expression and localization is, in particular, a well-established common feature in different neoplastic diseases [31], suggesting that APE1 may have prognostic and/or predictive significance in cancer. Fishel et al. [104] demonstrated that using siRNA technology that reduced levels of APE1 dramatically slows the growth of ovarian cancer cells, both in vitro and in vivo. In addition, a decrease in APE1 protein levels resulted in pancreatic cancer cell growth inhibition, increased apoptosis, and altered cell cycle progression [105].
APE1 localization is regulated, though the mechanism by APE1 compartmentalization is not clearly understood. In general, APE1 is preferentially expressed in nuclear, but the nuclear, cytoplasmic, and nuclear/cytoplasmic expressions were found in several types of cancer including epithelial ovarian cancer [106], thyroid carcinomas [107], and non-small-cell lung cancer [108].
The dysregulation of APE1 expression is considered as a potential marker to predict the sensitivity of the tumor against radio- or chemotherapy. An inverse relationship between radiosensitivity and the levels of APE1 was reported in cervical carcinoma [109], colorectal cancer [110], and pancreatic cancer [111]. Moreover, Robertson et al. [112] have suggested that the overexpressing APE1 observed in human testicular cancer might lead to cellular protection from bleomycin treatment.
APE1 polymorphism is also important in cancer susceptibility and development. Eighteen polymorphisms in APE1 have been reported [113], but the most extensively studied polymorphism is D148E (rs1130409) [114, 115]. Although the D148E polymorphism of APE1 is frequent and does not impart a reduction in AP endonuclease efficiency [114], numerous reports have suggested that D148E polymorphism is associated with increased susceptibility to colorectal, gastric, and prostate cancer, as well as cutaneous melanoma [116–120]. The APE1 −656T > G in the promoter region is another widely studied polymorphism which is suggested to influence the gene expression at the transcriptional level. The functionally significant −656T > G polymorphism of APE1 contributes to the susceptibility to breast and cervical cancers [121, 122]. In contrast, the −656T > G polymorphism shows a decreased risk of bladder cancer [123]. The polymorphism of APE1 D148E and −656T > G has also been extensively studied in lung cancer [80, 124–127], but confusing results demand further elucidation.
3.2. Neuropathology
APE1 is highly expressed in the central nervous system (CNS), albeit it varies in different cell types and regions of the human brain [128]. Vasko et al. [129] demonstrated that decreased APE1 level in primary rat hippocampal or sensory neuronal cell caused the inhibition of cell viability and enhanced apoptosis and DNA damage under hydrogen peroxide treatment. In particular, the inherited defect in DNA repair pathways was suggested as one of the main causes for diverse neurological disorders in human.
Numerous studies have suggested chronic oxidative stress as a cause of neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS), implying the potential importance of DNA repair genes, including APE1, as risk factors. AD causes functional impairments through neuron losses in the cerebral cortex, whereas PD is characterized by motor deficits and cell death in the substantia nigra. ALS is a debilitating disease that causes muscle weakness and atrophy throughout the body due to the degeneration of the upper and lower motor neurons.
The hippocampus and surrounding temporal cortex of patients with AD showed an increased expression of APE1 levels relative to matched controls in senile plaques and plaque-like structures [130]. Recently, Marcon et al. [131] have reported that an increased nuclear expression of APE1/Ref-1 in neuronal and glial cells of the cerebral cortex in both familial and sporadic AD brains might be associated with the cellular adaptive response to the oxidative stress condition. In contrast, the frontal cortical levels and activity of APE1 were significantly lower in 11 patients with sporadic ALS than in controls [132].
Recently, Gencer et al. [133] have suggested that APE1 genetic variant (D148E) might be a risk factor by causing the loss of dopaminergic cells in the substantia nigra and locus caeruleus, and, ultimately, the development of PD. In addition, APE1 mutations including the missense variants L104R, E126D, D148E, D283G, and G306A were found in eight of 11 patients with ALS and familial ALS [134]. Although the correlations between APE1 malfunction and various neuropathologies have been widely accepted, few studies have focused on the mechanism of the protective effect of APE1 as a target for future therapeutic development.
3.3. Age-Associated Disorders
The accumulation of oxidative DNA damage, which leads to cellular malfunctions, has been considered as a main cause of aging [135]. Consequently, it has been suggested that a decrease of the repair capacity to remove oxidatively damaged DNA correlates with age-related disease. Indeed, Intano et al. [136] showed that an 85% decline in BER activity was observed in brain nuclear extracts and a 50% decrease in liver nuclear extracts prepared from old mice compared with 6-day-old mice. Recently, Swain and Rao [137] have demonstrated that the APE1 activity in rat brain decreased significantly with age. Since APE1 decreases intracellular ROS by inhibiting rac1 regulated NAD(P)H oxidase [138], its expression change can also affect the aging. The gene expression of mRNA and APE1 enzyme decreased with age in the lenses of rats, resulting in a decrease in the repair capabilities and an accumulation of damaged DNA [139]. In addition to the alteration of APE1 expression and activity, the age-dependent redistribution of APE1 in the nucleus and mitochondria was found in mouse liver [140]. Moreover, recent study has reported that APE1 was an essential factor stabilizing telomeric DNA and its deficiency was associated with telomere dysfunction and segregation [141], suggesting the deeper molecular mechanism of APE1 in aging because dysregulated or shorter telomeres are thought to be a cause of aging.
3.4. Other Diseases
Other than in the disorders mentioned above, APE1 deregulation has also been demonstrated in other pathologies. APE1 polymorphism significantly increased the risk of myocardial infarcts [142] and ulcerative colitis [143]. Moreover, Jiang et al. [144] have demonstrated that the inhibition of APE1 redox activity using a small molecule inhibitor APX3330 blocked retinal angiogenesis in vitro and in vivo. The inhibited APE1 redox function blocks the ability of HIF-1a to bind to various downstream target promoters including angiogenic molecules [145, 146]. This evidence suggests that APE1 may have potential as a therapeutic target for antiangiogenesis treatment in retinal neovascular disorders.
4. APE1 as a Prediction Marker of Environmental Carcinogenesis Risk
Almost all diseases result from complex interactions between an individual's genetic make-up and environmental factors. There is great significance of gene-environment interaction in the development of diseases, especially lethal diseases including cancer. In particular, several reports suggested that genetically based variability (silencing, polymorphism) of the proteins involved in cancer metabolism may influence susceptibility to environmental carcinogens [147]. Here, we summarize the interference of APE1 expression and function with environmental factors including heavy metals, smoking, and radiations, with emphasis on the gene-environment interaction studies focused on APE1 in terms of human pathologies (Table 1).
Table 1.
Interference of APE1 upon environmental carcinogen exposure using in vitro and in vivo mammalian models and human population samples.
| Environmental factor | Subject | Dose | Effect on APE1 | Reference |
|---|---|---|---|---|
| Heavy metal | ||||
| Arsenic | Human skin cell line | 0.005–5 μM | Decrease of APE1 mRNA expression | [66] |
| Human lung cell line | 5 μM | Decrease of APE1 mRNA expression | [67] | |
| Human skin cell line | 0.1–5 μM | Increase of APE1 protein expression (short-term) | [68] | |
| 0.1 or 0.5 μM | Decrease of APE1 protein expression (long-term) | |||
| >1 μM | Dose-dependent decrease of APE1 mRNA expression | |||
| Human lung and skin cell lines | <1 μM | Increase of APE1 protein expression | [69] | |
| 5–100 μM | Dose-dependent increase of APE1 mRNA expression | |||
| Mouse embryo cell line | 10–75 μM | Dose-dependent increase of APE1 protein expression | [70] | |
| 10–100 μM | Dose-dependent increase of APE1 activity | |||
| Human population | — | Induction of APE1 polymorphism (D148E) | [71] | |
| Human population | — | Induction of skin lesions with APE1 polymorphism (D148E) | [72]∗ | |
| Human kidney cell line | 100 μM | No effect on APE1 activity | [73] | |
|
| ||||
| Cadmium | Human colon cell line | >10 μM | Decrease of APE1 mRNA expression | [74] |
| >25 μM | Decrease of APE1 activity | |||
| Human kidney cell line | 100 μM | Decrease of APE1 activity | [73] | |
| Human population | — | Decrease of APE1 mRNA expression | [75] | |
| Human breast and cervix cell lines | 20–80 μM | No effect on APE1 protein expression and activity | [76] | |
|
| ||||
| Lead | Mouse brain cell | 10 μM | APE1 accumulation in nucleus | [77] |
| Hamster ovary cell line | 0.5–500 μM | Dose-dependent accumulation of AP sites and decrease of APE1 activity | [4] | |
| Human kidney cell line | 100 μM | Decrease of APE1 activity | [73] | |
| Human population | — | Induction of APE1 polymorphism (D148E) | [78] | |
| Human population | — | No effect on APE1 mRNA expression | [75] | |
|
| ||||
| Smoking | ||||
| Swiss ICR albino mice | 119, 292, 438, 631 mg/m3 (TSM§) | Decrease of APE1 protein expression in brain tissue | [79] | |
| Human population | — | Induction of lung cancer with APE1 polymorphism (D148E and −656T > G) | [80–83]∗ | |
| Human population | — | Induction of bladder cancer with APE1 polymorphism (D148E) | [84]∗ | |
| Human population | — | No effect on the induction of lung cancer with APE1 polymorphism (D148E) | [85, 86] | |
| Human population | — | No effect on the induction of bladder cancer with APE1 polymorphism (D148E) | [87] | |
|
| ||||
| Radiation | ||||
| Ultraviolet | SKH-1 hairless mice | 5 days a week, 9 h a day at 10,000 lx (UVC) | Increase of APE1 mRNA expression | [88] |
| Human lymphocyte | 4 J/m2 (UVA) | Induction of APE1 polymorphism (D148E) | [89] | |
| Human cervix cell line | 0.2 J/cm2 (UVA) | Induction of APE1 relocalization to nuclear speckles | [90] | |
|
| ||||
| Ionizing radiation | Human population | — | Induction of APE1 polymorphism (D148E) | [91] |
| Human population | — | Induction of breast cancer with APE1 polymorphism (D148E) | [92] | |
| Human population | — | Induction of APE1 polymorphism (D148E) | [93] | |
| Human lung cell line | 200 cGy/min (X-ray) | Increase of APE1 protein expression | [94] | |
| Human lung cell line | 4, 16 Gy (X-ray) | Increase of APE1 protein expression | [95] | |
| Human blood culture | 80 cGy/min (X-ray) | Induction of APE1 polymorphism (D148E) | [89] | |
| Human lymphocyte | 1 Gy/min (X-ray) | Induction of APE1 polymorphism (D148E) | [96] | |
∗It represents study of gene-environment interaction; §TSM is abbreviation of total suspended matter in average after burning cigarette.
4.1. Heavy Metals
4.1.1. Arsenic
Arsenic (As) is a common environmental contaminant that enters humans through drinking water. In 2002, the International Agency for Research on Cancer (IARC) has classified As in drinking water as a group 1 human carcinogen [148]. Besides the induction of different types of cancer (skin, bladder, liver, kidney, and lung) [149, 150], sublethal exposure to As can cause severe human health problems such as diabetes [151] and neurological diseases (Alzheimer and Parkinson's) [152, 153]. While epidemiological studies have clearly demonstrated the harmful effects of As with respect to the induction of human diseases, the mechanisms of toxicity remain largely unknown except ROS generation [154].
It is known that As can affect both the endonuclease and the redox functions of APE1 to increase oxidative stress and inhibit DNA repair. In particular, APE1 activity is affected indirectly by As through the changes in transcription levels [66, 68]. Sykora and Snow [69] have reported that APE1 mRNA exhibited significant dose-dependent downregulation in response to low, physiologically relevant doses of As. Similarly, through the analysis of gene expression profiling by As exposure in human lung cells, the exposure to sodium arsenite for 4 hours decreased APE1 mRNA level [67]. In contrast, several reports found the stimulated APE1 expression and activity in cultured cells exposed by submicromolar As doses [69, 70, 155].
APE1 is associated with the oxidative biomarker, 8-hydroxy-2′-deoxyguanosine (8-OHdG), which can be induced by As exposure. The repair of 8-OHdG lesions requires both apurinic/apyrimidinic endonuclease (APE1) and human 8-oxoguanine glycosylase (hOGG1) [156]. Indeed, the polymorphisms (D148E) in the APE1 gene led to the decreased repair ability of oxidative DNA damage [157] and affected the urinary 8-OHdG concentrations in Vietnamese exposed to As [71]. Moreover, Caucasians showed higher mutant frequencies in APE1 than those of African and Asian populations in response to As. Among Asian populations, the Bangladeshi population showed relatively higher mutant allele frequencies of the APE1 D148E [71]. Another study on an As-exposed population conducted in Bangladesh explored some polymorphisms in BER genes, providing evidence that APE1 was related to As-induced skin lesions [72]. These accumulated data emphasize the importance of the combination of APE1 polymorphism with heavy metal exposure in terms of human disease development.
4.1.2. Cadmium
Cadmium (Cd) is a hazardous heavy metal that induces cytotoxicity and carcinogenicity upon persistent environmental exposure. In 1993, the International Agency for Research on Cancer (IARC) classified Cd compounds as group 1 carcinogens in humans [158]. Chronic exposure to Cd causes a wide range of toxicity-related diseases, including cancer in the lung, prostate, kidney, liver, and testis in humans and other mammals [159, 160]. Although the molecular mechanisms of toxicity and carcinogenicity of Cd remain poorly understood, two commonly suggested mechanisms for toxicity are the induction of oxidative DNA damage and the inhibition of DNA repair [160–162]. BER triggered by oxidative DNA damage is one of the important target mechanisms against Cd genotoxicity [163]. In particular, Cd inhibits the initial steps of BER, including the removal of AP site by APE1 endonuclease activity [164].
McNeill et al. [73] reported that Cd selectively inhibited APE1 endonuclease activity in whole-cell extracts but had no significant effect on single nucleotide gap filling, 5′-flap endonuclease, and nick ligation activities. In addition, recent evidences have demonstrated that Cd also impaired APE1 mRNA level as well as its activity [74, 75]. With respect to the potential role of tumor suppressor p53 in mechanism study for cadmium-induced inhibition of APE1 activity, the genotoxic stress was induced by Cd activated p53 and led to a significant downregulation of APE1 by p53 in HCT116 p53+/+ cells [74]. However, another study has found that in vivo treatment of human cells with Cd at sublethal concentrations had no effect on the APE1 activity [76]. Although they suggested that the intracellular concentrations of free Cd do not reach the levels required for the inhibition of APE1 due to the complexity of Cd within the cells [76], further studies are needed to fully demonstrate the effects of Cd with respect to APE1 activity.
4.1.3. Lead
Lead (Pb) exposure is hazardous to human health because it is widely distributed and persists in the environment. International Agency for Research on Cancer (IARC) classified it as possible human carcinogen (group 2B) [165] and its inorganic compounds as probable human carcinogens (group 2A) [166]. A number of human epidemiological studies and animal experiments have reported the harmful effects of Pb when it causes serious damage to bone, kidney, lung, nervous system, and red blood cell function [167, 168]. Although the induction of oxidative stress, the inhibition of DNA repair, and the formation of DNA/protein crosslink have been suggested as main toxic mechanisms of Pb exposure [169], the results are still conflicting in terms of promoting genotoxicity [170].
The alterations of APE1 level and activity may be a potential target against Pb genotoxicity. Pb inhibited the APE1 endonuclease activity of whole-cell extracts in a concentration-dependent manner with selective inactivation [73, 171]. In addition, McNeill et al. [171] have demonstrated a dose-dependent accumulation of AP sites as well as inhibition of APE1 activity in AP site incision under Pb exposure, implying an underlying mechanism by which Pb promotes cocarcinogenesis. On the other hand, Scortegagna and Hanbauer [77] have reported the enhanced APE1 accumulation in the nucleus in response to Pb treatment. In fact, oxidative stress can increase the expression of APE1, which functions against the genotoxic responses in diverse mammalian cells [70, 172, 173]. Interestingly, Pb-exposed workers showed the induced APE1 D148E polymorphism on T-cell receptor mutation frequency, suggesting a role for APE1 polymorphism as a susceptibility biomarker in biomonitoring by occupational Pb exposure [78].
4.2. Smoking
Cigarette smoke contains many carcinogens including heavy metals and has been clearly identified as a direct cause of lung cancer [174]. Cigarette smoke can induce various types of DNA damage [175]; consequently, individuals with reduced DNA repair ability would be expected to accumulate more carcinogen-induced DNA adducts in their tissue [176]. Indeed, lung cancer patients, mainly smokers, may have a lower capacity of DNA repair compared with healthy subjects and this can be suggested as a target mechanism for risk prediction of lung cancer associated with smoking [177–179].
Several studies have reported that cigarette smoke condensate (CSC) increased expression of adenomatous polyposis coli (APC) and blocked long-patch base excision repair (LP-BER) in human breast epithelial cells [180, 181]. In addition, benzo[α]pyrene, a major constituent of CSC, enhanced APC-mediated accumulation of abasic DNA lesions, leading to increased neoplastic transformation of breast epithelial cells [182]. Smoking also leads to brain disorder because, as la Maestra et al. [79] demonstrated, cigarette smoke altered BER by decreasing APE1 expression in the hippocampus and cerebellum of neonatal mice.
A number of epidemiological studies have evaluated the relationship among polymorphisms of APE1, smoking, and the risk of lung cancer. Several reports have suggested that APE1 genotypes were not major determinants of lung cancer susceptibility among smokers [85, 86], while several authors have asserted that APE1 polymorphisms such as −656T > G located in the promoter region and D148E could modify the risk of developing lung cancer attributable to cigarette smoking exposure [80–83]. With regard to bladder cancer, Huang et al. [84] have found that the gene-environment interaction between smoking and APE1 D148E polymorphism was statistically significant for the risk of the cancer. In contradiction, Terry et al. [87] found no overall association between APE1 genotype and bladder cancer risk. Although the precise biological mechanisms of the interaction of APE1 phenotype and smoking still need to be clarified, these studies provide potential evidence of gene-environment interactions between APE1 polymorphisms and smoking in cancer development.
4.3. UV and Other Radiations
Ultraviolet (UV) can induce DNA repair process directly or indirectly through the photoproducts generation or the ROS accumulation. Indeed, Izzotti et al. [88] reported that the expression of genes involved in oxidative stress response and DNA repair mechanism (including APE1) were increased in the skin of SKH-1 hairless mice exposed to UVC. UVA is also a critical component of solar radiation that has been implicated in photocarcinogenesis. UVA irradiation induced APE1 polymorphisms on various chromosome aberration types and relocalization of this protein to nuclear speckles against oxidative stress [89, 90].
Ionizing radiation (IR) is a widely used therapy for treating various types of cancers. However, it causes damage to normal cells of living tissue vicinity to tumors, resulting in mutation, radiation sickness, cancer, and ultimately death [183, 184]. Several reports demonstrated that IR induced APE1 polymorphism through the analysis of blood in breast cancer patients or occupationally exposed workers [91–93]. In particular, diagnostic X-rays increase the risk of developmental abnormalities and cancer in the exposed individuals [185–187]. In vitro findings revealed that the mammalian cells exposed to X-rays caused change in the expression and activity as well as polymorphism of APE1 [89, 94–96]. Taken together, these studies suggest that APE1 can be a vital marker for risk prediction upon exposure of physical carcinogens in addition to chemical carcinogens.
5. Concluding Remarks
There is no doubt of critical roles of APE1 in maintenance of genomic stability against endogenous and exogenous stresses. APE1 possesses powerful biological capacities in various cellular processes such as DNA repair, redox regulation, cell cycle, and RNA modulation. Because of the widely known APE1 multifunction, there are many reports that deal with the importance of APE1 in human disease developments. In particular, previous cell-based, animal model, and epidemiological studies suggested that the genetic variation (e.g., polymorphism) of APE1 as well as the alteration of APE1 localization, expression, and activity can be possible causes of lethal disease, including cancer (Figure 2). Although APE1 is overexpressed in many tumors and its enhanced nuclear levels correlate with reduced sensitivity to anticancer drugs, our review emphasizes the possibility of APE1 of relatively low level as a protector in human diseases induced by environmental factors under normal conditions, not under the abnormal conditions of the APE1 level in the cancerous predisposition.
Figure 2.
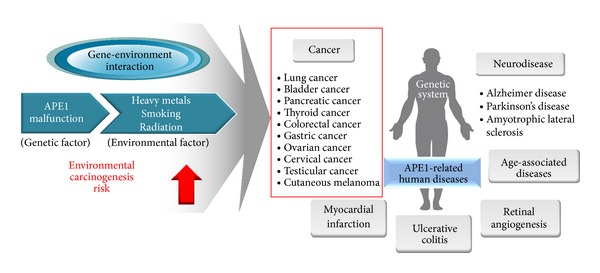
Scheme showing the potential of APE1 as an important modulator in human diseases. APE1 abnormality is known to be associated with the induction of various lethal diseases including cancer. In particular, gene-environment interaction between APE1 malfunction and several environmental factors increases the carcinogenic risk, leading to cancer development. Thus, APE1 can be suggested as one of the potential markers for risk prediction of environmental carcinogenesis.
Since several studies showed that APE1 malfunction enhances the risk of disease development when a person is exposed to harmful substances, we attempted to suggest APE1 as an important modulator in human disease caused by various environmental factors considering gene-environment interactions. Heavy metals such as arsenic, cadmium, and lead are widespread environmental pollutants and cause detrimental health effects including cancer, even at low doses. Although the molecular mechanisms of toxicity and carcinogenicity of these heavy metals remain poorly understood, commonly suggested mechanisms involve induction of oxidative DNA damage and the inhibition of DNA repair. It is well known that these heavy metals can change the expression and activity of APE1 and such APE1 malfunction is a vital factor related to severe human diseases. Nevertheless, as shown in Table 1, since the study on gene-environment interaction between APE1 and heavy metal in human pathologies is almost nonexistent, further studies are positively necessary. With regard to smoking of cigarette and radiation exposure, though the clear evidences of correlation among APE1, environmental factor, and related diseases are still required, the accumulated evidences suggest that APE1 may be a promising marker for risk prediction of environmental diseases including cancer.
Acknowledgments
This subject is supported by Korea Ministry of Environment as “The Ecoinnovation Project” (412-112-011) and by the Mid-Career Research Program (2012R1A2A2A01046228) of the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Science, and Technology.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Jae Sung Park and Hye Lim Kim equally contributed to this work and are considered as co-first authors.
References
- 1.Tell G, Fantini D, Quadrifoglio F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cellular and Molecular Life Sciences. 2010;67(21):3589–3608. doi: 10.1007/s00018-010-0486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xanthoudakis S, Miao GG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(1):23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo M, Delaplane S, Jiang A, et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxidants and Redox Signaling. 2008;10(11):1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McNeill DR, Wilson DM., III A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Molecular Cancer Research. 2007;5(1):61–70. doi: 10.1158/1541-7786.MCR-06-0329. [DOI] [PubMed] [Google Scholar]
- 5.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutation Research-DNA Repair. 2000;461(2):83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 6.Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis. 2008;643(1-2):54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fantini D, Vascotto C, Marasco D, et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Research. 2010;38(22):8239–8256. doi: 10.1093/nar/gkq691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park JY, Seo YR. The protective role of Nrf2 in cadmium-induced DNA damage. Molecular and Cellular Toxicology. 2011;7(1):61–66. [Google Scholar]
- 9.Kim HL, Seo YR. Synergistic genotoxic effect between gene and environmental pollutant: oxidative DNA damage induced by thioredoxin reductase 1 silencing under nickel treatment. Molecular and Cellular Toxicology. 2011;7(3):251–257. [Google Scholar]
- 10.Park JY, Lee YJ, Koedrith P, Seo YR. Protective role of thioredoxin reductase 1 in cadmium-induced DNA damage. Molecular and Cellular Toxicology. 2012;8(3):289–295. [Google Scholar]
- 11.Kim HL, Seo YR. Molecular and genomic approach for understanding the gene-environment interaction between Nrf2 defciency and carcinogenic nickel-induced DNA damage. Oncology Reports. 2012;28(6):1959–1967. doi: 10.3892/or.2012.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beernink PT, Segelke BW, Hadi MZ, Erzberger JP, Wilson DM, III, Rupp B. Two divalent metal ions in the active site of a new crystal form of human apurinic/apyrimidinic endonuclease, Ape1: implications for the catalytic mechanism. Journal of Molecular Biology. 2001;307(4):1023–1034. doi: 10.1006/jmbi.2001.4529. [DOI] [PubMed] [Google Scholar]
- 13.Li MX, Wilson DM. Human apurinic/apyrimidinic endonuclease 1. Antioxidants & Redox Signaling. 2014;20(4):678–707. doi: 10.1089/ars.2013.5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxidants and Redox Signaling. 2009;11(3):601–619. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flaherty DM, Monick MM, Hunninghake GW. AP endonucleases and the many functions of Ref-1. The American Journal of Respiratory Cell and Molecular Biology. 2001;25(6):664–667. doi: 10.1165/ajrcmb.25.6.f220. [DOI] [PubMed] [Google Scholar]
- 16.Izumi T, Brown DB, Naidu CV, et al. Two essential but distinct functions of the mammalian abasic endonuclease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(16):5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marenstein DR, Wilson DM, III, Teebor GW. Human AP endonuclease (APE1) demonstrates endonucleolytic activity against AP sites in single-stranded DNA. DNA Repair. 2004;3(5):527–533. doi: 10.1016/j.dnarep.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Wilson DM, III, Barsky D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutation Research/DNA Repair. 2001;485(4):283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 19.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annual Review of Genetics. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 20.Seeberg E, Eide L, Bjoras M. The base excision repair pathway. Trends in Biochemical Sciences. 1995;20(10):391–397. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 21.Winters TA, Henner WD, Russell PS, McCullough A, Jorgensen TJ. Removal of 3′-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Research. 1994;22(10):1866–1873. doi: 10.1093/nar/22.10.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suh D, Wilson DM, III, Povirk LF. 3′-Phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Research. 1997;25(12):2495–2500. doi: 10.1093/nar/25.12.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barzilay G, Mol CD, Robson CN, et al. Identification of critical active-site residues in the multifunctional human DNA repair enzyme HAP1. Nature Structural Biology. 1995;2(7):561–568. doi: 10.1038/nsb0795-561. [DOI] [PubMed] [Google Scholar]
- 24.Wilson DM, III, Takeshita M, Grollman AP, Demple B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. Journal of Biological Chemistry. 1995;270(27):16002–16007. doi: 10.1074/jbc.270.27.16002. [DOI] [PubMed] [Google Scholar]
- 25.Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Research. 1991;19(21):5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annual Review of Biochemistry. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 27.Wiederhold L, Leppard JB, Kedar P, et al. AP endonuclease-independent DNA base excision repair in human cells. Molecular Cell. 2004;15(2):209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Izumi T, Wiederhold LR, Roy G, et al. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193(1-2):43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 29.Flaherty DM, Monick MM, Carter AB, Peterson MW, Hunninghake GW. Oxidant-mediated increases in redox factor-1 nuclear protein and activator protein-1 DNA binding in asbestos-treated macrophages. Journal of Immunology. 2002;168(11):5675–5681. doi: 10.4049/jimmunol.168.11.5675. [DOI] [PubMed] [Google Scholar]
- 30.Hazra TK, Kow YW, Hatahet Z, et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. The Journal of Biological Chemistry. 2002;277(34):30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 31.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxidants and Redox Signaling. 2005;7(3-4):367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 32.Dianova II, Bohr VA, Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40(42):12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 33.Kim K, Biade S, Matsumoto Y. Involvement of flap endonuclease 1 in base excision DNA repair. The Journal of Biological Chemistry. 1998;273(15):8842–8848. doi: 10.1074/jbc.273.15.8842. [DOI] [PubMed] [Google Scholar]
- 34.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) The EMBO Journal. 1997;16(11):3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung HJ, Kim EH, Mun J-Y, et al. Base excision DNA repair defect in Gadd45a-deficient cells. Oncogene. 2007;26(54):7517–7525. doi: 10.1038/sj.onc.1210557. [DOI] [PubMed] [Google Scholar]
- 36.Kim HL, Kim SU, Seo YR. A novel role for Gadd45α in base excision repair: modulation of APE1 activity by the direct interaction of Gadd45α with PCNA. Biochemical and Biophysical Research Communications. 2013;434(2):185–190. doi: 10.1016/j.bbrc.2013.02.066. [DOI] [PubMed] [Google Scholar]
- 37.Jung HJ, Kim HL, Kim YJ, Weon J, Seo YR. A novel chemopreventive mechanism of selenomethionine: enhancement of APE1 enzyme activity via a Gadd45a, PCNA and APE1 protein complex that regulates p53-mediated base excision repair. Oncology Reports. 2013;30(4):1581–1586. doi: 10.3892/or.2013.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia. Cell. 2003;112(1):7–10. doi: 10.1016/s0092-8674(02)01247-3. [DOI] [PubMed] [Google Scholar]
- 39.Taylor RM, Moore DJ, Whitehouse J, Johnson P, Caldecott KW. A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Molecular and Cellular Biology. 2000;20(2):735–740. doi: 10.1128/mcb.20.2.735-740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vidal AE, Boiteux S, Hickson ID, Radicella JP. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. The EMBO Journal. 2001;20(22):6530–6539. doi: 10.1093/emboj/20.22.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burkart V, Liu H, Bellmann K, et al. Natural resistance of human beta cells toward nitric oxide is mediated by heat shock protein 70. Journal of Biological Chemistry. 2000;275(26):19521–19528. doi: 10.1074/jbc.M002265200. [DOI] [PubMed] [Google Scholar]
- 42.Mendez F, Sandigursky M, Franklin WA, Kenny MK, Kureekattil R, Bases R. Heat-shock proteins associated with base excision repair enzymes in HeLa cells. Radiation Research. 2000;153(2):186–195. doi: 10.1667/0033-7587(2000)153[0186:hspawb]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 43.Kenny MK, Mendez F, Sandigursky M, et al. Heat shock protein 70 binds to human apurinic/apyrimidinic endonuclease and stimulates endonuclease activity at abasic sites. Journal of Biological Chemistry. 2001;276(12):9532–9536. doi: 10.1074/jbc.M009297200. [DOI] [PubMed] [Google Scholar]
- 44.Mendez F, Sandigursky M, Kureekattil RP, Kenny MK, Franklin WA, Bases R. Specific stimulation of human apurinic/apyrimidinic endonuclease by heat shock protein 70. DNA Repair. 2003;2(3):259–271. doi: 10.1016/s1568-7864(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 45.Kelley MR, Parsons SH. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxidants and Redox Signaling. 2001;3(4):671–683. doi: 10.1089/15230860152543014. [DOI] [PubMed] [Google Scholar]
- 46.Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Current Molecular Pharmacology. 2012;5(1):36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. Ap-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Molecular and Cellular Biology. 1993;13(9):5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mol CD, Izumi T, Mitra S, Talner JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature. 2000;403(6768):451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 50.Luo MH, Zhang J, He HZ, et al. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012;51(2):695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. The EMBO Journal. 1992;11(2):653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao X, Kambe F, Ohmori S, Seo H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochemical and Biophysical Research Communications. 2002;297(2):288–293. doi: 10.1016/s0006-291x(02)02196-4. [DOI] [PubMed] [Google Scholar]
- 53.Ema M, Hirota K, Mimura J, et al. Molecular mechanisms of transcription activation by HLF and HIF1α in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. The EMBO Journal. 1999;18(7):1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirota K, Murata M, Sachi Y, et al. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-κB. Journal of Biological Chemistry. 1999;274(39):27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 55.Lando D, Pongratz I, Poellinger L, Whitelaw ML. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1α and the HIF-like factor. The Journal of Biological Chemistry. 2000;275(7):4618–4627. doi: 10.1074/jbc.275.7.4618. [DOI] [PubMed] [Google Scholar]
- 56.Tell G, Zecca A, Pellizzari L, et al. An “environment to nucleus” signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Research. 2000;28(5):1099–1105. doi: 10.1093/nar/28.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueno M, Masutani H, Arai RJ, et al. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. Journal of Biological Chemistry. 1999;274(50):35809–35815. doi: 10.1074/jbc.274.50.35809. [DOI] [PubMed] [Google Scholar]
- 58.Akamatsu Y, Ohno T, Hirota K, Kagoshima H, Yodoi J, Shigesada K. Redox regulation of the DNA binding activity in transcription factor PEBP2 the roles of two conserved cysteine residues: the roles of two conserved cysteine residues. The Journal of Biological Chemistry. 1997;272(23):14497–14500. doi: 10.1074/jbc.272.23.14497. [DOI] [PubMed] [Google Scholar]
- 59.Huang RP, Adamson ED. Characterization of the DNA-binding properties of the early growth response-1 (Egr-1) transcription factor: evidence for modulation by a redox mechanism. DNA and Cell Biology. 1993;12(3):265–273. doi: 10.1089/dna.1993.12.265. [DOI] [PubMed] [Google Scholar]
- 60.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. Journal of Cellular Biochemistry. 1993;11(9):3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abate C, Patel L, Rauscher FJ, III, Curran T. Redox regulation of Fos and Jun DNA-binding activity in vitro. Science. 1990;249(4973):1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- 62.Jayaraman L, Murthy KGK, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes and Development. 1997;11(5):558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 63.Hainaut P, Milner J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Research. 1993;53(18):4469–4473. [PubMed] [Google Scholar]
- 64.Luo M-H, He H-Z, Kelley MR, Georgiadis MM. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxidants & Redox Signaling. 2010;12(11):1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nature Reviews Molecular Cell Biology. 2005;6(1):44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- 66.Hamadeh HK, Trouba KJ, Amin RP, Afshari CA, Germolec D. Coordination of altered DNA repair and damage pathways in arsenite-exposed keratinocytes. Toxicological Sciences. 2002;69(2):306–316. doi: 10.1093/toxsci/69.2.306. [DOI] [PubMed] [Google Scholar]
- 67.Andrew AS, Warren AJ, Barchowsky A, et al. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environmental Health Perspectives. 2003;111(6):825–838. doi: 10.1289/ehp.111-1241504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu Y, Jin XM, Snow ET. Effect of arsenic on transcription factor AP-1 and NF-κB DNA binding activity and related gene expression. Toxicology Letters. 2002;133(1):33–45. doi: 10.1016/s0378-4274(02)00083-8. [DOI] [PubMed] [Google Scholar]
- 69.Sykora P, Snow ET. Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicology and Applied Pharmacology. 2008;228(3):385–394. doi: 10.1016/j.taap.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 70.Fung H, Liu P, Demple B. ATF4-dependent oxidative induction of the DNA repair enzyme Ape1 counteracts arsenite cytotoxicity and suppresses arsenite-mediated mutagenesis. Molecular and Cellular Biology. 2007;27(24):8834–8847. doi: 10.1128/MCB.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujihara J, Soejima M, Yasuda T, et al. Polymorphic trial in oxidative damage of arsenic exposed Vietnamese. Toxicology and Applied Pharmacology. 2011;256(2):174–178. doi: 10.1016/j.taap.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 72.Breton CV, Zhou W, Kile ML, et al. Susceptibility to arsenic-induced skin lesions from polymorphisms in base excision repair genes. Carcinogenesis. 2007;28(7):1520–1525. doi: 10.1093/carcin/bgm063. [DOI] [PubMed] [Google Scholar]
- 73.McNeill DR, Narayana A, Wong H, Wilson DM., III Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environmental Health Perspectives. 2004;112(7):799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamann I, König C, Richter C, Jahnke G, Hartwig A. Impact of cadmium on hOGG1 and APE1 as a function of the cellular p53 status. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2012;736(1-2):56–63. doi: 10.1016/j.mrfmmm.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 75.Al Bakheet SA, Attafi IM, Maayah ZH, Abd-Allah AR, Asiri YA, Korashy HM. Effect of long-term human exposure to environmental heavy metals on the expression of detoxification and DNA repair genes. Environmental Pollution. 2013;181:226–232. doi: 10.1016/j.envpol.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 76.Bravard A, Campalans A, Vacher M, et al. Inactivation by oxidation and recruitment into stress granules of hOGG1 but not APE1 in human cells exposed to sub-lethal concentrations of cadmium. Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis. 2010;685(1-2):61–69. doi: 10.1016/j.mrfmmm.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 77.Scortegagna M, Hanbauer I. Increased AP-1 DNA-binding activity and nuclear REF-1 accumulation in lead-exposed primary cultures of astrocytes. Neurochemical Research. 2000;25(6):861–866. doi: 10.1023/a:1007577710066. [DOI] [PubMed] [Google Scholar]
- 78.García-Lestón J, Roma-Torres J, Vilares M, et al. Genotoxic effects of occupational exposure to lead and influence of polymorphisms in genes involved in lead toxicokinetics and in DNA repair. Environment International. 2012;43(1):29–36. doi: 10.1016/j.envint.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 79.la Maestra S, Kisby GE, Micale RT, et al. Cigarette smoke induces DNA damage and alters base-excision repair and tau levels in the brain of neonatal mice. Toxicological Sciences. 2011;123(2):471–479. doi: 10.1093/toxsci/kfr187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lo Y, Jou Y, Hsiao C, et al. A polymorphism in the APE1 gene promoter is associated with lung cancer risk. Cancer Epidemiology Biomarkers and Prevention. 2009;18(1):223–229. doi: 10.1158/1055-9965.EPI-08-0749. [DOI] [PubMed] [Google Scholar]
- 81.Ito H, Matsuo K, Hamajima N, et al. Gene-environment interactions between the smoking habit and polymorphisms in the DNA repair genes, APE1 Asp148Glu and XRCC1 Arg399Gln, in Japanese lung cancer risk. Carcinogenesis. 2004;25(8):1395–1401. doi: 10.1093/carcin/bgh153. [DOI] [PubMed] [Google Scholar]
- 82.Ağaçhan B, Küçükhüseyin Ö, Aksoy P, et al. Apurinic/apyrimidinic endonuclease (APE1) gene polymorphisms and lung cancer risk in relation to tobacco smoking. Anticancer Research. 2009;29(6):2417–2420. [PubMed] [Google Scholar]
- 83.Osawa K, Miyaishi A, Uchino K, et al. APEX1 Asp148glu gene polymorphism is a risk factor for lung cancer in relation to smoking in Japanese. Asian Pacific Journal of Cancer Prevention. 2010;11(5):1181–1186. [PubMed] [Google Scholar]
- 84.Huang M, Dinney CP, Lin X, Lin J, Grossman HB, Wu X. High-order interactions among genetic variants in DNA base excision repair pathway genes and smoking in bladder cancer susceptibility. Cancer Epidemiology Biomarkers and Prevention. 2007;16(1):84–91. doi: 10.1158/1055-9965.EPI-06-0712. [DOI] [PubMed] [Google Scholar]
- 85.Misra RR, Ratnasinghe D, Tangrea JA, et al. Polymorphisms in the DNA repair genes XPD, XRCC1, XRCC3, and APE/ref-1, and the risk of lung cancer among male smokers in Finland. Cancer Letters. 2003;191(2):171–178. doi: 10.1016/s0304-3835(02)00638-9. [DOI] [PubMed] [Google Scholar]
- 86.Ryk C, Kumar R, Thirumaran RK, Hou S. Polymorphisms in the DNA repair genes XRCC1, APEX1, XRCC3 and NBS1, and the risk for lung cancer in never- and ever-smokers. Lung Cancer. 2006;54(3):285–292. doi: 10.1016/j.lungcan.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 87.Terry PD, Umbach DM, Taylor JA. APE1 genotype and risk of bladder cancer: evidence for effect modification by smoking. International Journal of Cancer. 2006;118(12):3170–3173. doi: 10.1002/ijc.21768. [DOI] [PubMed] [Google Scholar]
- 88.Izzotti A, Cartiglia C, Longobardi M, et al. Alterations of gene expression in skin and lung of mice exposed to light and cigarette smoke. The FASEB Journal. 2004;18(13):1559–1561. doi: 10.1096/fj.04-1877fje. [DOI] [PubMed] [Google Scholar]
- 89.Au WW, Salama SA, Sierra-Torres CH. Functional characterization of polymorphisms in DNA repair genes using cytogenetic challenge assays. Environmental Health Perspectives. 2003;111(15):1843–1850. doi: 10.1289/ehp.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Campalans A, Amouroux R, Bravard A, Epe B, Radicella JP. UVA irradiation induces relocalisation of the DNA repair protein hOGG1 to nuclear speckles. Journal of Cell Science. 2007;120(1):23–32. doi: 10.1242/jcs.03312. [DOI] [PubMed] [Google Scholar]
- 91.Hu JJ, Smith TR, Miller MS, Mohrenweiser HW, Golden A, Case LD. Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis. 2001;22(6):917–922. doi: 10.1093/carcin/22.6.917. [DOI] [PubMed] [Google Scholar]
- 92.Hu JJ, Smith TR, Miller MS, Lohman K, Case LD. Genetic regulation of ionizing radiation sensitivity and breast cancer risk. Environmental and Molecular Mutagenesis. 2002;39(2-3):208–215. doi: 10.1002/em.10058. [DOI] [PubMed] [Google Scholar]
- 93.Andreassi MG, Foffa I, Manfredi S, Botto N, Cioppa A, Picano E. Genetic polymorphisms in XRCC1, OGG1, APE1 and XRCC3 DNA repair genes, ionizing radiation exposure and chromosomal DNA damage in interventional cardiologists. Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis. 2009;666(1-2):57–63. doi: 10.1016/j.mrfmmm.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 94.Gu X, Cun Y, Li M, et al. Human apurinic/apyrimidinic endonuclease siRNA inhibits the angiogenesis induced by X-Ray irradiation in lung cancer cells. International Journal of Medical Sciences. 2013;10(7):870–882. doi: 10.7150/ijms.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Z, Yang X, Wang D, et al. Nm23-H1 protein binds to APE1 at AP sites and stimulates AP endonuclease activity following ionizing radiation of the human lung cancer A549 cells. Cell Biochemistry and Biophysics. 2011;61(3):561–572. doi: 10.1007/s12013-011-9238-9. [DOI] [PubMed] [Google Scholar]
- 96.Gdowicz-Klosok A, Widel M, Rzeszowska-Wolny J. The influence of XPD, APE1, XRCC1, and NBS1 polymorphic variants on DNA repair in cells exposed to X-rays. Mutation Research—Genetic Toxicology and Environmental Mutagenesis. 2013;755(1):42–48. doi: 10.1016/j.mrgentox.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 97.Berquist BR, McNeill DR, Wilson DM., III Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. Journal of Molecular Biology. 2008;379(1):17–27. doi: 10.1016/j.jmb.2008.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu PF, Demple B. DNA repair in mammalian mitochondria: much more than we thought? Environmental and Molecular Mutagenesis. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 99.Barnes T, Kim W, Mantha AK, et al. Identification of Apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Research. 2009;37(12):3946–3958. doi: 10.1093/nar/gkp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guikema JEJ, Linehan EK, Tsuchimoto D, et al. APE1- And APE2-dependent DNA breaks in immunoglobulin class switch recombination. Journal of Experimental Medicine. 2007;204(12):3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Molecular and Cellular Biology. 2013;33(7):1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiarini LB, Freitas FG, Petrs-Silva H, Linden R. Evidence that the bifunctional redox factor/AP endonuclease Ref-1 is an anti-apoptotic protein associated with differentiation in the developing retina. Cell Death and Differentiation. 2000;7(3):272–281. doi: 10.1038/sj.cdd.4400639. [DOI] [PubMed] [Google Scholar]
- 103.Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treatment Reviews. 2010;36(5):425–435. doi: 10.1016/j.ctrv.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 104.Fishel ML, He Y, Reed AM, et al. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair. 2008;7(2):177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jiang YL, Zhou SY, Sandusky GE, Kelley MR, Fishel ML. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Investigation. 2010;28(9):885–895. doi: 10.3109/07357907.2010.512816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moore DH, Michael H, Tritt R, Parsons SH, Kelley MR. Alterations in the expression of the DNA repair/redox enzyme APE/ref-1 in epithelial ovarian cancers. Clinical Cancer Research. 2000;6(2):602–609. [PubMed] [Google Scholar]
- 107.Tell G, Pellizzari L, Pucillo C, et al. TSH controls Ref-1 nuclear translocation in thyroid cells. Journal of Molecular Endocrinology. 2000;24(3):383–390. doi: 10.1677/jme.0.0240383. [DOI] [PubMed] [Google Scholar]
- 108.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Molecular Cancer Therapeutics. 2004;3(6):679–686. [PubMed] [Google Scholar]
- 109.Herring CJ, West CML, Wilks DP, et al. Levels of the DNA repair enzyme human apurinic/apyrimidinic endonuclease (APE1, APEX, Ref-1) are associated with the intrinsic radiosensitivity of cervical cancers. British Journal of Cancer. 1998;78(9):1128–1133. doi: 10.1038/bjc.1998.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xiang D-B, Chen Z-T, Wang D, et al. Chimeric adenoviral vector Ad5/F35-mediated APE1 siRNA enhances sensitivity of human colorectal cancer cells to radiotherapy in vitro and in vivo . Cancer Gene Therapy. 2008;15(10):625–635. doi: 10.1038/cgt.2008.30. [DOI] [PubMed] [Google Scholar]
- 111.Chen S, Xiong G, Wu S, Mo J. Downregulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 enhances the sensitivity of human pancreatic cancer cells to radiotherapy in vitro. Cancer Biotherapy and Radiopharmaceuticals. 2013;28(2):169–176. doi: 10.1089/cbr.2012.1266. [DOI] [PubMed] [Google Scholar]
- 112.Robertson KA, Bullock HA, Xu Y, et al. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Research. 2001;61(5):2220–2225. [PubMed] [Google Scholar]
- 113.Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83(6):970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 114.Hadi MZ, Coleman MA, Fidelis K, Mohrenweiser HW, Wilson DM., III Functional characterization of Ape1 variants identified in the human population. Nucleic Acids Research. 2000;28(20):3871–3879. doi: 10.1093/nar/28.20.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hung RJ, Hall J, Brennan P, Boffetta P. Genetic polymorphisms in the base excision repair pathway and cancer risk: a huge review. The American Journal of Epidemiology. 2005;162(10):925–942. doi: 10.1093/aje/kwi318. [DOI] [PubMed] [Google Scholar]
- 116.Canbay E, Agachan B, Gulluoglu M, et al. Possible associations of APE1 polymorphism with susceptibility and HOGG1 polymorphism with prognosis in gastric cancer. Anticancer Research. 2010;30(4):1359–1364. [PubMed] [Google Scholar]
- 117.Canbay E, Cakmakoglu B, Zeybek U, et al. Association of APE1 and hOGG1 polymorphisms with colorectal cancer risk in a Turkish population. Current Medical Research and Opinion. 2011;27(7):1295–1302. doi: 10.1185/03007995.2011.573544. [DOI] [PubMed] [Google Scholar]
- 118.Chen L, Ambrosone CB, Lee J, Sellers TA, Pow-Sang J, Park JY. Association between polymorphisms in the DNA repair genes XRCC1 and APE1, and the risk of prostate cancer in white and black Americans. Journal of Urology. 2006;175(1):108–112. doi: 10.1016/S0022-5347(05)00042-X. [DOI] [PubMed] [Google Scholar]
- 119.Li C, Liu Z, Wang L, et al. Genetic variants of the ADPRT, XRCC1 and APE1 genes and risk of cutaneous melanoma. Carcinogenesis. 2006;27(9):1894–1901. doi: 10.1093/carcin/bgl042. [DOI] [PubMed] [Google Scholar]
- 120.Li Y, Li S, Wu Z, et al. Polymorphisms in genes of APE1, PARP1, and XRCC1: risk and prognosis of colorectal cancer in a Northeast Chinese population. Medical Oncology. 2013;30(2, article no. 505) doi: 10.1007/s12032-013-0505-z. [DOI] [PubMed] [Google Scholar]
- 121.Kang HF, Dai ZJ, Ma XB, et al. A genetic variant in the promoter of APE1 gene (−656 T > G) is associated with breast cancer risk and progression in a Chinese population. Gene. 2013;531(1):97–100. doi: 10.1016/j.gene.2013.08.052. [DOI] [PubMed] [Google Scholar]
- 122.Wang M, Chu H, Wang S, et al. Genetic variant in APE1 gene promoter contributes to cervical cancer risk. American Journal of Obstetrics and Gynecology. 2013;209(4):360.e1–360.e7. doi: 10.1016/j.ajog.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 123.Wang ML, Qin C, Zhu JA, et al. Genetic variants of XRCC1, APE1, and ADPRT genes and risk of bladder cancer. DNA and Cell Biology. 2010;29(6):303–311. doi: 10.1089/dna.2009.0969. [DOI] [PubMed] [Google Scholar]
- 124.Lu J, Zhang SY, Chen D, et al. Functional characterization of a promoter polymorphism in APE1/Ref-1 that contributes to reduced lung cancer susceptibility. The FASEB Journal. 2009;23(10):3459–3469. doi: 10.1096/fj.09-136549. [DOI] [PubMed] [Google Scholar]
- 125.Li Z, Guan W, Li M, et al. Genetic polymorphism of DNA base-excision repair genes (APE1, OGG1 and XRCC1) and their correlation with risk of lung cancer in a Chinese population. Archives of Medical Research. 2011;42(3):226–234. doi: 10.1016/j.arcmed.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 126.Ji Y, Zhan P, Wang J, Qiu L, Yu L. APE1 Asp148Glu gene polymorphism and lung cancer risk: a meta-analysis. Molecular Biology Reports. 2011;38(7):4537–4543. doi: 10.1007/s11033-010-0584-2. [DOI] [PubMed] [Google Scholar]
- 127.Deng Q, Sheng L, Su D, et al. Genetic polymorphisms in ATM, ERCC1, APE1 and iASPP genes and lung cancer risk in a population of southeast China. Medical Oncology. 2011;28(3):667–672. doi: 10.1007/s12032-010-9507-2. [DOI] [PubMed] [Google Scholar]
- 128.Wilson DM, III, McNeill DR. Base excision repair and the central nervous system. Neuroscience. 2007;145(4):1187–1200. doi: 10.1016/j.neuroscience.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 129.Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair. 2005;4(3):367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 130.Tan Z, Sun N, Schreiber SS. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer’s hippocampus. NeuroReport. 1998;9(12):2749–2752. doi: 10.1097/00001756-199808240-00012. [DOI] [PubMed] [Google Scholar]
- 131.Marcon G, Tell G, Perrone L, et al. APE1/Ref-1 in Alzheimer’s disease: An immunohistochemical study. Neuroscience Letters. 2009;466(3):124–127. doi: 10.1016/j.neulet.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 132.Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. NeuroReport. 1997;8(6):1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- 133.Gencer M, Dasdemir S, Cakmakoglu B, et al. DNA repair genes in Parkinson’s disease. Genetic Testing and Molecular Biomarkers. 2012;16(6):504–507. doi: 10.1089/gtmb.2011.0252. [DOI] [PubMed] [Google Scholar]
- 134.Olkowski ZL. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. NeuroReport. 1998;9(2):239–242. doi: 10.1097/00001756-199801260-00012. [DOI] [PubMed] [Google Scholar]
- 135.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radical Biology and Medicine. 2002;32(9):804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 136.Intano GW, Cho EJ, McMahan CA, Walter CA. Age-related base excision repair activity in mouse brain and liver nuclear extracts. Journals of Gerontology A. 2003;58(3):205–211. doi: 10.1093/gerona/58.3.b205. [DOI] [PubMed] [Google Scholar]
- 137.Swain U, Rao KS. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mechanisms of Ageing and Development. 2011;132(8-9):374–381. doi: 10.1016/j.mad.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 138.Angkeow P, Deshpande SS, Qi B, et al. Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death and Differentiation. 2002;9(7):717–725. doi: 10.1038/sj.cdd.4401025. [DOI] [PubMed] [Google Scholar]
- 139.Zhang Y, Zhang L, Zhang L, Bai J, Ge HY, Liu P. Expression changes in DNA repair enzymes and mitochondrial DNA damage in aging rat lens. Molecular Vision. 2010;16:1754–1763. [PMC free article] [PubMed] [Google Scholar]
- 140.Szczesny B, Mitra S. Effect of aging on intracellular distribution of abasic (AP) endonuclease 1 in the mouse liver. Mechanisms of Ageing and Development. 2005;126(10):1071–1078. doi: 10.1016/j.mad.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 141.Madlener S, Strobel T, Vose S, et al. Essential role for mammalian apurinic/apyrimidinic (AP) endonuclease Ape1/Ref-1 in telomere maintenance. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(44):17844–17849. doi: 10.1073/pnas.1304784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tekeli A, Isbir S, Ergen A, et al. APE1 and XRCC3 polymorphisms and myocardial infarction. In Vivo. 2008;22(4):477–480. [PubMed] [Google Scholar]
- 143.Bardia A, Tiwari SK, Gunisetty S, et al. Functional polymorphisms in XRCC-1 and APE-1 contribute to increased apoptosis and risk of ulcerative colitis. Inflammation Research. 2012;61(4):359–365. doi: 10.1007/s00011-011-0418-2. [DOI] [PubMed] [Google Scholar]
- 144.Jiang A, Gao H, Kelley MR, Qiao X. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vision Research. 2011;51(1):93–100. doi: 10.1016/j.visres.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 146.Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438(7070):960–966. doi: 10.1038/nature04482. [DOI] [PubMed] [Google Scholar]
- 147.d’Errico A, Malats N, Vineis P, Boffetta P. Review of studies of selected metabolic polymorphisms and cancer. IARC scientific publications. 1999;(148):323–393. [PubMed] [Google Scholar]
- 148.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some drinking-water disinfectants and contaminants, including arsenic. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 2004;84:1–477. [PMC free article] [PubMed] [Google Scholar]
- 149.Kligerman AD, Tennant AH. Insights into the carcinogenic mode of action of arsenic. Toxicology and Applied Pharmacology. 2007;222(3):281–288. doi: 10.1016/j.taap.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 150.Szymańska-Chabowska A, Antonowicz-Juchniewicz J, Andrzejak R. The concentration of selected cancer markers (TPA, TPS, CYFRA 21-1, CEA) in workers occupationally exposed to arsenic (As) and some heavy metals (Pb, Cd) during a two-year observation study. International Journal of Occupational Medicine and Environmental Health. 2007;20(3):229–239. doi: 10.2478/v10001-007-0026-0. [DOI] [PubMed] [Google Scholar]
- 151.Díaz-Villaseñor A, Burns AL, Hiriart M, Cebrián ME, Ostrosky-Wegman P. Arsenic-induced alteration in the expression of genes related to type 2 diabetes mellitus. Toxicology and Applied Pharmacology. 2007;225(2):123–133. doi: 10.1016/j.taap.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 152.Schmuck EM, Board PG, Whitbread AK, et al. Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of Omega class glutathione transferase variants: Implications for arsenic metabolism and the age-at-onset of Alzheimer’s and Parkinson’s diseases. Pharmacogenetics and Genomics. 2005;15(7):493–501. doi: 10.1097/01.fpc.0000165725.81559.e3. [DOI] [PubMed] [Google Scholar]
- 153.Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity—a review. Human and Experimental Toxicology. 2007;26(10):823–832. doi: 10.1177/0960327107084539. [DOI] [PubMed] [Google Scholar]
- 154.Huang C, Ke QD, Costa M, Shi X. Molecular mechanisms of arsenic carcinogenesis. Molecular and Cellular Biochemistry. 2004;255(1-2):57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- 155.Snow ET, Sykora P, Durham TR, Klein CB. Arsenic, mode of action at biologically plausible low doses: what are the implications for low dose cancer risk? Toxicology and Applied Pharmacology. 2005;207(2) supplememet 1:557–564. doi: 10.1016/j.taap.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 156.Lunec J, Holloway KA, Cooke MS, Faux S, Griffiths HR, Evans MD. Urinary 8-oxo-2′-deoxyguanosine: redox regulation of DNA repair in vivo? Free Radical Biology and Medicine. 2002;33(7):875–885. doi: 10.1016/s0891-5849(02)00882-1. [DOI] [PubMed] [Google Scholar]
- 157.Vodicka P, Stetina R, Polakova V, et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis. 2007;28(3):657–664. doi: 10.1093/carcin/bgl187. [DOI] [PubMed] [Google Scholar]
- 158.Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. Working Group views and expert opinions, Lyon, 9–16 February 1993. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 1993;58:1–415. [PMC free article] [PubMed] [Google Scholar]
- 159.Waalkes MP. Cadmium carcinogenesis. Mutation Research—Fundamental and Molecular Mechanisms of Mutagenesis. 2003;533(1-2):107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 160.Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192(2-3):95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 161.Bertin G, Averbeck D. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review) Biochimie. 2006;88(11):1549–1559. doi: 10.1016/j.biochi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 162.Joseph P. Mechanisms of cadmium carcinogenesis. Toxicology and Applied Pharmacology. 2009;238(3):272–279. doi: 10.1016/j.taap.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 163.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Research. 2008;18(1):27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Giaginis C, Gatzidou E, Theocharis S. DNA repair systems as targets of cadmium toxicity. Toxicology and Applied Pharmacology. 2006;213(3):282–290. doi: 10.1016/j.taap.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 165.Lead and lead compounds: lead and inorganic lead compounds. IARC Monographs Evaluation Carcinogenic Risks to Humans. 1987;23(supplement 7):1–729. [Google Scholar]
- 166.Inorganic and organic lead compounds. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 2006;87:1–471. [PMC free article] [PubMed] [Google Scholar]
- 167.Järup L. Hazards of heavy metal contamination. British Medical Bulletin. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- 168.Patocka J, Cerný K. Inorganic lead toxicology. Acta Medica. 2003;46(2):65–72. [PubMed] [Google Scholar]
- 169.Méndez-Gómez J, García-Vargas G, López-Carrillo L, et al. Genotoxic effects of environmental exposure to arsenic and lead on children in Region Lagunera, Mexico. Annals of the New York Academy of Sciences. 2008;1140:358–367. doi: 10.1196/annals.1454.027. [DOI] [PubMed] [Google Scholar]
- 170.Garcia-Leston J, Mendez J, Pasaro E, Laffon B. Genotoxic effects of lead: an updated review. Environment International. 2010;36(6):623–636. doi: 10.1016/j.envint.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 171.McNeill DR, Wong H, Narayana A, Wilson DM., III Lead promotes abasic site accumulation and co-mutagenesis in mammalian cells by inhibiting the major abasic endonuclease Ape1. Molecular Carcinogenesis. 2007;46(2):91–99. doi: 10.1002/mc.20196. [DOI] [PubMed] [Google Scholar]
- 172.Grösch S, Fritz G, Kaina B. Apurinic endonuclease (Ref-1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Research. 1998;58(19):4410–4416. [PubMed] [Google Scholar]
- 173.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(9):5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Boyle P. Cancer, cigarette smoking and premature death in Europe: a review including the Recommendations of European Cancer Experts Consensus Meeting, Helsinki, October 1996. Lung Cancer. 1997;17(1):1–60. doi: 10.1016/s0169-5002(97)00648-x. [DOI] [PubMed] [Google Scholar]
- 175.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. The Lancet Oncology. 2002;3(8):461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 176.Wei Q, Spitz MR. The role of DNA repair capacity in susceptibility to lung cancer: a review. Cancer and Metastasis Reviews. 1997;16(3-4):295–307. doi: 10.1023/a:1005852211430. [DOI] [PubMed] [Google Scholar]
- 177.Spitz MR, Wu X, Wang Y, et al. Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Research. 2001;61(4):1354–1357. [PubMed] [Google Scholar]
- 178.Wei Q, Cheng L, Hong WK, Spitz MR. Reduced DNA repair capacity in lung cancer patients. Cancer Research. 1996;56(18):4103–4107. [PubMed] [Google Scholar]
- 179.Shen H, Spitz MR, Qiao Y, et al. Smoking, DNA repair capacity and risk of nonsmall cell lung cancer. International Journal of Cancer. 2003;107(1):84–88. doi: 10.1002/ijc.11346. [DOI] [PubMed] [Google Scholar]
- 180.Kundu CN, Balusu R, Jaiswal AS, Gairola CG, Narayan S. Cigarette smoke condensate-induced level of adenomatous polyposis coli blocks long-patch base excision repair in breast epithelial cells. Oncogene. 2007;26(10):1428–1438. doi: 10.1038/sj.onc.1209925. [DOI] [PubMed] [Google Scholar]
- 181.Mohapatra P, Satapathy SR, Das D, et al. Resveratrol mediated cell death in cigarette smoke transformed breast epithelial cells is through induction of p21Waf1/Cip1 and inhibition of long patch base excision repair pathway. Toxicology and Applied Pharmacology. 2014;275(3):221–231. doi: 10.1016/j.taap.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 182.Jaiswal AS, Panda H, Pampo CA, et al. Adenomatous polyposis coli- mediated accumulation of abasic dna lesions lead to cigarette smoke condensate- induced neoplastic transformation of normal breast epithelial cells. Neoplasia. 2013;15(4):454–460. doi: 10.1593/neo.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ron E. Ionizing radiation and cancer risk: evidence from epidemiology. Radiation Research. 1998;150(5):S30–S41. [PubMed] [Google Scholar]
- 184.Brenner DJ, Doll R, Goodhead DT, et al. Cancer risks attributable to low doses of ionizing radiation: assessing what we really know. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):13761–13766. doi: 10.1073/pnas.2235592100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hall EJ, Brenner DJ. Cancer risks from diagnostic radiology. British Journal of Radiology. 2008;81(965):362–378. doi: 10.1259/bjr/01948454. [DOI] [PubMed] [Google Scholar]
- 186.Brenner DJ. Should we be concerned about the rapid increase in CT usage? Reviews on Environmental Health. 2010;25(1):63–67. doi: 10.1515/reveh.2010.25.1.63. [DOI] [PubMed] [Google Scholar]
- 187.de Santis M, Cesari E, Nobili E, Straface G, Cavaliere AF, Caruso A. Radiation effects on development. Birth Defects Research C: Embryo Today: Reviews. 2007;81(3):177–182. doi: 10.1002/bdrc.20099. [DOI] [PubMed] [Google Scholar]