

Abstract
We sequenced the whole exome of 35 cases and 7 controls from 9 age-related macular degeneration (AMD) families in whom known common genetic risk alleles could not explain their high disease burden and/or their early-onset advanced disease. Two families harbored novel rare mutations in CFH (R53C and D90G). R53C segregates perfectly with AMD in 11 cases (heterozygous) and 1 elderly control (reference allele) (LOD = 5.07, P = 6.7 × 10−7). In an independent cohort, 4 out of 1676 cases but none of the 745 examined controls or 4300 NHBLI Exome Sequencing Project (ESP) samples carried the R53C mutation (P = 0.0039). In another family of six siblings, D90G similarly segregated with AMD in five cases and one control (LOD = 1.22, P = 0.009). No other sample in our large cohort or the ESP had this mutation. Functional studies demonstrated that R53C decreased the ability of FH to perform decay accelerating activity. D90G exhibited a decrease in cofactor-mediated inactivation. Both of these changes would lead to a loss of regulatory activity, resulting in excessive alternative pathway activation. This study represents an initial application of the whole-exome strategy to families with early-onset AMD. It successfully identified high impact alleles leading to clearer functional insight into AMD etiopathogenesis.
INTRODUCTION
With the emerging understanding of the strong genetic underpinnings of age-related macular degeneration (AMD) (MIM 603075) based on genome-wide association studies and more recently targeted sequencing with the discovery of rare, penetrant variants (1,2), we are approaching an era of applying these genetic discoveries for personalized medicine. Common variants collectively allow us to predict increased risk of AMD and progression of disease on a population level (3–5), but may not be applicable for an individual family. Rare and penetrant variants, which are strongly related to AMD, can have a major impact on disease risk in an individual and a family. For example, the CFH R1210C variant, with a gene frequency of 0.02% in the European population, carries a 20-fold increased risk of AMD for an individual with the heterozygous mutation, the strongest genetic risk factor for AMD to date. These individuals are also diagnosed with advanced AMD at an earlier age (1). More recently, rare variants in C3, CFI and C9 have also been discovered that influence disease risk (2,6,7). These rare variants open up new avenues for family counseling, selective and possibly more informative clinical trials, and eventually for personalized targeted therapies.
We previously demonstrated that some families, despite a high density of affected individuals, have a lower than expected genotypic load based on known common variants, suggesting they may harbor undiscovered risk variants (8). It has been reported that rare, high penetrant mutations that co-segregate with early-onset or severe phenotypes may exist among the distribution of a common trait (e.g. high-density lipoprotein cholesterol) (9). They can be transmitted through generations like a dominant Mendelian disease, yet remain hidden in the population given the high frequency of AMD. Since very rare (private) mutations occur in only a few families, we performed state-of-the-art whole-exome sequencing in families most likely to harbor novel or rare, highly penetrant variants. Such variants in CFH were discovered to segregate with disease in two of the nine families reported here.
To understand the impact of these variants, we performed functional studies to characterize the effect(s) these mutations have on protein function. Functional studies are crucial to elucidate the relationship between rare variants and disease given the limited power to associate these variants in all but the largest families and cohorts.
RESULTS
We sequenced the exomes of 42 samples from 9 families with unusually low common variant AMD risk (Methods). The coverage was >10× for a median of 97.25% of the targeted regions (Supplementary Material, Fig. S1A). For the 18 783 RefSeq genes captured by the Nimblegen Human exome library, 16 717 genes were sequenced at >10× depth for 90% of the regions targeted per gene (Supplementary Material, Fig. S1B). After strict quality control, we identified 153 534 high quality variants with no missing genotypes. To test the overall quality of the sequence data, we compared the genotypes of variants found in the sequence data to variants derived from genotyping via an exome array (Illumina Infinium Human Exome BeadChip v1.0). There were 20 454 variants that were captured by sequencing and that were also genotyped using the exome array. The concordance between sequencing and genotyping data was 99.76%. Allelic dosages were almost perfectly correlated (r > 0.99) for 93.5% of common variants (f > 0.01) and 99.2% of rare variants (Supplementary Material, Fig. S1C and D); only 1.3 and 0.7% of common and rare SNPs, respectively, had modestly correlated dosages (r < 0.9).
To screen for rare functional variants in the selected families, we filtered the variants in each step by several criteria (Table 1). First, we included only coding SNPs of high and moderate impact on protein sequence/structure (missense, nonsense, read-through or splice variants). We excluded SNPs with minor allele frequency greater than 0.1% in the 1000 genomes project or the NHBLI exome sequencing project (ESP). There was only one variant exome-wide (R53C in CFH) remaining in pedigree V that segregated with advanced AMD (Table 1). Remarkably, this was a missense variant, R53C, in the well known AMD gene, CFH.
Table 1.
Number of SNPs identified in each family by filtering on their function and rareness in population databases
| I | II | III | IV | V | VI | VII | VIII | IX | |
|---|---|---|---|---|---|---|---|---|---|
| Case: control ratio per family | 3:1 | 5:1 | 4:1 | 3:1 | 5:1 | 4:0 | 4:1 | 4:0 | 3:1 |
| High and moderate impact SNPsa | 20397 | 18414 | 21313 | 19437 | 22984 | 17904 | 20853 | 17796 | 17603 |
| MAF <0.1% in databasesb | 436 | 320 | 540 | 420 | 572 | 394 | 480 | 286 | 324 |
| Shared between cases but not in controls | 4 | 5 | 2 | 6 | 1 | 18 | 3 | 11 | 18 |
| Probably damaging (Polyphen-2) | 1 | 1 | 1 | 3 | 1 | 3 | 0 | 3 | 7 |
| Deleterious (SIFT) | 2 | 2 | 1 | 3 | 1 | 5 | 3 | 6 | 9 |
| Probably damaging or deleterious | 2 | 2 | 2 | 3 | 1 | 5 | 3 | 6 | 10 |
| Probably damaging and deleterious | 1 | 1 | 0 | 3 | 1 | 3 | 0 | 3 | 6 |
aHigh and moderate impact SNPs include missense, nonsense, read-through or splice variants.
bVariants with minor allele frequency (MAF) greater than 0.1% in the data sets of the 1000 genomes project or the NHLBI GO ESP were filtered out. Polyphen-2 is a tool that predicts possible impact of an amino acid substitution on the structure and function of a human protein using physical and comparative considerations. SIFT is a tool that predicts whether an amino acid substitution affects protein function. SNPs, single nucleotide polymorphisms.
For other families with multiple candidate SNPs remaining, we included SNPs that were annotated as loss of function or probably damaging by PolyPhen-2 or were assigned ‘deleterious’ by SIFT. Using functional prediction, pedigree II yielded a novel variant that segregated with disease and was predicted to be deleterious: D90G in CFH (Table 1). To further narrow down the list of candidate SNPs remaining after filtering (rare, segregating in exome sequenced individuals and predicted to be altered by either Sift or Polyphen-2), we recruited additional relatives and obtained their genotypes and analyzed segregation (Table 2 and Supplementary Material, Table S1). Table 2 shows the genomic locations, amino acid change and minor allele frequencies in ESP of the SNPs that passed filtering and segregated in each family with LOD scores > 1.0. The results for the candidate SNPs with no evidence of segregation when using data from sequenced and genotyped members of each family are shown in Supplementary Material, Table S1.
Table 2.
Potential pathogenic variants identified in each family with LOD score ≥1.0
| FAM | Gene | Location | Ref | Alt | rsID | Amino acid | Allele frequency (ESP Europeans) | LOD score |
|---|---|---|---|---|---|---|---|---|
| II | CFH* | chr1:196643011 | A | G | D90G | 0 | 1.22 | |
| V | CFH* | chr1:196642206 | C | T | R53C | 0 | 5.07 | |
| VIII | CCDC75* | chr2:37315562 | C | A | S9Y | 0 | 1.50 | |
| IX | TARS2* | chr1:150477420 | C | A | P620H | 0 | 1.22 | |
| IX | WNT2 | chr7:116918397 | G | A | rs148046128 | R299W | 0.00093 | 1.22 |
Variants predicted to be both probably damaging by Polyphen-2 and deleterious by SIFT. FAM, family; Ref, reference allele; Alt, alternative allele; rsID, rs number; ESP, exome sequencing project. Asterisks indicate genes expressed in retina tissue (10).
Because two of the new rare variants (R53C and D90G) are located in CFH, in which both a rare causal mutation (R1210C) (1) and common alleles have been previously associated (11–13), we conducted follow-up analyses by recruiting additional family members and a replication analysis in independent samples. We performed Sanger sequencing of the CFH gene in six additionally affected relatives in pedigree V using the same approach as our previous targeted sequencing project (2). These additional cases also carried the R53C mutation. There were a total of 11 cases heterozygous for R53C and 1 unaffected individual (90 years old) without the mutation in pedigree V (Fig. 1B). Complement Factor H R53C was significantly linked to advanced AMD in this pedigree (LOD = 5.07, P = 6.7 × 10−7). We separately sequenced CFH in our study targeting 681 genes in 1676 cases and 745 controls (2). We identified four cases with the R53C mutation (unrelated to individuals in pedigree V). Combining the ESP data set of European ancestry as shared controls (4300 samples) with our targeted sequencing data set, R53C was associated with AMD (Fisher's exact test, P = 0.0039).
Figure 1.

Rare CFH mutations segregate with AMD in families. (A) Pedigree of a family with AMD carrying the D90G mutation in CFH. (B) Pedigree of a large multigenerational family with AMD explained by the R53C mutation in CFH. Family members in the generation of the proband (arrow) and their descendants are displayed except for individuals not enrolled in the study including some unaffected individuals. Common antecedents are also displayed although deceased and not enrolled. Squares represent male and circles are female family members. Black symbols indicate affected persons and white symbols represent unaffected persons. Slashes indicate deceased family members. The genotype information of common risk SNPs in CFH and R53C or D90G mutation allele (in red) are shown below each genotyped individual. Blue star indicates a whole-exome sequenced sample. Red cross indicates individual with renal disease or low kidney function.
Since some of the original families were selected for advanced AMD of early onset, we tested whether cases with R53C allele in the independent cohort also tended to be diagnosed with advanced AMD at an early age. We compared the age of diagnosis of advanced AMD in the four patients with R53C mutations who were unrelated to family V and in 1832 unrelated patients without rare mutations in CFH, C3 or CFI genes. As seen previously with the rare R1210C variant of CFH (1), patients with R53C mutations had a significantly earlier onset of advanced AMD (median of 71 versus 76 years, P = 0.03, the rank-sum test).
Based on the exome sequencing of pedigree II, there were five affected individuals with D90G and one unaffected individual without this variant, resulting in a LOD score of 1.22 (P = 0.009). We did not find any additional samples with the D90G mutation in our targeted sequencing project and there were no additional relatives available for sequencing in this pedigree (Fig. 1A). The proband of this family has ‘low renal function’ and is on dialysis and her brother has chronic kidney disease stage III with creatinine clearance of 41. Their father also had kidney disease (specific diagnosis unknown).
Although the R53C and D90G risk alleles reside on the CFH haplotype containing the rs1061170(Y402H) and rs10737680 common risk alleles (Fig. 1A and B), the common risk alleles cannot explain the extraordinary large effect size and the early-onset cases in these families. Therefore, we carried out functional studies to investigate the potential impact of R53C and D90G risk alleles on the activation of the complement pathway.
We measured serum FH levels in the 22 individuals carrying the R53C and D90G variants and the levels of secreted FH protein were normal. We then generated wild-type (FH1-4WT) and mutant (FH1-4R53C; FH1-4D90G) in a recombinant fragment, comprising the functional domains CCPs 1–4 of FH. These proteins were expressed in both a yeast system and a mammalian system. We saw the same effects in proteins produced in either system.
Effect of R53C and D90G on C3b binding
We used SPR to examine the binding interaction between FH1-4 (0.04–40 µm) to immobilized C3b. Overlaying sensograms show the steady-state response for the concentration range. We calculated the dissociation constant (KD) using a steady-state affinity model. The affinity of FH1-4WT and FH1-4D90G for C3b was similar, 7.6 and 8.3 μm, respectively. The affinity of FH1-4R53C for C3b is slightly weaker than FH1-4WT. The KD was 12.2 μm for the FH1-4R53C–C3b interaction versus 8.3 μm for FH1-4WT (Fig. 2). In addition, we identified a similar trend if the FH1-4 proteins were immobilized on a sensor chip. FH1-4WT and FH1-4D90G had a KD of 8.6 and 8.3 μm, respectively, while the KD of FH 1-4R53C for C3b was 34 μm (Supplementary Material, Fig. S2).
Figure 2.

R53C binds C3b with a lower affinity than WT. Overlaying sensograms show the steady-state response for the binding of CFH1-4 (Pichia) (0.4–40 µM) to immobilized C3b on the surface of a CM5 sensor chip for (A and B) WT, (C and D) R53C and (E and F) D90G. The steady-state response was plotted against concentration and an affinity curve was fitted.
Decay accelerating activity (DAA)
We then examined decay accelerating activity (DAA) of FH1-4R53C, FH1-4D90G and FH1-4WT using SPR (Fig. 3). In the SPR assay, the FH1-4R53C showed a marked loss of DAA, while FH1-4D90G exhibited normal DAA (Fig. 3). To further investigate the DAA defect in R53C, we used a sheep erythrocyte lysis assay. Fifty percent inhibition of lysis was achieved using 6.0 nm FH1-4WT; however, 2000 nm (a >300-fold difference) was required to obtain the same inhibition using FH1-4R53C. (Supplementary Material, Fig. S3).
Figure 3.
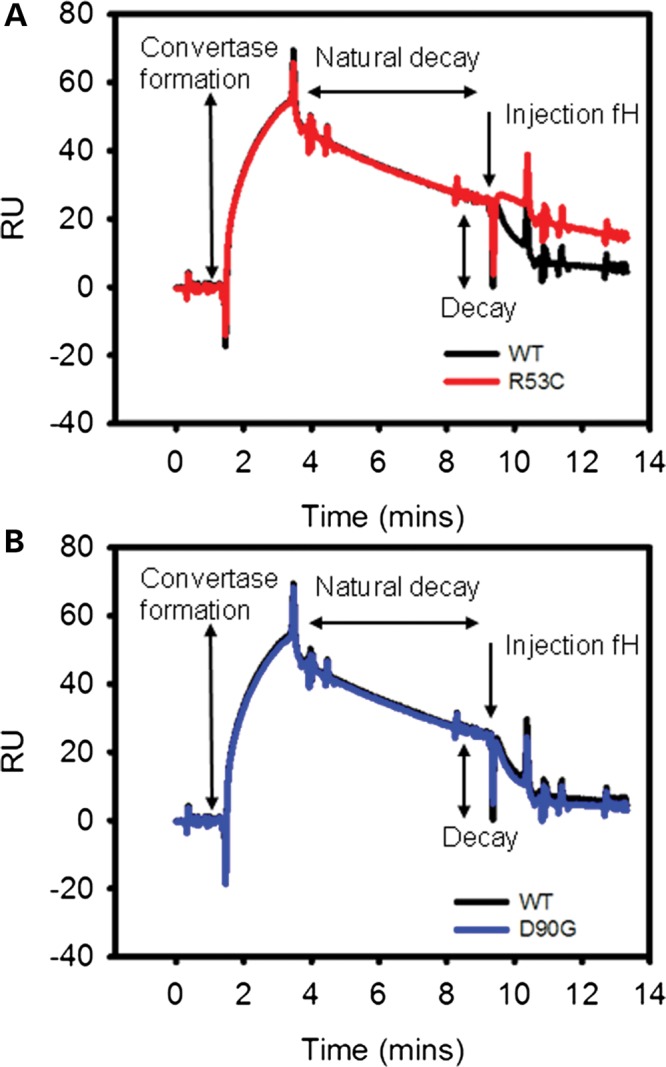
R53C has a profound decay accelerating defect. SPR decay accelerating assay for (A) R53C and (B) D90G. R53C fails to facilitate decay of the C3b:Bb complex in the same way that WT and D90G do (representative trace of three replicates).
Both R53C and D90G affect cofactor function
Using a fluid phase assay in which FH1-4 supplies cofactor activity (CA) for the protease CFI, we saw decreased CA with both mutants. As C3b is cleaved to iC3b, the C3 α′ chain is consumed and α1 and α40 fragments are generated (Supplementary Material, Fig. S4). Both mutants lag behind FH1-4WT in terms of α′ remaining (Fig. 4) over a 30-min time course compared with the original amount of α′. FH1-4D90G's activity is significantly less than FH1-4WT at all time points (Fig. 4A, P < 0.01, the two-tailed t-test). FH1-4R53C also had a consistent and reproducible trend toward less activity than FH1-4WT (Fig. 4B, P > 0.05). To confirm that FH1-4R53C's CA was less than FH1-4WT, we also used an hemolysis based assay; ∼8 µm of FH1-4WT was required to achieve 50% inhibition of lysis, while at any given concentration of FH1-4R53C, less protection was seen (Fig. 4C).
Figure 4.
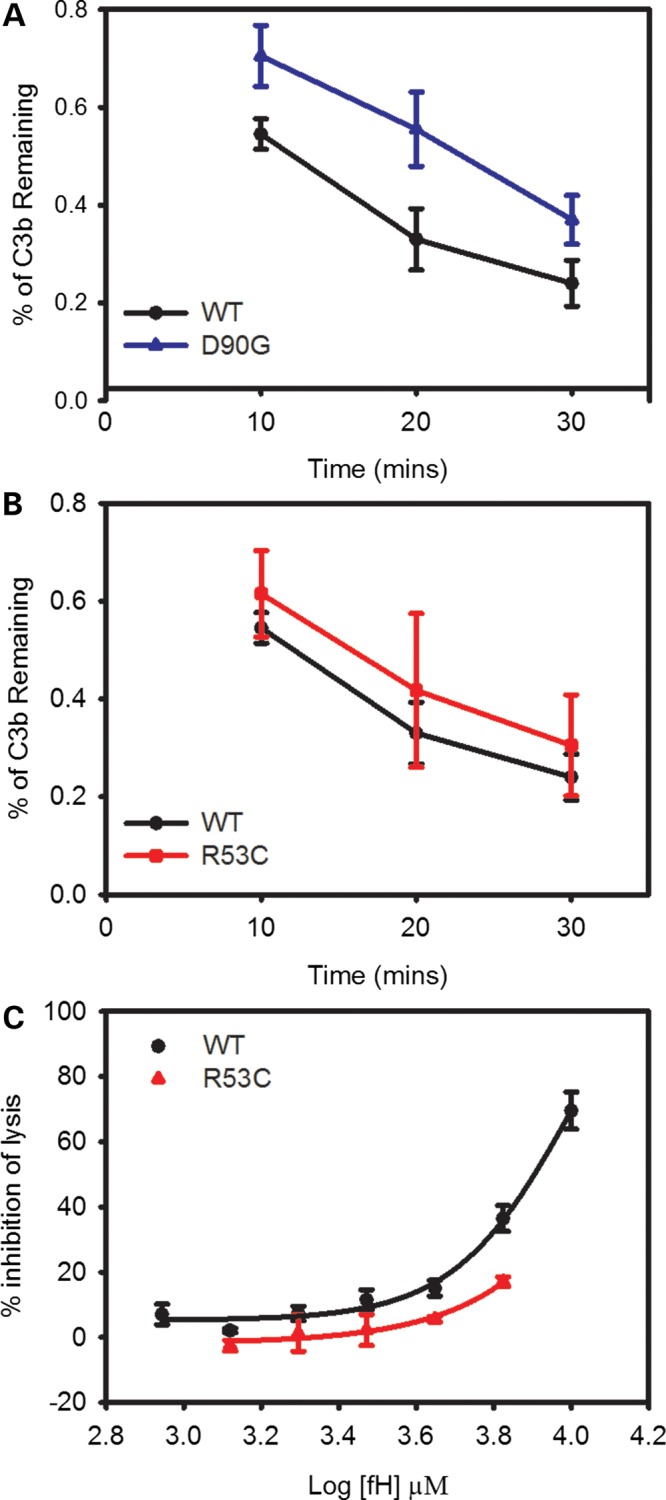
Both D90G and R53C have defects in CA. (A) D90G has a significant defect in co-factor activity (CA) (P < 0.01 at all time points, two-tailed t-test). (B) R53C has lower CA in the fluid phase assay (P > 0.05 at all time points, two-tailed t-test). (C) R53C also has a significant defect in CA in a hemolysis based assay. For (A) and (B), quantitation of the α′ produced over time relative to the amount of α′ present originally. These are the average of four independent experiments; error bars represent one standard deviation. Quantification was done by densitometrically scanning the α′ bands and normalizing to the amount of C3b in that lane (β chain).
DISCUSSION
Common variants explain a large part of the genetic burden related to AMD. However, the observation of densely affected families with early onset cases of advanced AMD lacking a significant contribution from common alleles suggested that rare, more Mendelian-like subtypes of disease exist, being hidden within the otherwise common phenotype. These rare, highly penetrant alleles can be detected only by sequencing, so we applied the strategy of whole-exome sequencing in these AMD families. To our knowledge, cohorts and families with AMD have never been thoroughly investigated previously on the whole-exome level. This approach successfully identified two rare mutations, R53C and D90G, in the CFH gene. These two newly identified CFH mutations are highly penetrant, similar to the previously identified R1210C mutation in the same gene (1) but with lower frequencies. The strong evidence of linkage and association with these very rare variants observed in this study indicates that our family selection strategy leveraged the inherent power of the family structure and effectively increased the power to detect such mutations. Our functional studies of these new high impact alleles provide clearer insight into the mechanisms underlying the development of AMD.
Other than these two new rare variants in CFH, several other variants passed our filtering analysis in selected families. Compared with the other families, the two pedigrees for whom a causal allele was identified in this report had more affected individuals as well as an unaffected individual sequenced, thus allowing identification and investigation of a smaller number of variants that followed the assumed dominant inheritance pattern. Other pedigrees did not have additional members available so we resorted to bioinformatic means to narrow the list of variants that could explain their disease. Such tools are not 100% accurate and additional causal variants may exist which are not predicted to be damaging or deleterious or that were not well covered by the exome sequencing. As more affected pedigrees are interrogated, we expect more overlap will occur between ‘causal variants’ in terms of genes and pathways. While availability of functional assays and known relationship of CFH to AMD made interpretation of new CFH variants more straightforward, some of these additional prioritized variants may also be relevant but await replication in additional families and unrelated cases as well as functional characterization.
Several rare risk alleles in genes of the complement pathway have been identified recently, including R1210C in CFH (1), G119R in CFI (14), K155Q in C3 (2) and P167S in C9 (2), as well as multiple rare variants in CFI (2). Most of these rare alleles were found in genes where a common AMD risk locus had been previously associated, but they are independent of the common risk haplotypes in the disease gene. In this family-based study, the two newly identified risk alleles, R53C and D90G, reside on the common risk haplotype but have larger effects. While these rare variants might be tagging a different causal mutation, we consider this possibility unlikely given the functional alterations conferred by these mutations. It is worthwhile to continue searching for other rare, causal, function-altering variants that are likely to unveil the underlying roles of the associated genes in AMD pathogenesis and to provide new targets for screening, prevention and treatment for AMD.
The molecular mechanisms of R53C and D90G are different from the previously identified R1210C substitution in CFH, which disrupts FH binding to C3b and heparin (15,16), and has been demonstrated to circulate in serum complexed covalently to albumin (15,17,18). R1210C and the common allele Y402H both affect localization of FH onto surfaces (19–22). These new rare variants illustrate that, even when localized, FH must have intact regulatory function to prevent AMD.
The R53C variant shows only a minor decrease in binding affinity for C3b. A co-crystal structure of C3b/FH demonstrates that R53 does not oppose C3b, but is on the exposed face of FH, in keeping with the minor differences in binding (Fig. 5). The R53C variant, however, severely impairs DAA of FH1-4 and reduces its CA.
Figure 5.

FH1-4-C3b co-crystal structure demonstrating the position of the R53C and D90G mutations. An X-ray-derived co-crystal structure of FH-C3b (23) was used to model the mutation and displayed with Pymol (Delano Scientific). The location of the two mutated amino acids (red spheres) are shown within the co-crystal structure of a FH1-4 (dark grey):C3b (light grey) complex. The R53 amino acid is on the opposite face to the FH-C3b interface, while the D90 amino acid is in direct opposition to C3b (Protein Data Base ID code 2WII) (23).
R53C is an example of the pleiotropic effects mutations in the alternative pathway (AP) can have. Generally, rare, highly penetrant variants in this pathway have been linked to a variety of kidney diseases, which can vary widely in their presentation and pathologic findings. The R53C variant has been detected in three such patients with glomerular disease. One patient was heterozygous for this variant and had moderately low C3 levels and C3 glomerulopathy (24). Another patient was heterozygous for R53C, had moderately low C3 levels and developed atypical hemolytic uremic syndrome (aHUS) (25). A third patient was homozygous for the variant and had very low C3 levels and mesangioproliferative glomerulonephritis type 1 (24). Interestingly, this amino acid has also been found mutated to histidine in another patient with aHUS (26). Functional analysis of the R53H variant demonstrated only a minor perturbation of binding to C3b, consistent with this amino acid not playing a major role in the interaction between C3b and FH. As with the R53C variant, the R53H variant also profoundly impaired DAA and reduced CA. None of the individuals carrying R53C in pedigree V reported renal disease. Both R53C and R1210C highlight the phenotypic heterogeneity possible with variants leading to dysregulation of the AP. There are no reports in the published literature of associations between D90G and kidney disease, to our knowledge. In our pedigree with the D90G mutation, two affected individuals with the variant reported having chronic kidney disease or reduced kidney function. It remains to be seen if individuals with such variants that manifest with renal disease will go on to develop AMD at higher rates.
A model of FI docked in a groove between FH and C3b would suggest that R53 would oppose FI in keeping with the impaired CA (27). Additionally, the loss of DAA suggests that an interaction with FB occurs on this face; overlapping binding sites for FI and FB have previously been suggested (26).
Despite residing in the interface between FH and C3b, the D90G variant does not impair the binding to C3b or disrupt DAA at levels detectable in our assays. D90G does, however, affect CA. In aHUS, CA is the critical modality of regulation required for AP homeostasis. This variant has not been identified previously in normal individuals (NHLBI ESP and 1000 Genome Project) or patient populations (aHUS or C3 glomerulopathies). The D90G phenotype of normal C3b binding to one of its regulators, but reduced CA has previously been described for complement inhibitors. Specifically, mutations in the C3b binding site of MCP (CD46) and CR1 (CD35) have been characterized with this type of a dysfunctional pattern (28). CA requires the transient formation of a tri-molecular complex (C3b:Cofactor:Factor I), which is dominated by low affinity interactions. While in most cases, dysfunction moves in parallel (i.e. alterations in C3b binding mimic those observed in CA), the requirement for FI to join the party indicates that some variants may solely affect CA (29–31).
Small losses in regulatory activity may lead to excessive AP activation over decades that manifest itself as the degenerative process of AMD. Thus, rare mutations such as D90G, with a more subtle loss of function, may not be fully penetrant for kidney disease, but are likely to be causative in AMD.
The identification of such rare variants with large effect has implications for clinical trial design and the medical care of carriers and their relatives. Currently, complement inhibitors are available for a variety of complementopathies (32,33) and promising results have come for a phase II trial in AMD (34). Carriers of variants such as those described here in CFH, and already described in CFI, would facilitate proving the concept that such a therapeutic intervention is efficacious.
MATERIALS AND METHODS
Diagnosis and family selection
All family members included in this study were evaluated by board-certified ophthalmologists. Individuals were evaluated (i) by clinical examination with visual acuity measurements, dilated slit-lamp biomicroscopy and stereoscopic color fundus photography or (ii) by reviewing ophthalmologic medical records. All individuals were graded using the Clinical Age-Related Maculopathy Grading System (CARMS) (35). Advanced AMD patients had either geographic atrophy (advanced dry AMD) or neovascular disease (wet AMD). Phenotypes were based on fundus photography or dilated fundus examination using standardized protocols.
Among a collection of 591 families we recruited over the past two decades, 364 families have at least two members with advanced AMD. Using an algorithm we previously published, we selected three families with a genotypic load that was less than expected given the density of disease in their family (3,8,36). We also included one family with more than two cases with a low genetic risk score (< −1.0) derived from a general linear model of the genotype dosages of known common alleles (37). Only 5% of the advanced cases in our GWAS cohort had such a low genetic risk score (<−1.0). The chance of observing two or more cases with low genetic risk score in a family by random chance is rare. Finally, we included five families with at least two advanced AMD cases diagnosed before age 60. Demonstrating the effect of the R1210C variant, nine other families identified using these criteria had the high-risk CFH R1210C allele and were excluded from exome sequencing (1).
For each selected family, all advanced AMD cases with sufficient DNA and an unaffected relative with CARMS grade 1 (if available) were selected for whole exome sequencing. The number of cases and controls in each family are shown in Table 1.
Exome capture and sequencing
The samples were sequenced at the Genome Institute at Washington University. Genomic DNA extracted from blood samples was used to construct Illumina libraries with indexing barcodes to uniquely identify each sequenced individual. We sequenced family members by combining the indexed libraries at an equimolar mixture and then hybridizing to the Nimblegen 3.0 human exome reagent, designed to capture a total 60 Mb consisting of the coding exons and splice junctions of genes cataloged in the human RefSeq database, non-repetitive 3′ and 5′ UTRs, and selected non-coding RNA sequences. We isolated the hybridized library fragments, quantitated with qPCR and then sequenced with the Illumina HiSeq2000™ platform. We produced 2 × 100 bp reads on the HiSeq2000.
Read mapping, variant detection and annotation
Following deconvolution of the barcodes from each Illumina lane, we aligned individual reads for each sample to the human reference genome (NCBI build 37.3, hg19) using Burrows-Wheeler Aligner (BWA, v0.59) (38). We called the consensus genotypes in the target regions with SAMtools (39) and VarScan 2 (40) in the genomic regions targeted by the capture probe set. We filtered variants with ‘PASS’ and high quality (Phred-like quality score ≥30 and an allele balance ≥35) to remove systematic false positives followed by annotation with the VEP tool (41). All sequenced samples were required to have over 10× coverage at greater than 90% of the targeted regions and over 20× coverage at greater than 80% of the targeted regions.
Data analyses
To distinguish potentially pathogenic mutations from other variants, we examined variants that altered coding sequences (missense, nonsense, read-through or splice variants). We predicted that the variants responsible for advanced AMD in the selected families would be very rare and therefore likely to be previously unidentified or have low frequency in public databases. Consequently, we filtered out variants with allele frequency >0.1% in the data sets of the 1000 genomes project (42) or the NHLBI GO ESP (43). Since we were searching for mutations that have a high penetrance and follow a dominantly inherited pattern, we prioritized candidate variants that occur in all cases in a pedigree and none of the sequenced controls. Finally, when evaluating candidate variants, we applied one additional filter to include only variants predicted to be loss of function or probably damaging by PolyPhen-2 (44) or predicted to be deleterious by SIFT (45). These analyses were performed by xBrowse, developed at ATGU at the Massachusetts General Hospital (http://atgu.mgh.harvard.edu/xbrowse). We performed linkage analysis with the LINKAGE programs (46). Fisher's exact test was used to test the association in case-control samples.
TaqMan genotyping of selected SNPs
To confirm segregation within families, we genotyped candidate variants within additional samples within the same families at the Johns Hopkins Genotyping Core Laboratory using a custom made TaqMan genotyping assay by Applied Biosystems and with the ABI 7900 Real-Time PCR system. For the R53C and D90G variants in CFH, we sequenced the additional cases and controls using a targeted-sequencing approach described previously (2).
Production and purification of proteins
We generated and purified wild-type and mutant complement factor H (FH) in the setting of complement control protein (CCPs) 1-4 (FH1–4) using two distinct methods. The first production system utilized a Pichia pastoris system as previously described (26). Briefly, for wild-type we used a pPICZαB (Invitrogen) vector containing residues 19–263 of CFH (which encodes CCPs 1-4 of mature FH; residues 1–18 are the mammalian signal peptide) with an N-terminal myc tag and a C-terminal 6× His tag. We generated the R53C and D90G point mutations using QuikChange site-directed mutagenesis kit (Stratagene) with the following primers: R53C—(f) cccaggctatctataaatgc <T> gccctggatatagatctcttgg; (r) ccaagagatctatatccagggc <A> gcatttatagatagcctggg and D90G—(f) ggccctgtggacatcctggag <G> tactccttttggtacttttaccc; (r) gggtaaaagtaccaaaaggagta <C> ctccaggatgtccacagggcc. We confirmed fidelity with bi-directional Sanger sequencing and transformed these vectors into Pichia pastoris (KM71H cells) by electroporation. Clones producing wild-type (FH1-4WT) or mutant (FH1-4MUT) FH1-4 were selected by zeocin and checked for protein expression.
We expressed protein in a 3 liter BioFlo 115 Biofermenter (New Brunswick) by transferring a starter culture in buffered minimal glycerol into 1 liter of basal fermentor salts [0.095% (w/v) calcium sulphate, 1.82% (w/v) potassium sulphate, 1.5% (w/v) magnesium sulphate heptahydrate, 0.42% (w/v) potassium hydroxide, 2.7% (v/v) phosphoric acid and 2.5% (v/v) glycerol, enriched with 1% (w/v) casein amino acids, 0.5% (w/v) PTM1 salts and 0.5% (v/v) antifoam A (Sigma)]. After the initial batch fed glycerol was exhausted, glycerol feeds were maintained for 24 h at 30°C. The cells were allowed to starve for 4 h before recombinant protein expression was induced with 0.75% methanol containing 0.5% (w/v) PTM1 salts. After 3 days at 15°C with methanol feeds, the supernatant was removed and filtered and its pH adjusted to 7.4.
We applied the supernatant to a 5 ml His trap column (GE-healthcare) at 4°C and eluted the protein with 500 mm imidazole followed by size exclusion chromatography on Superdex 200 (GE Healthcare). The protein concentrations were determined using absorbance at 280 nm and calculated extinction coefficients (47870 M.cm−1).
The second method for production of FH1−4 was performed in human 293-T cells. The first 265 amino acids, including the signal peptide of FH, were cloned by PCR and a C-terminal 6× His Tag was added. This was inserted into a pSG5 vector using EcoR1. R53C was introduced using the following primers:
Forward: ACC CAG GCT ATC TAT AAATGC <T>GC CCT GGATAT AGATCT CTT
Reverse: AAG AGATCT ATATCC AGG GC<A> GCATTT ATA GAT AGC CTG GGT. D90G was introduced with forward: GGACATCCTGGAG<G>TACTCCTTTTGGT; reverse: ACCAAAAGGAGTA<C>CTCCAGGATGTCC. PCR with Phusion (NEB) in the presence of 1 m betaine was used to introduce mutations and they were confirmed with bidirectional Sanger sequencing.
Transfection of 293T cells was performed with XtremeGene-9 (Roche) in serum-free Optimem for 3 days. Supernatants were harvested and recombinant FH1-4 was purified using a His-Trap column (GE). Binding to the column was carried out at 50 mm imidazole with elution at 95 mm imidazole. Protein concentration was determined using an ELISA relative to full-length purified FH (CompTech). A monoclonal capture antibody (H2, #A254, Quidel) was coated in ELISA wells at 500 μg/ml. A goat polyclonal antibody (#A312, Quidel) was used for detection. Relative concentration was confirmed by subjecting FH 1-4 constructs to SDS–PAGE and subsequent silver stain or fluorescent stain with Krypton dye (Thermo).
Binding affinity for C3b by surface plasmon resonance
We monitored the binding affinity of FH1-4WT, FH1-4R53C and FH1-4D90G by surface plasmon resonance (SPR) with Biacore X100 and Biacore 2000 instruments (GE Healthcare). These measurements were performed in two orientations. First, we immobilized 850 resonance units (RU) of human C3b (CompTech) on a Biacore series CM5 sensor chip (GE Healthcare) using standard amine coupling. The reference surface of the chip was prepared by performing a mock coupling in the absence of any protein. We performed the experiments at 25°C and 30 μl/min flow rate in duplicate using FH1-4 produced in Pichia [concentrations 0.04–40 μm in 10 mm HEPES-buffered saline, 3 mm EDTA, 0.05% (v/v) surfactant p20 (GE Healthcare)]. In the second method, we attached the FH1-4 recombinant proteins (produced in mammalian cells) to the CM5 sensor chips via standard amine coupling (∼1000 RU). Purified C3b was injected in duplicate using multiple concentrations (500 nm–5.6 μm). In both cases, a contact time of 90 s was used (sufficient for achieving steady-state conditions) followed by a dissociation period of 45 s. We regenerated the chip surface between sample injections with one 45 s injection of 1 m NaCl (pH 7.0). We processed data using the BIAevaluation 4.1 software (GE Healthcare). The data from the reference cell and a blank (buffer) injection were subtracted and we calculated dissociation constants using a steady-state affinity model from the background-subtracted traces.
Measurement of decay accelerating activity by SPR
We measured DAA in real-time using a Biacore X100 instrument as described previously (26). Briefly, we immobilized 850 RU of C3b using standard amine coupling Biacore series CM5 sensor chip (GE Healthcare). We injected a mixture of 500 nm complement factor B (fB), and 60 nm complement factor D (fD), at 10 μl/min over the surface for 120 s to form the AP C3 convertase.
We allowed the convertase to decay naturally for 210 s and then injected 0.5 μm FH1-4WT or FH1-4MUT produced in Pichia [in running buffer, HEPES-buffered saline containing 0.5% (v/v) surfactant P20 and 1 mm MgCl2, 25°C] across the surface for 60 s and C3 convertase decay was visualized in real time. Between injections, we regenerated the chip surface using a 45 s injection of 10 μm FH1-4WT followed by a 45 s injection of 1 m NaCl, pH 7.0. We evaluated data using BIAevaluation 4.1 (GE Healthcare). As a control, the construct was also flowed over the bare surface and binding data subtracted from the decay sensogram.
Measuring DAA on sheep erythrocytes
We prepared C3b-coated sheep erythrocytes (EA-C3b) as described previously (26). The cells were resuspended to 2% (v/v) in AP buffer (5 mm sodium barbitone, pH 7.4, 150 mm NaCl, 7 mm MgCl2, 10 mm EGTA) and then the AP C3 convertase was formed on the cell surface by incubating 50 μl of the cell preparation with an equal volume of AP buffer containing fB (40 μg/ml; purified from serum by Dr C. Harris, Cardiff) and fD (0.4 μg/ml; CompTech) at 37°C for 15 min. We incubated cells with 50 μl of a concentration range of CFH1-4WT and CFH1-4R53C in PBS/20 mm EDTA for 15 min (both produced in Pichia). We developed lysis by adding 50 μl of 4% (v/v) normal human serum depleted of fB and CFH (NHS-ΔBΔH; prepared by Dr C. Harris, Cardiff) (47) in PBS/20 mm EDTA and incubating at 37°C for 60 min. To determine the amount of lysis, we pelleted cells by centrifugation and hemoglobin release was measured at 410 nm (A410). We used buffer only (no FH1-4) to define maximum lysis by the addition of serum-A410 (buffer only). Percentage of inhibition of lysis in the presence of increasing concentrations of FH was defined as [A410(buffer only)−A410(CFH)/A410(buffer only)*100].
Cofactor assay in fluid phase
Fluid phase assays were carried out with the following concentrations of components: FH1-4 (produced in mammalian cells), 23 nm (0.67 ng/μl); C3b, (Comptech), 35.1 nm (6.67 ng/μl) and factor I (FI) (Comptech), 25 nm (2.2 ng/μl). All dilutions were made in 150 mm NaCl Tris-buffered saline. A positive control used an equivalent mass of full-length purified FH (Comptech). A negative control used full-length FH but no CFI. Reactions were prepared on ice and with a refrigerated centrifuge. Time began when reactions were placed in a 37°C water bath. Samples were removed at 10, 20 and 30 min. Reactions were stopped with 3× reducing Laemmeli buffer, mixed and heated at 95° C for 5 min. A volume of reaction equivalent to 45 ng of C3b was electrophoresed by SDS–PAGE and stained with Krypton dye. Gels were scanned on a Typhon variable mode imager with excitation at 530 and a 580 nm filter. Quantification was performed in ImageQuantTL. Krypton stain was found to be equally sensitive and more robust than silver stain or western blot using polyclonal anti-C3 antibodies.
Measuring CA on sheep erythrocytes
To test for CA, we resuspended washed EA-C3b cells to 2% (v/v) in AP buffer and incubated with an equal volume of a range of concentrations of CFH1-4WT and FH1-4R53C (produced in Pichia) and 2.5 μg/ml FI (CompTech) for 8 min at 25°C. After three washes in AP buffer, we mixed a 50 μl aliquot of cells (2%) with 50 μl AP buffer containing FB (40 μg/ml) and fD (0.4 μg/ml) and then incubated for 15 min at 25°C to form the AP C3 convertase on the remaining C3b. We developed lysis by adding 50 μl of 4% (v/v) NHS-ΔBΔH in PBS/20 mm EDTA and incubating at 37°C for 10 min. We again calculated the percentage of inhibition of lysis in the presence of increasing concentrations of FH using [A410(buffer only)−A410(FH)/A410(buffer only)*100].
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This research was supported in part by NIH grants R01-EY11309 (J.M.S.), K08AR055688 (S.R.), U01HG0070033 (S.R.), F30HL103072 (M.T.), R01-AI041592 (J.P.A. and E.C.S.), U54HG00307910 (E.M.), U54 HL112303 (J.P.A.); Edward N. & Della L.Thome Memorial Foundation (J.P.A.); the Doris Duke Clinical Scientist Development Award; the Facility of the Rheumatic Diseases Core supported by NIH-Arthritis and Musculoskeletal and Skin Diseases P30 AR48335 (J.P.A.), Massachusetts Lions Eye Research Fund, Inc. (J.M.S.); the Foundation Fighting Blindness (J.M.S.); the Macular Vision Research Foundation (J.M.S.); Research to Prevent Blindness Challenge Grant to the New England Eye Center, Department of Ophthalmology, Tufts University School of Medicine; American Macular Degeneration Foundation (J.M.S.); The Arnold and Mabel Beckman Initiative for Macular Research (E.M., S.R., J.M.S.) and the Macular Degeneration Research Fund of the Ophthalmic Epidemiology and Genetics Service, New England Eye Center, Tufts Medical Center, Tufts University School of Medicine. D.K. is a Wellcome Intermediate Clinical Fellow. E.K.S.W. is a Medical Research Council clinical research training fellow. M.T. is a Ruth L. Kirschstein National Research Service Award recipient (National Heart, Lung and Blood Institute). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
REFERENCES
- 1.Raychaudhuri S., Iartchouk O., Chin K., Tan P.L., Tai A.K., Ripke S., Gowrisankar S., Vemuri S., Montgomery K., Yu Y., et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011;43:1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seddon J.M., Yu Y., Miller E.C., Reynolds R., Tan P.L., Gowrisankar S., Goldstein J.I., Triebwasser M., Anderson H.E., Zerbib J., et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013;45:1366–1370. doi: 10.1038/ng.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seddon J.M., Reynolds R., Maller J., Fagerness J.A., Daly M.J., Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest. Ophthalmol. Vis. Sci. 2009;50:2044–2053. doi: 10.1167/iovs.08-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seddon J.M., Reynolds R., Yu Y., Daly M.J., Rosner B. Risk models for progression to advanced age-related macular degeneration using demographic, environmental, genetic, and ocular factors. Ophthalmology. 2011;118:2203–2211. doi: 10.1016/j.ophtha.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Y., Reynolds R., Rosner B., Daly M.J., Seddon J.M. Prospective assessment of genetic effects on progression to different stages of age-related macular degeneration using multistate Markov models. Invest. Ophthalmol. Vis. Sci. 2012;53:1548–1556. doi: 10.1167/iovs.11-8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helgason H., Sulem P., Duvvari M.R., Luo H., Thorleifsson G., Stefansson H., Jonsdottir I., Masson G., Gudbjartsson D.F., Walters G.B., et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat. Genet. 2013;45:1371–1374. doi: 10.1038/ng.2740. [DOI] [PubMed] [Google Scholar]
- 7.Zhan X., Larson D.E., Wang C., Koboldt D.C., Sergeev Y.V., Fulton R.S., Fulton L.L., Fronick C.C., Branham K.E., Bragg-Gresham J., et al. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013;45:1375–1379. doi: 10.1038/ng.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobrin L., Maller J.B., Neale B.M., Reynolds R.C., Fagerness J.A., Daly M.J., Seddon J.M. Genetic profile for five common variants associated with age-related macular degeneration in densely affected families: a novel analytic approach. Eur. J. Hum. Genet. 2010;18:496–501. doi: 10.1038/ejhg.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison A.C., Voorman A., Johnson A.D., Liu X., Yu J., Li A., Muzny D., Yu F., Rice K., Zhu C., et al. Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat. Genet. 2013;45:899–901. doi: 10.1038/ng.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farkas M.H., Grant G.R., White J.A., Sousa M.E., Consugar M.B., Pierce E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics. 2013;14:486. doi: 10.1186/1471-2164-14-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klein R.J., Zeiss C., Chew E.Y., Tsai J.Y., Sackler R.S., Haynes C., Henning A.K., SanGiovanni J.P., Mane S.M., Mayne S.T., et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haines J.L., Hauser M.A., Schmidt S., Scott W.K., Olson L.M., Gallins P., Spencer K.L., Kwan S.Y., Noureddine M., Gilbert J.R., et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 13.Edwards A.O., Ritter R., 3rd, Abel K.J., Manning A., Panhuysen C., Farrer L.A. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 14.van de Ven J.P., Nilsson S.C., Tan P.L., Buitendijk G.H., Ristau T., Mohlin F.C., Nabuurs S.B., Schoenmaker-Koller F.E., Smailhodzic D., Campochiaro P.A., et al. A functional variant in the CFI gene confers a high risk of age-related macular degeneration. Nat. Genet. 2013;45:813–817. doi: 10.1038/ng.2640. [DOI] [PubMed] [Google Scholar]
- 15.Manuelian T., Hellwage J., Meri S., Caprioli J., Noris M., Heinen S., Jozsi M., Neumann H.P., Remuzzi G., Zipfel P.F. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Invest. 2003;111:1181–1190. doi: 10.1172/JCI16651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira V.P., Herbert A.P., Cortes C., McKee K.A., Blaum B.S., Esswein S.T., Uhrin D., Barlow P.N., Pangburn M.K., Kavanagh D. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J. Immunol. 2009;182:7009–7018. doi: 10.4049/jimmunol.0804031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez-Corral P., Perez-Caballero D., Huarte O., Simckes A.M., Goicoechea E., Lopez-Trascasa M., de Cordoba S.R. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am. J. Hum. Genet. 2002;71:1285–1295. doi: 10.1086/344515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jozsi M., Heinen S., Hartmann A., Ostrowicz C.W., Halbich S., Richter H., Kunert A., Licht C., Saunders R.E., Perkins S.J., et al. Factor H and atypical hemolytic uremic syndrome: mutations in the C-terminus cause structural changes and defective recognition functions. J. Am. Soc. Nephrol. 2006;17:170–177. doi: 10.1681/ASN.2005080868. [DOI] [PubMed] [Google Scholar]
- 19.Clark S.J., Ridge L.A., Herbert A.P., Hakobyan S., Mulloy B., Lennon R., Wurzner R., Morgan B.P., Uhrin D., Bishop P.N., et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J. Immunol. 2013;190:2049–2057. doi: 10.4049/jimmunol.1201751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark S.J., Perveen R., Hakobyan S., Morgan B.P., Sim R.B., Bishop P.N., Day A.J. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J. Biol. Chem. 2010;285:30192–30202. doi: 10.1074/jbc.M110.103986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weismann D., Hartvigsen K., Lauer N., Bennett K.L., Scholl H.P., Charbel Issa P., Cano M., Brandstatter H., Tsimikas S., Skerka C., et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjoberg A.P., Trouw L.A., Clark S.J., Sjolander J., Heinegard D., Sim R.B., Day A.J., Blom A.M. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J. Biol. Chem. 2007;282:10894–10900. doi: 10.1074/jbc.M610256200. [DOI] [PubMed] [Google Scholar]
- 23.Wu J., Wu Y.Q., Ricklin D., Janssen B.J., Lambris J.D., Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 2009;10:728–733. doi: 10.1038/ni.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Servais A., Noel L.H., Roumenina L.T., Le Quintrec M., Ngo S., Dragon-Durey M.A., Macher M.A., Zuber J., Karras A., Provot F., et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012;82:454–464. doi: 10.1038/ki.2012.63. [DOI] [PubMed] [Google Scholar]
- 25.Fakhouri F., Roumenina L., Provot F., Sallee M., Caillard S., Couzi L., Essig M., Ribes D., Dragon-Durey M.A., Bridoux F., et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J. Am. Soc. Nephrol. 2010;21:859–867. doi: 10.1681/ASN.2009070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pechtl I.C., Kavanagh D., McIntosh N., Harris C.L., Barlow P.N. Disease-associated N-terminal complement factor H mutations perturb cofactor and decay-accelerating activities. J. Biol. Chem. 2011;286:11082–11090. doi: 10.1074/jbc.M110.211839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roversi P., Johnson S., Caesar J.J., McLean F., Leath K.J., Tsiftsoglou S.A., Morgan B.P., Harris C.L., Sim R.B., Lea S.M. Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc. Natl Acad. Sci. USA. 2011;108:12839–12844. doi: 10.1073/pnas.1102167108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang C.J., Fremeaux-Bacchi V., Liszewski M.K., Pianetti G., Noris M., Goodship T.H., Atkinson J.P. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood. 2008;111:624–632. doi: 10.1182/blood-2007-04-084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liszewski M.K., Leung M., Cui W., Subramanian V.B., Parkinson J., Barlow P.N., Manchester M., Atkinson J.P. Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46) J. Biol. Chem. 2000;275:37692–37701. doi: 10.1074/jbc.M004650200. [DOI] [PubMed] [Google Scholar]
- 30.Krych M., Hauhart R., Atkinson J.P. Structure-function analysis of the active sites of complement receptor type 1. J. Biol. Chem. 1998;273:8623–8629. doi: 10.1074/jbc.273.15.8623. [DOI] [PubMed] [Google Scholar]
- 31.Krych M., Clemenza L., Howdeshell D., Hauhart R., Hourcade D., Atkinson J.P. Analysis of the functional domains of complement receptor type 1 (C3b/C4b receptor; CD35) by substitution mutagenesis. J. Biol. Chem. 1994;269:13273–13278. [PubMed] [Google Scholar]
- 32.Legendre C.M., Licht C., Muus P., Greenbaum L.A., Babu S., Bedrosian C., Bingham C., Cohen D.J., Delmas Y., Douglas K., et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013;368:2169–2181. doi: 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 33.Hillmen P., Hall C., Marsh J.C., Elebute M., Bombara M.P., Petro B.E., Cullen M.J., Richards S.J., Rollins S.A., Mojcik C.F., et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2004;350:552–559. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 34.Yehoshua Z., Rosenfeld P.J., Albini T.A. Current clinical trials in dry AMD and the definition of appropriate clinical outcome measures. Semin. Ophthalmol. 2011;26:167–180. doi: 10.3109/08820538.2011.577132. [DOI] [PubMed] [Google Scholar]
- 35.Seddon J.M., Sharma S., Adelman R.A. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology. 2006;113:260–266. doi: 10.1016/j.ophtha.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Fritsche L.G., Chen W., Schu M., Yaspan B.L., Yu Y., Thorleifsson G., Zack D.J., Arakawa S., Cipriani V., Ripke S., et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013;45:433–439. doi: 10.1038/ng.2578. 439e431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu Y., Bhangale T.R., Fagerness J., Ripke S., Thorleifsson G., Tan P.L., Souied E.H., Richardson A.J., Merriam J.E., Buitendijk G.H., et al. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 2011;20:3699–3709. doi: 10.1093/hmg/ddr270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tennessen J.A., Bigham A.W., O'Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sim N.L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P.C. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lathrop G.M., Lalouel J.M., Julier C., Ott J. Strategies for multilocus linkage analysis in humans. Proc. Natl Acad. Sci. USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tortajada A., Montes T., Martinez-Barricarte R., Morgan B.P., Harris C.L., de Cordoba S.R. The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum. Mol. Genet. 2009;18:3452–3461. doi: 10.1093/hmg/ddp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.