

Abstract
To facilitate accurate histone variant and post-translational modification (PTM) quantification via mass spectrometry, we present a library of 93 synthetic peptides using Protein-Aqua™ technology. The library contains 55 peptides representing different modified forms from histone H3 peptides, 23 peptides representing H4 peptides, 5 peptides representing canonical H2A peptides, 8 peptides representing H2A.Z peptides, and peptides for both macroH2A and H2A.X. The PTMs on these peptides include lysine mono- (me1), di- (me2), and tri-methylation (me3); lysine acetylation; arginine me1; serine/threonine phosphorylation; and N-terminal acetylation. The library was subjected to chemical derivatization with propionic anhydride, a widely employed protocol for histone peptide quantification. Subsequently, the detection efficiencies were quantified using mass spectrometry extracted ion chromatograms. The library yields a wide spectrum of detection efficiencies, with more than 1700-fold difference between the peptides with the lowest and highest efficiencies. In this paper, we describe the impact of different modifications on peptide detection efficiencies and provide a resource to correct for detection biases among the 93 histone peptides. In brief, there is no correlation between detection efficiency and molecular weight, hydrophobicity, basicity, or modification type. The same types of modifications may have very different effects on detection efficiencies depending on their positions within a peptide. We also observed antagonistic effects between modifications. In a study of mouse trophoblast stem cells, we utilized the detection efficiencies of the peptide library to correct for histone PTM/variant quantification. For most histone peptides examined, the corrected data did not change the biological conclusions but did alter the relative abundance of these peptides. For a low-abundant histone H2A variant, macroH2A, the corrected data led to a different conclusion than the uncorrected data. The peptide library and detection efficiencies presented here may serve as a resource to facilitate studies in the epigenetics and proteomics fields.
In eukaryotes, histones package and order DNA into nucleosomes. Histones are critical for the structural organization and transcriptional activities of chromatin. These proteins are highly conserved in eukaryotes, but they have very dynamic functions and are subject to many different regulatory mechanisms (1). One of the major mechanisms is enacted via the use of different histone variants and post-translational modifications (PTMs).1 Over the past two decades, accumulating evidence has suggested that levels and PTM compositions of many histone variants play important roles in cellular processes and human diseases. There are increasing needs for the precise quantification of histone variants and PTMs in biomedical studies. Mass spectrometry (MS) has proved to be a robust and powerful tool for analyzing histones, as it can be used to simultaneously identify and quantify histone variants and PTMs (2).
Histones are rich in basic residues and can be highly modified, resulting in an abundance of different isoforms. As an example, the H3 N-terminal tail carries more than two dozen residues that may be modified (3). Many histone lysine residues can be acetylated or mono-, di-, or tri-methylated. These features make histone analysis challenging for the analytical chemistry field. To overcome these challenges, we previously developed an efficient chemical derivatization method for histone peptides that utilizes propionic anhydride to react with the primary amine on lysine residues (4, 5). This method has been adopted by research groups worldwide (6–17) and enables the production of (i) tryptic peptides of a reasonable length appropriate for mass spectrometry, (ii) increased hydrophobicity of histone peptides to obtain better separation and retention on reverse phase (RP) HPLC columns, and (iii) improved fragmentation of lesser charged histone peptides.
Our protocol of chemical derivatization combined with high-resolution bottom-up nano-LC-MS/MS has been highly referenced in the literature (6–17). In the majority of these studies, a relative abundance strategy was applied (8, 13, 14, 18, 19): histones were extracted from two or more biological samples and processed in parallel. The relative percentage, rather than the absolute amount of one or more particular histone PTMs, is quantified and compared. For a given histone peptide with multiple isoforms, including the unmodified peptide and the peptide with various PTMs and combinations of PTMs, the raw abundance of each isoform is quantified using each MS extracted ion chromatogram. The relative percentage of a specific isoform of the same peptide is calculated from the raw abundance of this particular isoform over the sum of all possible forms. This strategy eliminates systematic bias caused by sample preparation or instrumentation. Furthermore, stable isotope labeling in cell culture or at the chemical derivazation step may be introduced to enable more precise quantification (5). However, this strategy cannot eliminate the bias in the peptide detection efficiency in bottom-up MS. A well-known example of this bias is that phosphorylated peptides generally have lower detection efficiencies than their unphosphorylated counterparts, as recently systematically illustrated by Kuster and colleagues (20). This bias would potentially cause problems in the interpretation of histone PTM data, especially when there is a need for absolute quantification. It would also cause a problem for relative quantifications—because the data are normalized to the total signal from all isoforms of a peptide (8, 13, 14, 18, 19), bias in quantification of one particular form would affect all the other forms. To overcome these issues, we present here a library of 93 synthetic histone peptides and comprehensive analyses of these peptides using bottom-up nano-LC-MS/MS and correction factors generated through normalization of their detection efficiencies. We hope these data will become a useful resource for improved accurate quantitative analyses of histone proteins.
EXPERIMENTAL PROCEDURES
Synthetic Peptide Preparation
Ninety-three synthetic peptides representing various histone tryptic peptides were synthesized via Cell Signaling Technology® Protein-Aqua™ (Table I). As previously described (21–23), these peptides were purified by means of RP-HPLC and analyzed via MALDI-TOF-MS and nanospray tandem MS. The peptide concentration was measured via amino acid analysis. The stock solution was 25 pmol/μl for all peptides. For each sample preparation, equal amounts (5 μl) of each peptide were mixed and aliquoted, resulting in a concentration of 0.27 pmol/μl (25/93 = 0.27) for each peptide. For each sample preparation, 50 to 100 μl of the mixture were subjected to two rounds of propionylation as previously described (5). Samples were then desalted by C18-based STAGE tips and injected onto an online nano-LC-MS setup as described in Ref. 5.
Table I. Synthetic peptide sequences and relative quantifications.
| Peptide number | CST product number | Protein and peptide positions | Synthetic peptide sequence and PTMs | Mass shift (Da) | Endogenous peptide (propionylated) MH+ mass | Synthetic peptide (propionylated) MH+ mass | Relative abundance (%) |
|---|---|---|---|---|---|---|---|
| 1 | 11188 | Histone H3 1–8 | ARTKQTAR | 8.014 | 1043.596 | 1051.610 | 0.305 |
| 2 | 11189 | ARme1TKQTAR | 8.014 | 1057.612 | 1065.626 | 0.375 | |
| 3 | 11190 | Histone H3 3–8 | TKQTAR | 4.007 | 816.458 | 820.465 | 0.0326 |
| 4 | 11191 | TKme1QTAR | 4.007 | 830.474 | 834.481 | 0.178 | |
| 5 | 11192 | TKme2QTAR | 4.007 | 788.463 | 792.470 | 0.0018 | |
| 6 | 11193 | TKme3QTAR | 4.007 | 802.478 | 806.485 | 0.0019 | |
| 7 | 11194 | TphKQTAR | 4.007 | 896.424 | 900.431 | 0.0055 | |
| 8 | 11195 | Histone H3 9–17 | KSTGGKAPR | 6.014 | 1069.601 | 1075.614 | 0.571 |
| 9 | 11197 | Kme1STGGKAPR | 6.014 | 1083.616 | 1089.630 | 1.295 | |
| 10 | 11198 | Kme2STGGKAPR | 6.014 | 1041.606 | 1047.619 | 0.303 | |
| 11 | 11199 | Kme3STGGKAPR | 12.021 | 1055.621 | 1067.642 | 0.252 | |
| 12 | 11196 | KacSTGGKAPR | 6.014 | 1055.584 | 1061.598 | 0.314 | |
| 13 | 11200 | KSTGGKacAPR | 9.018 | 1055.584 | 1064.602 | 0.378 | |
| 14 | 11201 | Kme1STGGKacAPR | 6.014 | 1069.600 | 1075.614 | 1.014 | |
| 15 | 11202 | Kme2STGGKacAPR | 6.014 | 1027.589 | 1033.603 | 0.177 | |
| 16 | 11203 | Kme3STGGKacAPR | 10.021 | 1041.605 | 1051.625 | 0.156 | |
| 17 | 11204 | KacSTGGKacAPR | 6.014 | 1041.568 | 1047.582 | 0.276 | |
| 18 | 11205 | KSphTGGKAPR | 6.014 | 1149.566 | 1155.580 | 0.238 | |
| 19 | 11206 | Kme1SphTGGKAPR | 6.014 | 1163.582 | 1169.596 | 0.622 | |
| 20 | 11207 | Kme2SphTGGKAPR | 6.014 | 1121.571 | 1127.585 | 0.204 | |
| 21 | 11208 | Kme3SphTGGKAPR | 6.014 | 1135.587 | 1141.600 | 0.160 | |
| 22 | 11209 | Histone H3 18–26 | KQLATKAAR | 7.017 | 1154.690 | 1161.707 | 1.608 |
| 23 | 11211 | Kme1QLATKAAR | 7.017 | 1168.705 | 1175.723 | 1.210 | |
| 24 | 11213 | KQLATKme1AAR | 11.024 | 1168.705 | 1179.730 | 1.591 | |
| 25 | 11210 | KacQLATKAAR | 7.017 | 1140.674 | 1147.691 | 1.553 | |
| 26 | 11212 | KQLATKacAAR | 15.031 | 1140.674 | 1155.705 | 1.307 | |
| 27 | 11214 | KacQLATKacAAR | 7.017 | 1126.657 | 1133.675 | 1.711 | |
| 28 | 11215 | Histone H3 27–40 | KSAPATGGVKKPHR | 6.014 | 1657.939 | 1663.953 | 1.885 |
| 29 | 11217 | Kme1SAPATGGVKKPHR | 6.014 | 1671.955 | 1677.968 | 2.081 | |
| 30 | 11218 | Kme2SAPATGGVKKPHR | 6.014 | 1629.944 | 1635.958 | 3.201 | |
| 31 | 11219 | Kme3SAPATGGVKKPHR | 10.021 | 1643.959 | 1653.980 | 3.176 | |
| 32 | 11216 | KacSAPATGGVKKPHR | 6.014 | 1643.923 | 1649.937 | 2.305 | |
| 33 | 11220 | KSAPATGGVKme1KPHR | 9.018 | 1671.955 | 1680.972 | 1.870 | |
| 34 | 11221 | KSAPATGGVKme2KPHR | 9.018 | 1629.944 | 1638.962 | 1.678 | |
| 35 | 11222 | KSAPATGGVKme3KPHR | 9.018 | 1643.959 | 1652.977 | 1.762 | |
| 36 | 11223 | Kme1SAPATGGVKme1KPHR | 12.028 | 1685.970 | 1697.998 | 1.377 | |
| 37 | 11224 | Kme1SAPATGGVKme2KPHR | 12.028 | 1643.959 | 1655.987 | 1.377 | |
| 38 | 11225 | Kme1SAPATGGVKme3KPHR | 6.014 | 1657.975 | 1663.989 | 1.670 | |
| 39 | 11226 | Kme2SAPATGGVKme1KPHR | 15.031 | 1643.959 | 1658.990 | 2.288 | |
| 40 | 11227 | Kme2SAPATGGVKme2KPHR | 9.018 | 1601.949 | 1610.967 | 1.619 | |
| 41 | 11228 | Kme2SAPATGGVKme3KPHR | 6.014 | 1615.964 | 1621.978 | 2.119 | |
| 42 | 11229 | Kme3SAPATGGVKme1KPHR | 12.028 | 1657.975 | 1670.002 | 2.840 | |
| 43 | 11230 | Kme3SAPATGGVKme2KPHR | 9.018 | 1615.964 | 1624.982 | 1.653 | |
| 44 | 11231 | Kme3SAPATGGVKme3KPHR | 6.014 | 1629.979 | 1635.993 | 1.746 | |
| 45 | 11232 | KSphAPATGGVKKPHR | 6.014 | 1737.905 | 1743.919 | 1.647 | |
| 46 | 11233 | Kme1SphAPATGGVKKPHR | 6.014 | 1751.920 | 1757.934 | 1.438 | |
| 47 | 11234 | Kme2SphAPATGGVKKPHR | 6.014 | 1709.910 | 1715.924 | 2.894 | |
| 48 | 11235 | Kme3SphAPATGGVKKPHR | 6.014 | 1723.925 | 1729.939 | 2.222 | |
| 49 | 11242 | Histone H3 41–49 | YRPGTVALR | 7.017 | 1088.622 | 1095.639 | 0.654 |
| 50 | 11240 | Histone H3 54–63 | YQKSTELLIR | 7.017 | 1362.763 | 1369.780 | 0.217 |
| 51 | 11241 | YQKacSTELLIR | 7.017 | 1348.747 | 1355.764 | 0.512 | |
| 52 | 11236 | Histone H3 73–83 | EIAQDFKTDLR | 7.017 | 1447.743 | 1454.760 | 0.125 |
| 53 | 11237 | EIAQDFKme1TDLR | 7.017 | 1461.759 | 1468.776 | 0.069 | |
| 54 | 11238 | EIAQDFKme2TDLR | 7.017 | 1419.748 | 1426.765 | 0.548 | |
| 55 | 11239 | EIAQDFKme3TDLR | 7.017 | 1433.763 | 1440.781 | 0.668 | |
| 56 | 11243 | Histone H4 1–17 | acSGRGKGGKGLGKGGAKR | 7.017 | 1837.040 | 1844.057 | 1.227 |
| 57 | 11244 | acSGRme1GKGGKGLGKGGAKR | 7.017 | 1851.056 | 1858.073 | 1.878 | |
| 58 | 11245 | Histone H4 4–17 | GKGGKGLGKGGAKR | 7.017 | 1550.902 | 1557.919 | 1.872 |
| 59 | 11246 | GKGGKGLGKGGAKacR | 6.008 | 1536.886 | 1542.893 | 1.500 | |
| 60 | 11247 | GKGGKGLGKacGGAKR | 9.011 | 1536.886 | 1545.897 | 1.788 | |
| 61 | 11248 | GKGGKacGLGKGGAKR | 11.024 | 1536.886 | 1547.910 | 1.357 | |
| 62 | 11249 | GKacGGKGLGKGGAKR | 14.028 | 1536.886 | 1550.914 | 1.950 | |
| 63 | 11250 | GKacGGKGLGKGGAKacR | 6.008 | 1522.869 | 1528.877 | 2.011 | |
| 64 | 11251 | GKGGKacGLGKGGAKacR | 7.017 | 1522.869 | 1529.887 | 1.023 | |
| 65 | 11252 | GKGGKGLGKacGGAKacR | 9.011 | 1522.869 | 1531.881 | 1.401 | |
| 66 | 11253 | GKacGGKGLGKacGGAKR | 11.024 | 1522.869 | 1533.894 | 1.431 | |
| 67 | 11254 | GKGGKacGLGKacGGAKR | 13.025 | 1522.869 | 1535.894 | 1.633 | |
| 68 | 11255 | GKacGGKacGLGKGGAKR | 14.028 | 1522.869 | 1536.897 | 1.000 | |
| 69 | 11256 | GKGGKacGLGKacGGAKacR | 6.008 | 1508.853 | 1514.861 | 1.644 | |
| 70 | 11257 | GKacGGKGLGKacGGAKacR | 9.011 | 1508.853 | 1517.864 | 1.317 | |
| 71 | 11258 | GKacGGKacGLGKGGAKacR | 11.024 | 1508.853 | 1519.877 | 1.315 | |
| 72 | 11259 | GKacGGKacGLGKacGGAKR | 14.028 | 1508.853 | 1522.881 | 1.589 | |
| 73 | 11260 | GKacGGKacGLGKacGGAKacR | 7.017 | 1494.837 | 1501.854 | 1.399 | |
| 74 | 11261 | Histone H4 20–23 | KVLR | 7.017 | 627.419 | 634.436 | 0.946 |
| 75 | 11262 | Kme1VLR | 7.017 | 641.435 | 648.452 | 1.412 | |
| 76 | 11263 | Kme2VLR | 7.017 | 599.424 | 606.442 | 0.078 | |
| 77 | 11264 | Kme3VLR | 7.017 | 613.440 | 620.457 | 0.075 | |
| 78 | 11265 | Histone H4 68–78 | DAVTYTEHAKR | 6.014 | 1402.697 | 1408.710 | 0.411 |
| 79 | 11274 | Canonical histone H2A 4–11 | GKQGGKAR | 7.011 | 969.548 | 976.559 | 0.329 |
| 80 | 11275 | GKQGGKacAR | 7.011 | 955.532 | 962.543 | 0.123 | |
| 81 | 11276 | GKacQGGKAR | 10.015 | 955.532 | 965.546 | 0.070 | |
| 82 | 11277 | GKacQGGKacAR | 7.011 | 941.516 | 948.526 | 0.030 | |
| 83 | 11273 | Canonical histone H2A 82–88 | HLQLAIR | 7.017 | 906.552 | 913.570 | 1.338 |
| 84 | 11278 | Histone H2A.Z 1–19 | AGGKAGKDSGKAKTKAVSR | 6.014 | 2153.193 | 2159.207 | 0.720 |
| 85 | 11279 | AGGKacAGKDSGKAKTKAVSR | 6.014 | 2139.177 | 2145.190 | 0.754 | |
| 86 | 11280 | AGGKAGKacDSGKAKTKAVSR | 9.018 | 2139.177 | 2148.194 | 0.422 | |
| 87 | 11281 | AGGKAGKDSGKacAKTKAVSR | 12.021 | 2139.177 | 2151.198 | 0.616 | |
| 88 | 11282 | AGGKAGKacDSGKacAKTKAVSR | 6.014 | 2125.160 | 2131.174 | 0.338 | |
| 89 | 11283 | AGGKacAGKDSGKacAKTKAVSR | 9.018 | 2125.160 | 2134.178 | 0.944 | |
| 90 | 11284 | AGGKacAGKacDSGKAKTKAVSR | 12.021 | 2125.160 | 2137.182 | 0.756 | |
| 91 | 11285 | AGGKacAGKacDSGKacAKTKAVSR | 6.014 | 2111.144 | 2117.158 | 0.731 | |
| 92 | 11286 | Histone macroH2A 44–55 | SAKAGVIFPVGR | 6.014 | 1313.758 | 1319.772 | 0.856 |
| 93 | 11287 | Histone H2A.X 4–11 | GKTGGKAR | 7.011 | 942.537 | 949.548 | 0.235 |
93 histone peptides with various PTMs were synthesized with stable isotope (C13 and N15)-labeled amino acids (underlined in the column “Synthetic Peptide Sequence and PTMs”). me1, -2, and -3 denote mono-, di-, and tri-methylation; ph, phosphorylation; ac, acetylation. The peptide number used in Fig. 2 is shown in the first column. Cell Signaling Technology Inc. product numbers are shown in the second column. The averaged relative abundance in the last column represents the detection efficiencies of these peptides.
Nano–Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry
The samples were loaded onto one of the three instrument setups shown in Table II, all at 300 nL/min. Histone peptides were resolved on a two-step gradient from 2% acetonitrile to 30% acetonitrile in 0.1% formic acid over 40 min, then from 30% acetonitrile to 95% acetonitrile in 0.1% formic acid over 20 min. The mass spectrometers were operated in the data-dependent mode with dynamic exclusion enabled (repeat count: 1; exclusion duration: 0.5 min). MS instrument methods were set up as previously reported (2). Settings for resolution, automatic gain control, and normalized collision energy are listed in Table II. Every cycle, one full MS scan (m/z 290 to 1600) was collected, followed by 10 MS/MS scans using either high-energy C-trap dissociation or collision-induced dissociation in the ion trap (Table II). All isolation windows were set at 2.0 m/z. Ions with a charge state of 1 and a rejection list of common contaminant ions (including keratin, trypsin, and BSA) (exclusion width = 10 ppm) were excluded from MS/MS.
Table II. Nano-LC-MS/MS instruments.
| Mass spectrometer | Q Exactive (Q-E) | Orbitrap Elite | Orbitrap Velos Pro |
|---|---|---|---|
| HPLC | Thermo EASY-nLC 1000 | Eksigent nanoLC Ultra-2D plus | Thermo EASY-nLC 1000 |
| Pre-column | Acclaim® PepMap 100 pre-column | 75 μm × 2 cm, 3 μm ReproSil-Pur 120 C18-AQ (Dr. Maisch GmbH, Germany) | |
| Analytical column | Acclaim® PepMap RSLC column | 75 μm × 15 cm, 1.9 μm ReproSil-Pur 120 C18-AQ (Dr. Maisch GmbH, Germany) | 75 μm × 15 cm, 3 μm ReproSil-Pur 120 C18-AQ (Dr. Maisch GmbH, Germany) |
| MS resolution | 70,000 | 60,000 | 60,000 |
| MS automatic gain control target | 1e6 | 1e6 | 1e6 |
| MS/MS HCD resolution | 17,500 | 15,000 | 15,000 |
| MS/MS automatic gain control target | 1e5 | HCD, 5e4; CID, 1e4 | HCD, 5e4; CID, 3e4 |
| MS/MS normalized collision energy | 35.0 | HCD, 37.5; CID, 40.0 | HCD, 36.0; CID, 40.0 |
Listed are three instrument setups and settings for MS runs. For Elite and Velos, both high-energy C-trap dissociation (HCD) and collision-induced dissociation (CID) were used for MS/MS scans.
MS2 Spectra Extraction
MS2 spectra of 93 synthetic peptides (supplemental Fig. S1) were produced using a modified version of X! Tandem (version Sledgehammer 2013.09.01.1) (24) along with the Trans-Proteomics Pipeline (25). The Lorikeet viewer was used to generate MS spectra figures (26). Both X! Tandem and the Lorikeet viewer were enhanced to allow customer-defined amino acid residues with stable isotope labeling. The search was conducted against an in-house-generated database including the 17 peptides in the peptide library (Table I), as well as 17 corresponding decoy sequences. The mass tolerance for precursor ions was set at 0.007 m/z. Masses of the heavy residues were supplied to X! Tandem, being defined by otherwise unused single-letter abbreviations as follows: B for heavy A, J for heavy G, O for heavy L, U for heavy P, and Z for heavy V. Propionylation (+56.0262 Da) was defined as a static modification on the N termini and on unmodified and mono-methylated lysine residues. Other PTMs were defined as dynamic modifications.
Peptide Quantification
A three-step quantification protocol was conducted. First, the mono-isotopic raw abundance of each peptide was manually extracted from the Xcalibur Qual Browser, based on the area underneath extracted ion chromatograms at the full MS level. All detectable charge states were summed together, typically MH+, MH2+, MH3+, and MH4+. For examples, please see Ref. 5. Second, for some co-eluting peptides, isotope masking was observed and taken into account. The histone H4 4–17, canonical H2A 4–11, and H2A.Z 1–19 peptide families have isobaric peptides that co-elute because they carry identical numbers of acetylation groups. In addition, two co-eluting peptides in the histone H3 27–40 family also have partial isotope overlap (peptides 41 and 43). To overcome the interference of neighboring ion isotope envelopes in quantifications, the raw abundance of peptides 43, 60, 61, 62, 64, 65, 66, 67, 68, 70, 71, 72, 81, 86, 87, 89, and 90 was corrected based on theoretical distributions of naturally occurring heavy isotopes using emass (27) (supplemental Table S1). For instance, histone H4 4–17 peptides that carry one acetylated lysine (peptides 59–62) co-elute and have sequential overlapping isotopic peaks in the mass spectrometer (supplemental Fig. S2). Taking the isotope interference between peptides 60 and 61 as an example, the MH+ of peptide 60 is 1545.897, and the third isotopic peak of this peptide is 1547.911, which overlaps with peptide 61 (MH+ = 1547.910) (Table I). The theoretical abundance of peptide 60's third isotopic peak is 33.7% of the first peak (27) (supplemental Table S1). Therefore, for each individual MS run, we subtracted 33.7% of peptide 60's raw amount from peptide 61's raw amount. Supplemental Fig. S2 demonstrates that isotope correction was necessary to eliminate the isotope interference effects. Third, in order to compare the detection efficiencies across all 93 species of peptides, we took into account multiple isotopes of each peptide. The higher a peptide's molecular weight is, the more naturally occurring heavy isotopes it may have. Because the first quantification step was based on only the monoisotopic peaks, the quantifications were biased toward smaller peptides. To correct for this bias, we used emass (27) to calculate each peptide's isotope distribution and recalculated each peptide's raw amount based on the isotope-correction factors presented in supplemental Table S1.
Statistics
All t tests were performed with two tails. For p values shown in Figs. 4 to 7B and supplemental Tables S5 to S15, pair-wise p values were calculated. For p values used to generate q values (see below) and those shown in Figs. 7C to 7H and supplemental Tables S16 and S17, unpaired p values were calculated.
Fig. 4.

Lysine acetylation on four histone peptides. Bar graphs showing relative abundance of each peptide within its own peptide family (same peptide backbone). Relative standard deviation values represent variation within each peptide family. Error bars represent standard error across 16 MS runs. The lines across each bar graph show the calculated solution amount of each peptide. p values were calculated between Unmod forms of each peptide and the modified forms. A, histone H3 54–63 peptide family with two peptides. B, canonical histone H2A 4–11 peptide family with four peptides. C, D, histone H2A.Z 1–19 peptide family with eight peptides. C, each individual peptide; D, averages of peptide abundance based on the number of acetylation groups. E, F, histone H4 4–17 peptide family with sixteen peptides. E, each individual peptide; F, averages of peptide abundance based on the number of acetylation groups.
Fig. 7.

Application of the synthetic peptide library in TS cells. Bar graphs showing relative abundance of histone H2A variants and H4 K20 methylations in undifferentiated (undiff) and differentiated (diff) TS cells. White boxes, original data; light gray boxes, internal corrected (Int_corr) data; dark gray boxes, external corrected (Ext_corr) data. The undiff TS cell data are in solid boxes, and the diff TS cell data are in hatched boxes. A, B, comparison of original data and corrected data, using undiff TS cell data. p values show difference between the original data and corrected data. C–E, different compositions of histone H2A variants between undiff and diff TS cells. p values represent difference between the two cell types. The numbers above each diff TS-cell bar show the fold change (either increase or decrease) of averaged data between the corresponding diff and undiff TS cells. F–H, histone H4 K20 methylation changes between undiff and diff TS cells. p values represent difference between the two cell types. The numbers above each diff TS-cell bar show the fold change (either increase or decrease) of averaged data between the corresponding diff and undiff TS cells.
To correct for multiple testing, we employed the q value method developed by Storey and colleagues (28, 29). First, p values were generated using the quantification of each peptide between sample preparations 1 and 2 (Fig. 2A, supplemental Table S2). Subsequently, using the R package qvalue, a list of q values were generated based on the 93 p values, with a false discovery rate of 1% (supplemental Table S2) (28, 29). The Smoother method was applied. Degrees of freedom were chosen as 9 based on the best fit of pi0 curves to the raw data (supplemental Fig. S3).
Fig. 2.

Reproducibility between two sample preparations and across three instrument setups. A, B, heat-maps showing 16 MS runs. The relative abundance of each of the 93 peptides in each individual MS run is shown in each column (blue: low abundance; red: high abundance). The sum of all 93 peptides was set to be 100%. Arrowheads show the solution percentage of all peptides (100%/93 = 1.08%). The injection amount is noted at the bottom of each heat-map. A, heat-map representing reproducibility between two sample preparations (prep). Nine runs from sample prep #1 and seven runs from sample prep #2 are shown. No significant differences between the two sample preparations were seen (false discovery rate = 0.01, pi0 = 0.850, all q values > 0.01; supplemental Fig. S3, supplemental Table S2) (28, 29). B, heat-map representing similarity among three MS instrument setups: Q-E (Q-Exactive coupled with EASY-nLC 1000), Elite (Orbitrap Elite couple with Eksigent nanoLC Ultra-2D plus), and Velos (Orbitrap Velos Pro coupled with EASY-nLC 1000). C, large detection efficiency variation across the 93 peptides. Bar graph showing relative peptide abundance averaged from all 16 runs. The error bars represent standard error. The red line shows the calculated solution amount of all peptides (1.08%). All peptides are listed in Table I.
In order to further assess the contribution of different experimental conditions to the technical variance, namely, the two sample preparations and the equipment, we performed a permutation test of unsupervised hierarchical clustering. Each iteration of hierarchical clustering was performed on a random subsample of 60 (out of 93) peptides. The result was then compared to results from the known clusters of samples from either the two sample preparations or the three different instruments (Q-E, Elite, and Velos). p values were generated from a 10,000-repetition run.
GRAVY Scores and Isoelectric Points
GRAVY (grand average of hydropathy) scores of 16 peptides were calculated using an online GRAVY calculator. The GRAVY score is widely used to measure the hydrophobicity of peptides and is calculated by summing the hydropathy values for each amino acid residue and dividing by the length of the sequence, as defined in Ref. 30.
Isoelectric points were calculated using an online tool, “computer PI/Mw” from ExPASy Bioinfomatics Resource Portal (31).
Tissue Culture and Histone Processing
Trophoblast stem (TS) cells were kindly provided by Dr. Marisa S. Bartolomei (University of Pennsylvania, Perelman School of Medicine). TS cells were cultured and maintained as previously described (32). In brief, TS cells were plated onto feeder mouse embryonic fibroblast cells (inactivated by mitomycin C) supplemented with 0.1% 25 μg/ml FGF4 (Product No. 235-F4, R&D Systems, Minneapolis, MN) and 0.1% 1.5 mg/ml heparin (Product No. H3393, Sigma) in TS cell media. The TS cell media was composed of 20% FBS, 1% penicillin (500 U/ml) and streptomycin (5 mg/ml), 1% 200 mm l-glutamine, 1% 100 mm sodium pyruvate and 100 μm β-mercaptoethanol in RPMI 1640 (pH 7.2). To collect mouse embryonic fibroblast–free TS cells, cells were trypsinized and re-plated for 45 min. The supernatant was transferred to a new plate for another 45 min. The new supernatant contained mostly TS cells and was used for protein analysis. To differentiate TS cells, TS cells were separated from mouse embryonic fibroblast cells and cultured in TS cell media without FGF4 and heparin for 5 to 6 days.
Histones were acid extracted from TS cells and processed with two runs of chemical derivatization, trypsin digestion, and desalting as previously described (5). For each MS run, 100 fmol of the synthetic peptide library was mixed with 0.4 to 1 μg of endogenous histone peptides. Four and three MS runs of undifferentiated and differentiated TS cell histones, respectively, were conducted in Q-Exactive, using parameters shown in Table II. Endogenous peptide quantifications were done in the same manner as for the synthetic peptides, except for isobaric species. As previously illustrated in Ref. 5, we targeted the m/z for the isobaric peptides and quantified the relative abundance of their unique b or y ions at the MS/MS level. Subsequently we determined the relative abundance at the MS1 level based on the ratios we obtained.
Endogenous Histone Peptide Quantification and Correction
Endogenous histone peptides were quantified using three strategies: original data, internal-correction using spiked-in heavy peptide standards, and external correction using detection efficiencies generated from the 16 independent runs conducted earlier in the study. For each MS run (see above), the endogenous histone peptides (from TS cells) and the injected synthetic peptides were manually quantified based on extracted ion chromatograms. Using the same quantification strategies described above, we quantified using the monoisotopic peak for each peptide mass and across multiple charge states and corrected for underrepresentation of large peptides (supplemental Tables S1 and S3). We define the raw abundance of each peptide's monoisotopic peak as R and the isotope correction factors as ICF. For both endogenous (E) and synthetic peptides (Si for internal controls), the abundance of each peptide was achieved by dividing R by ICF. For external controls (Se), we used the relative abundance values from Table I, which were calculated using data from the 16 MS runs presented in Fig. 2.
For a given peptide family or histone variant group, a total number of N peptides was included in the quantification. The total abundance of each peptide family was set as 100%, and the relative abundance of each peptide (j) was normalized to the total abundance. The formulas used to calculate the three strategies are presented below.
First, for the original data, the relative abundance of each peptide is calculated as
 |
Second, for internal corrections, the following formula was used:
 |
Third, for external corrections, the following formula was used:
 |
RESULTS
Peptide Library Design
To facilitate the expanding needs of histone PTM analyses in biomedical studies and to provide more quantitative histone PTM characterization, we designed a peptide library standard utilizing the Protein-Aqua™ technology by Cell Signaling Technology® (Table I). The guiding principles in the design of the library were to (i) represent the most biologically important and commonly seen histone PTMs, (ii) have both unmodified and modified forms of the same peptide, (iii) make PTMs distinguishable from the endogenous peptides at the MS1 level, and (iv) make PTMs distinguishable from each other when they have the same peptide backbone. This study focused on histone proteins that generate suitable peptides for bottom-up MS after our chemical derivatization protocol, namely, histone H3, H4, and H2A variants. We included the following modifications in the library: lysine mono-, di-, and tri-methylation (me1, me2, and me3); lysine acetylation (ac); arginine me1; threonine phosphorylation (ph); serine ph; and N-terminal acetylation (Nac). 93 synthetic peptides were designed, including 55 peptides representing different forms of eight histone H3 peptides, 23 peptides representing four histone H4 peptides, 5 peptides representing two canonical histone H2A peptides, 8 peptides representing one histone H2A.Z peptide, and one peptide each for histone macroH2A and H2A.X. Each peptide had one to three C13- and N15-labeled amino acid residues (G, A, P, V, and L) (Table I), making these peptides at least 4 Da heavier than the endogenous peptides. For the same peptide family (peptides that have the same backbone but different PTMs), different combinations of heavy residues were used for each individual peptide.
An example of the peptide design is given in Fig. 1. The histone H4 4–17 peptide contains four lysine residues that may be acetylated. Multiple acetylation has been observed in many different organisms in vivo and is strongly correlated with active transcription (33, 34). Fig. 1A shows endogenous histone H4 4–17 peptides from mouse TS cells: unmodified peptide (Unmod), one-ac, two-ac, three-ac, and four-ac. These peptides were extracted and processed based on our standard bottom-up MS protocol (5). Acetylation on lysine residues blocks the chemical derivatization with propionic anhydride; therefore the more acetylations a peptide has, the lighter and more hydrophilic it is (5). Taking the one-ac population as an example, it contains four different forms: acetylation on K5, K8, K12, or K16. These isobaric peptides have very close retention times in RP-HPLC and can be distinguished only by their MS2 spectra. Fig. 1B shows 16 synthetic peptides of the histone H4 4–17 family, all having distinct m/z values due to different designs of heavy residues (Table I, peptides 58–73). The one-ac peptides are highlighted in different colors. Fig. 1C shows a mixture of endogenous and synthetic peptides. We can accurately differentiate and quantify all five forms of one-ac peptides (see below). To be noted, K16ac has a very small delay in elution relative to K5/K8/K12ac (Fig. 1B), and the endogenous one-ac peptide peak is wider than those of the individual synthetic peptides, possibly because of the co-eluting isoforms (Fig. 1C).
Fig. 1.

Effective separation between endogenous and synthetic histone peptides. Shown are MS1 chromatograms and corresponding averaged isotope distribution of histone H4 4–17 peptides from three runs: (A) endogenous histone peptides only, (B) synthetic peptides only, and (C) a mixture of endogenous and synthetic peptides. The (M+2H)2+ ions are shown. The one-ac population is highlighted with various colors: endogenous one-ac in maroon, synthetic K16ac in green, synthetic K12ac in navy, synthetic K8ac in olive, and synthetic K5ac in purple. A, the one-ac peak includes four populations (K5/K8/K12/K16 ac) that cannot be separated by standard RP-HPLC. B, the four one-ac synthetic peptides contain different C13 and N15 labeled residues (highlighted in red) and therefore are distinguishable from each other at the MS1 level. Because all of these peptides co-elute, some overlap of isotopes was observed (right panel). This can be easily corrected at the peptide quantification step (see “Experimental Procedures” and supplemental Table S1). C, approximately 400 ng of endogenous histone peptides were mixed with 100 fmol of synthetic peptide library. The MS1 chromatograms of five m/z representing the endogenous one-ac peptides and four heavy peptides are shown.
Reproducible Technical Replicates among Two Sample Preparations and Three Instruments
All 93 peptides were mixed at the same concentration and chemically derivatized with propionic anhydride following our standard bottom-up MS approach (5). To achieve an optimized peptide injection amount, we tested a range of injection amounts from 20 fmol to 100 fmol per peptide on the Q-Exactive. Results showed that 50-fmol and 100-fmol injections gave stable and similar quantifications, whereas runs with an injection amount of less than 50 fmol did not (supplemental Fig. S4). This was further confirmed by runs conducted on two other instruments using up to 150 fmol of sample (Fig. 2). A very similar optimal peptide standard injection amount was obtained in another recent study (35). Moreover, when we injected these peptides as internal controls with endogenous histone peptides, 100 fmol of the synthetic peptide mixture was approximately equivalent to our optimal endogenous histone concentration (0.4 to 1 μg) (Fig. 1C). Therefore, we decided to focus on this concentration range in our analyses.
To ensure the reproducibility of our experimental procedures, two batches of peptides were prepared and processed independently by two researchers (sample preparations 1 and 2). The library does not contain any overlapping monoisotopic m/z values among peptides that may co-elute; therefore we can easily extract the raw abundance of each peptide by measuring the peak area in the MS chromatogram. A total of 16 runs with injection amounts from 50 to 150 fmol were conducted on three different mass spectrometers (“Experimental Procedures” and Fig. 2). Figs. 2A and 2B show heat maps representing relative abundances of 93 peptides in each run and a comparison between sample preparations 1 and 2, as well as among the three instruments. We do not observe significant variation between the two sample preparations, measured by q values with a false discovery rate of 0.01 (Fig. 2A, supplemental Table S2, supplemental Fig. S3) (28, 29). Additionally, with a null hypothesis that all runs were the same between the two sample preparations, we performed a permutation test of unsupervised hierarchical clustering. Each iteration of hierarchical clustering was performed on a random subsample of 60 (out of 93) peptides. The result was then compared against those from the two known clusters (sample preparation 1 versus 2). We asked how likely it was for the data to cluster non-randomly into the two clusters over 10,000 repetitions. Proper clustering did not occur significantly, and we obtained a p value of 0.0843. This test further supports the result that no significant difference was generated by the two independent sample preparations (Fig. 2A). After combining data from both sample preparations, we also accessed the contribution of the instruments to the technical variance by means of unsupervised hierarchical clustering. For a strict test, we asked how likely it was for the data to cluster non-randomly into the three known clusters along the different instrument setups (Q-E, Elite, and Velos) over 10,000 repetitions. Proper clustering did not occur in most of the repetitions, and we got a p value of 0.9929. For a less strict test, we asked how often any two of the three groups clustered correctly, and we obtained a p value of 0.4943. These results indicate that technical replicates from different machines are not significantly different. This is illustrated by the heat maps in Fig. 2B.
Wide-spread Dynamic Differences in Detection Efficiencies
We observed a large dynamic range in relative abundance across 93 peptides in each individual MS run, even though all of the peptides were present at the same concentration. The average relative abundance ranged from 0.0018% to 3.20% (Fig. 2C, Table I), whereas the solution value should have been 1.08% (100%/93) for each peptide. The averaged value for the peptide with the highest intensity (peptide 30, Kme2SAPATGGVKKPHR, histone H3 27–40) was more than 1700-fold as high as the averaged value for the lowest peptide (5, TKme2QTAR, histone H3 3–8) (Table I). It is well known that variations in peptide length, primary sequence, and modifications may all contribute to differences in (i) peptide hydrophobicity, (ii) binding affinity with reverse phase resins, (iii) peptide basicity, and (iv) ionization efficiency at the liquid–gas interphase, thus resulting in the huge gap observed. In this study we aimed to describe the observed detection efficiencies of histone peptides and to provide quantitative normalization methods based on our quantifications.
The peptide length and primary sequence should be the major factors contributing to the detection efficiency of the 16 unmodified peptides in our library. Consistent with the general observations in the field, there was no clear correlation between peptide molecular weight and detection efficiency in the mass spectrometer for the size range of peptides in our study, as illustrated in Fig. 3A. Similarly, neither the GRAVY scores (30) (Fig. 3B) nor the isoelectric points (Fig. 3C) of these peptides correlated with the detection efficiency. To test for any influence of the chemical derivatization on the detection efficiencies, we also analyzed unpropionylated peptides. Because they are in general more hydrophilic than the propionylated peptides and are not retained well on C18 columns, we could reliably detect and quantify only 56 peptides (data not shown), 7 of which were unmodified peptides (28, 49, 50, 52, 78, 83, and 92). Consistent with the propionylated peptides, no correlation was observed between the peptides' detection efficiencies and their molecular weight, GRAVY scores, or isoelectric points (supplemental Fig. S5).
Fig. 3.
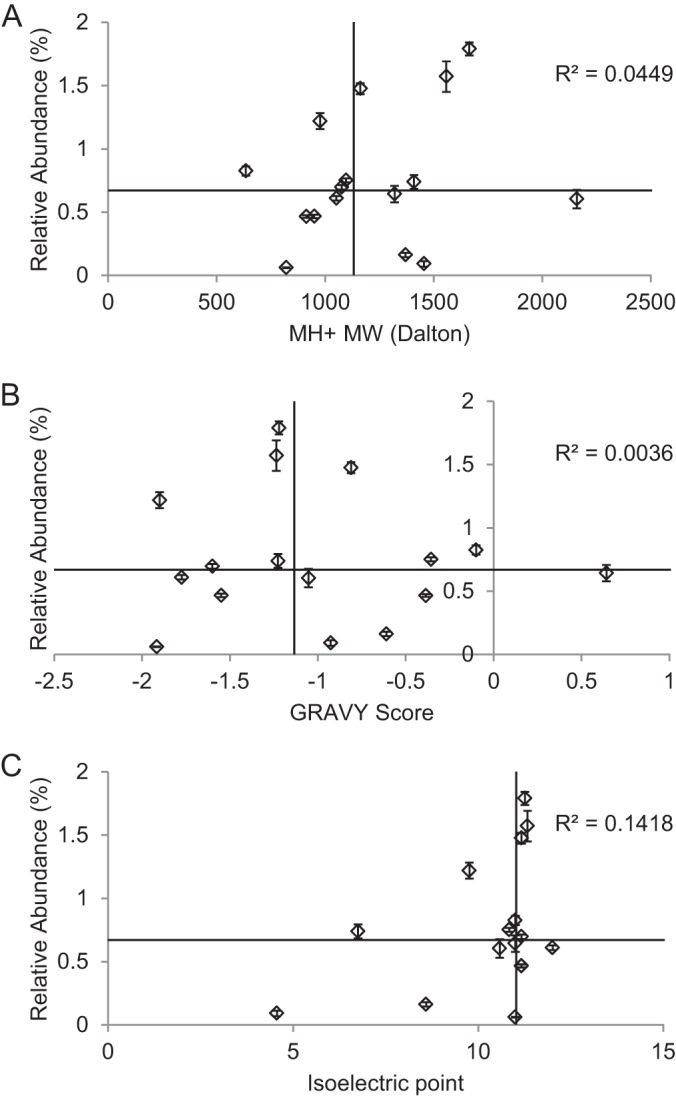
Primary peptide sequence affects the detection efficiency. Relative abundance values of 16 unmodified peptides from 16 MS runs were plotted against the (A) molecular weight (MW), (B) GRAVY scores (30), and (C) isoelectric points of the peptides. Error bars represent standard error. The horizontal and vertical lines represent median values of the relative abundance and MW/GRAVY/isoelectric point values of these peptides, respectively. R2 values generated from linear regression of each panel are also shown. No correlation was observed between the relative abundance and the three parameters measured.
Effects of Modifications on Detection Efficiency
Our library contains 87 peptides that carry at least one modification. To assess whether a particular modification affected the detection efficiency, we compared each modified peptide to its unmodified form (Unmod). The total amount of each peptide family was set at 100%, as shown in Figs. 4 to 6. We observed some trends in how different modifications may affect the detection efficiency. Surprisingly, the same modifications had very different effects when added on different peptides. To be noted, in order to provide relevant information for biological samples, we focused on derivatized synthetic peptides in this study. The propionyl groups were mainly added to primary amine groups, namely, the N termini of peptides, as well as lysine residues with no modifications or mono-methylation. Lysine residues that are acetylated or di- and tri-methylated are not propionylated.
Fig. 6.

Three histone peptide families have multiple combinatorial modifications. Bar graphs showing relative abundance of each peptide within its own peptide family. RSD values represent variation within each peptide family. Error bars represent standard error across 16 MS runs. The lines across each bar graph show the calculated solution amount of each peptide. p values were calculated between Unmod forms of each peptide and the modified forms. A, histone H3 18–26 peptide family with six peptides. B, histone H3 9–17 peptide family with 14 peptides. C, histone H3 27–40 peptide family with 21 peptides.
Lysine Acetylation
In our histone peptide library, four peptides (H3 54–63, canonical H2A 4–11, H2A.Z 1–19, and H4 4–17) carry mostly lysine acetylation when they are modified in vivo. Histone H3 54–63 peptide can be acetylated on K56, and this modification is related to chromatin assembly and DNA damage (36). As shown in Fig. 4A, 70.4% of the signal came from the acetylated form, which is a 2.4-fold increase relative to Unmod (p < 0.01). In contrast, when the canonical histone H2A 4–11 peptide was acetylated on either the K5 or the K9 position, the detection efficiency was significantly lower than for Unmod (4.7-fold and 2.7-fold decrease, respectively) (Fig. 4B, supplemental Table S5). The detection efficiency of double acetylation on K5 and K9 was further reduced, with an 11.2-fold decrease relative to Unmod (Fig. 4B). Intriguingly, K5ac and K9ac were also significantly different from each other (p < 0.01, supplemental Table S5), suggesting a positional effect of lysine acetylation.
Similarly, on histone H2A.Z 1–19 peptides, the position of the acetylations played an important role in determining the detection efficiency (Fig. 4C, supplemental Table S6). This peptide can be acetylated on K4, K7, and K11 in vivo (37, 38). For the mono-acetylated species, both K7ac and K11ac affected the detection efficiency negatively, showing a 1.6-fold and 1.1-fold decrease relative to Unmod, respectively. When two acetylations were present, K4acK11ac and K7acK11ac had opposite effects: a 1.4-fold increase and a 2.1-fold decrease, respectively. When all three lysines were acetylated, the detection efficiency was not different from that of Unmod (Fig. 4C, supplemental Table S6). These results suggest that multiple lysine acetylations on the same peptide may have antagonistic effects on detection efficiency. Supporting this idea, when we plotted the relative abundance against the number of acetylation events (Fig. 4D), only the averaged one-ac peptide was significantly lower than other species; Unmod, two-ac, and three-ac were not different from one or the other (supplemental Table S6).
As previously mentioned, the histone H4 4–17 peptide may have up to four acetylated lysine residues, and therefore a total of 16 isoforms have been observed in many biological models (Figs. 1 and 4E, supplemental Table S7). Our quantifications suggest that K8ac and K16ac had negative effects on detection efficiency, whereas K5ac and K12ac were not different from Unmod. Overall the peptides of the H4 4–17 family were more similar to each other (relative standard deviation (RSD) = 19.9%) than the other three families shown in Fig. 4 (RSD = 57.7%, 96.7%, and 30.0%). On this peptide, acetylations generally reduce detection efficiency (Fig. 4F). The averaged one-ac, two-ac, three-ac, and four-ac peptides were all significantly lower than Unmod (1.1-, 1.3-, 1.2-, and 1.3-fold, respectively). This is consistent with a previous observation in a study by Kelleher and colleagues using a middle-down approach (39).
Lysine and Arginine Methylation
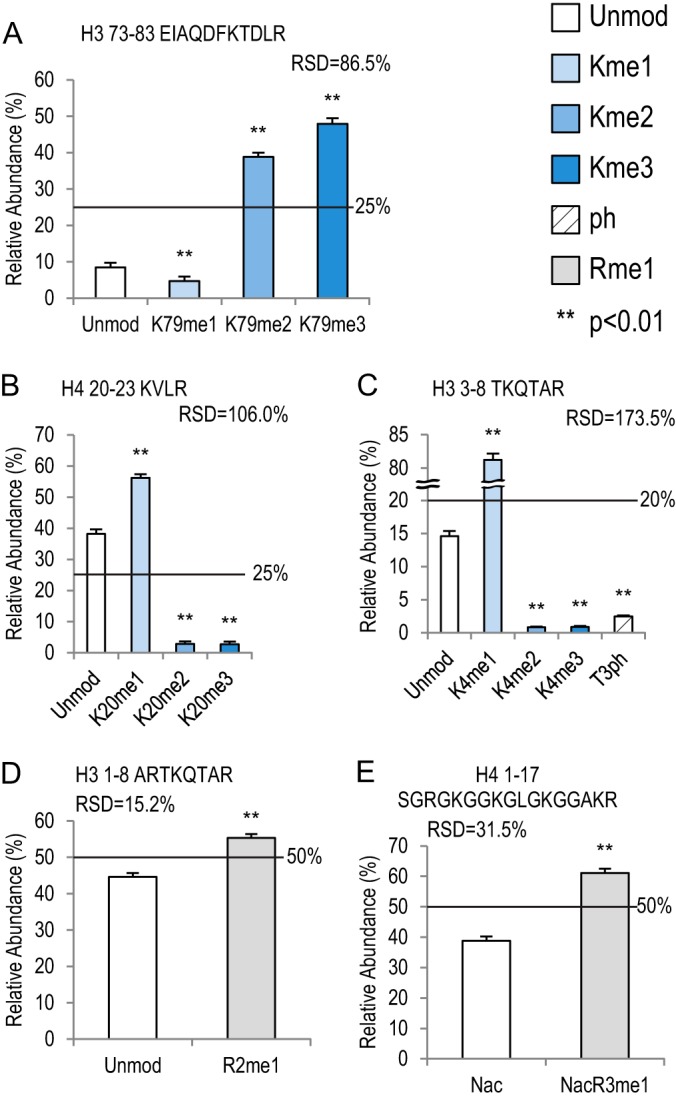
Both lysine and arginine residues can be methylated. Lysine has a primary amine that may be mono-, di-, or tri-methylated (me1, -2, and -3) (40), whereas arginine can only have me1 or me2 at the terminal amines (41). Among the three peptides that can have one methylatable lysine, two distinguishable patterns were observed (Fig. 5A versus Figs. 5B and 5C). The histone H3 73–83 peptide with K79me1 showed a 1.9-fold decreased relative to Unmod, whereas the levels of K79me2 and -me3 forms were much higher than in Unmod (4.6- and 5.6-fold increase, respectively) (Fig. 5A, supplemental Table S8). Conversely, on two other peptides, histones H4 20–23 and H3 3–8, me1 was higher than in Unmod, whereas me2 and me3 were both lower than in Unmod (Figs. 5B and 5C, supplemental Tables S9 and S10). Histone H4K20me1 showed a 1.5-fold increase relative to Unmod. K20me2 and K20me3 had 12.6- and 13.2-fold decreases, respectively. On H3 3–8, K4me1 was 5.6-fold higher than in Unmod, and K4me2 and K4me3 were 17.6- and 16.3-fold lower than in Unmod, respectively. In fact, the K4me2 and K4me3 peptides (peptides 5 and 6) also had the lowest detection efficiencies in the entire peptide library (Table I).
Fig. 5.
Lysine methylation and acetylation on five histone peptides. Bar graphs showing relative abundance of each peptide within its own peptide family. RSD values represent variation within each peptide family. Error bars represent standard error across 16 MS runs. The lines across each bar graph show the calculated solution amount of each peptide. p values were calculated between Unmod forms of each peptide and the modified forms. A, histone H3 73–83 peptide family with four peptides. B, histone H4 20–23 peptide family with four peptides. C, histone H3 3–8 peptide family with five peptides, including a phosphorylated form (T3ph). D, histone H3 1–8 peptide family with two peptides. E, histone H4 1–17 peptide family with two peptides, which are both acetylated on the N termini.
In contrast to the complicated effects of lysine acetylation and methylation, Rme1 seemed to positively affect the detection efficiencies, based on two peptides in our library. On the histone H3 1–8 peptide, R2me1 showed a 1.2-fold increase relative to Unmod (Fig. 5D) (p < 0.01), and R3me1 on histone H4 1–17 peptide showed a 1.6-fold increase (Fig. 5E) (p < 0.01).
Threonine and Serine Phosphorylation
As previously mentioned, phosphorylation (ph) on T, S, and Y residues usually has a negative effect on ionization efficiency, which may be due to (i) the suppressive effect that negative charges on phosphate groups have on ionization efficiency in positive ion mode or (ii) low retention of phosphorylated peptides in RP-HPLC resulting from their increased hydrophilicity (42). Therefore, the negative effects of phosphorylation should be more pronounced on shorter peptides. Three peptides in our peptide library consistently behaved as predicted. Relative to their unmodified counterparts, T3ph on histone H3 3–8, S10ph on H3 9–17, and S28ph on H3 27–40 exhibited 5.9-fold, 2.4-fold, and 1.1-fold decreases, respectively (Figs. 5C, 6B, and 6C; supplemental Tables S10, S12, and S13). To be noted, our chemical derivatization removes positive charges on unmodified lysine residues; therefore, the number of lysine residues on each peptide would not have as much of an impact on the overall charge of the peptides.
Peptides with Multiple Modifications
Three peptide families of the histone H3 N-terminal tail have multiple residues that may be modified differently on their own and in combination. We have included the most commonly seen PTMs on these peptides in our library.
The H3 18–26 peptide has two lysine residues that can be mono-methylated or acetylated in vivo (43). In our experiments this peptide exhibited the least variation (RSD = 13.1%) among its different isoforms (Fig. 6A, supplemental Table S11). K18me1 and K23ac had similar negative effects on detection efficiency (1.3-fold and 1.2-fold decreases, respectively). It seemed that me1 only had an effect when it was on the K18 position, whereas ac had an effect on the K23 position. This observation further supports the idea that the positions of the PTMs have a great effect on detection efficiency.
The H3 9–17 peptide and 27–40 peptides harbor some of the most important histone PTMs, including K9me2/3, K27me2/3, and K36me3, as well as acetylated K9, K14, and K27. K9me2/3 and K27me2/3 are tightly correlated with heterochromatin, genome architecture and stability, gene silencing, etc. K36me3, K9ac, K14ac, and K27ac are markers indicating active transcription. In addition, S10ph is a mitotic marker, and both S10ph and S28ph are involved in gene activation. For review, please see Refs. 40, 44, and 45.
As shown in Fig. 6B and supplemental Table S12, all acetylations on the H3 9–17 peptide reduced the signal intensity, similar to the canonical H2A 4–11 peptide (Fig. 4B). K9ac, K14ac, and K9acK14ac exhibited 1.8-, 1.5-, and 2.1-fold decreases relative to Unmod, respectively. K9acK14ac levels were also significantly lower than in the singly acetylated peptides (supplemental Table S12). The methylation profile of this peptide was similar to those of the H4 20–23 and H3 3–8 peptides in that K9me1 had the most abundant signal (21.9%, 2.3-fold increase from Unmod), whereas K9me2 and K9me3 were much lower (1.9- and 2.3-fold decreases). Similar patterns of K9 methylation persisted in combination with K14ac or S10ph (Fig. 6B, supplemental Table S12). All forms of K14ac and S10ph showed consistent negative effects on the detection efficiencies relative to their unmodified counterparts.
Unlike the acetylations on H3 9–17 peptide, which had reduced intensities, K27ac on histone H3 27–40 peptide was 1.2-fold higher than Unmod (Fig. 6C, supplemental Table S13). Interestingly, the methylation pattern on this peptide was different from those of all previously discussed peptides. All forms of K27 methylation by themselves were significantly higher than Unmod: me1, 1.1-fold; me2, 1.7-fold; and me3, 1.7-fold. The pattern of K27 methylation (K27me2/3 > K27me1) was largely maintained when S28 or K36 was modified, except that K27me3K36me3 was not different from K27me1K36me3 (supplemental Table S13). Moreover, K27me1 in combination with any other modifications on S28 and K36 reduced signal intensity (Fig. 6C). For methylation on K36, only K36me2 showed a decrease of 1.1-fold relative to Unmod. However, when K36 methylation was coupled with any form of K27 methylation, it was significantly lower than the unmodified K36 counterpart (Fig. 6C, supplemental Table S13). For instance, the K27me2K36me1 isoform was much lower than the K27me2 isoform. S28ph had similar effects. These data suggest that combinatorial PTMs on histone H3 27–40 peptide generally lower detection efficiency.
Application of the Synthetic Peptides in Histone and Histone PTM Quantification
Our original protocol (5) did not consider the variations in peptide detection efficiencies. As mentioned, this might have led to bias in the peptide quantification and data interpretation. With the synthetic peptide library available, we employed it to investigate histone variant and PTM changes during mouse TS cell differentiation. These cells were derived from the trophoectoderm layer of mouse embryos 3.5 days post-coitum. During mammalian early development, the trophoectoderm lineage develops to vital extraembryonic placenta tissues, and therefore is critical for embryonic development. TS cells are widely used as a trophoblast model in vitro (46). Similar to embryonic stem cells, these cells can be differentiated into multiple lineages that mimic extra-embryonic tissues (47). We mixed endogenous histone peptides with the synthetic peptide library and analyzed the histone variants and histone PTMs.
First, we quantified the histone H2A variants in undifferentiated TS cells. Three histone H2A peptide pairs (endogenous and synthetic) were used: peptide 83 (HLQLAIR) is shared by the canonical H2A and H2A.Z, peptide 93 (GKTGGKAR) comes from H2A.X, and peptide 92 (SAKAGVIFPVGR) comes from macroH2A. As shown in Fig. 7A, the original data were obtained by quantifying the endogenous peptides in four MS runs. 91.6% of the signal came from canonical H2A/H2A.Z. We also quantified the synthetic peptides in the same runs and used the detection efficiency to correct for the measurement of each individual run. The raw abundance of each endogenous peptide was adjusted based on the relative detection efficiency of its synthetic counterpart in the same MS run (“Experimental Procedures”). After the internal correction (Int_corr), averaged canonical H2A/H2A.Z amounts dropped to 62.8%, whereas H2A.X increased from 8.0% to 36.8%. MacroH2A was very low and remained relatively unchanged (0.37% versus 0.42%). To test whether we could obtain comparable corrections using the detection efficiencies obtained from independent runs, we performed external corrections (Ext_corr) using data from the 16 runs of just the synthetic peptides, which were previously presented (Fig. 2 and Table I). The raw abundance of each H2A variant was corrected based on the relative detection efficiencies in Table I. Subsequently, the relative abundance of each variant was calculated based on the sum of all corrected H2A variants (set at 100%). The results of Ext_corr were very similar to those of Int_corr: 66.5%, 33.1%, and 0.42% for canonical H2A/H2A.X, H2AX, and macroH2A, respectively (Fig. 7A, supplemental Table S14). The corrections enabled us to accurately measure and compare the histone H2A variants.
We measured histone PTMs using the same method. Taking histone H4 K20 methylation as an example, we quantified both endogenous and synthetic H4 20–23 peptides as shown in Fig. 7B. Before correction, K20me1 seemed to be the most abundant form (47.5%, Fig. 7B). After either internal or external corrections, K20me2 became the most abundant form, increasing from 24.6% to more than 70%, whereas the K20me1 level dropped to around 10%. This result is very similar to that of a previous study using top-down MS, which showed that more than 80% of histone H4s carry me2 on the K20 position in HeLa cells (48). Two peptides in the H4 20–23 peptide family, Kme2VLR and Kme3VLR (peptides 76 and 77), had very low detection efficiencies (Table I). Therefore, they would be more easily affected by technical noise and variations, which may contribute to some difference between internal and external corrections in this family (supplemental Table S15). However, both internal and external corrections correct the data in the same direction and are more similar to each other than the original data.
Next, we applied this method to compare histone variant and PTM levels in undifferentiated and differentiated TS cells. Three datasets were generated using the original method of quantification, internal correction factors from spiked-in heavy peptides (Int_corr), and external correction based on detection efficiencies obtained from the 16 independent runs (Ext_corr). For most peptides examined, all three datasets showed a similar trend of isoform/modification changes after TS cell differentiation. However, the relative ratios of isoforms/modifications changed after data correction. For example, all three datasets showed increased levels of histone H2A.X and H4 K20me2/me3 after TS cell differentiation (Figs. 7C–7H, supplemental Tables S16 and S17). However, the corrections changed the degree to which H2A.X and H4 K20me2/me3 increased after differentiation relative to the original method. The original data showed that the differentiated TS cells had a 5.2-fold increase in H2A.X and a 2.6-fold increase in H4K20me2 relative to the undifferentiated cells, whereas both internal and external data corrections brought the fold change to a lower level (H2A.X: 1.9- and 2.4-fold, respectively; H4K20me2: 1.2- and 1.1-fold, respectively). In one case examined, the corrected data led to an opposite biological conclusion: In the original data macroH2A showed a 1.4-fold increase when TS cells were differentiated (Fig. 7C). However, after the data correction, differentiated TS cells actually had less macroH2A than the undifferentiated TS cells (more than a 1.5-fold decrease) (Figs. 7D and 7E). We think this occurred because the amount of macroH2A peptide is generally very low in the cell (<1% in our data), and its relative abundance depended strongly on levels of other H2A peptides, because we normalized the measurements by the sum of three H2A variants.
In summary, these results demonstrated that our original protocol without correction was robust enough to identify the critical biological changes, especially when the target peptides were abundant. However, the uncorrected data might over- or underestimate some changes between two biological conditions. Therefore, utilizing the detection efficiencies described here would greatly improve the accuracy in quantification of histone variants and PTMs. Furthermore, for peptides that exist at very low abundances (e.g. macroH2A), the proper corrections were critical for accurate interpretations in biology.
DISCUSSION
To our knowledge, we have created the largest synthetic peptide library for histones, and here we have presented quantitative and systematic analyses. A handful of other studies have also employed synthetic histone peptides, but typically only for individual or limited histone variants/PTMs. For example, Darwanto et al. used two pairs of histone H2B peptides (unubiquitinated and ubiquitinated) to generate standard curves for multiple-reaction-monitoring analyses of H2B ubiquitination (11). Recently, Jaffe and colleagues reported the usage of a histone peptide collection including five synthetic histone H3 peptide families. These peptides were labeled with heavy arginine residues and were used to analyze the specificity of histone antibodies in chromatin-immunoprecipitation experiments, and also to quantify histone PTMs (14). However, they only included histone H3 peptides and did not evaluate systematic effects of the PTMs. Additionally, because they used the only heavy arginine residue (6C13, 4N15) in each tryptic peptide, the isobaric PTM peptides remained isobaric in mass (e.g. histone H3K9ac versus H3K14ac). This same group used these peptides in a second study, but only as an external guide for retention time windows for MS/MS targeting (49). In the present study, we extensively tested 93 histone peptides on three different LC/MS-MS instrument setups and obtained very similar results (Fig. 2B). This suggests that the detection efficiency variations are consistent under different conditions when following our protocols. The Protein-Aqua™ technology designed the peptides by substituting different numbers of heavy amino acid residues; therefore, all the originally isobaric peptides had different m/z values and could be easily separated at the MS1 level. Moreover, these peptides could also be measured in the presence of the endogenous peptides (Fig. 1C). This design enabled us to fully evaluate and quantify different forms of histone peptides.
We examined various aspects of the synthetic peptide data by manually extracting ion chromatograms for the MS analyses, and we also manually validated MS/MS results (examples are given in supplemental Fig. S1). For each peptide, all detectable charge states were quantified. Because longer peptides have more naturally occurring heavy isotopes, we found that the relative amounts of these peptides were underestimated when we measured only the first isotopic peak (e.g. C12). We also corrected for this factor using calculated isotope distributions (supplemental Tables S1 to S3). Overall, we are very confident with MS peak assignments and peptide quantifications.
As previously mentioned, many factors contribute to the final detection efficiencies of peptides, including peptide hydrophobicity, basicity and binding affinity with reverse phase resins. We observed no clear relationship between the peptide hydrophobicity or basicity and the detection efficiency (Fig. 3). We did, however, observe a significant loss of highly hydrophilic peptides, namely, peptides 5 and 6 (histone H3 3–8 with K4me2/3), in our analyses, as they had the lowest detection efficiencies in the entire library (Table I). Experiments (data not shown) demonstrated that this was mainly due to the poor affinity of C18 resin for these two small, very hydrophilic peptides. Usage of a different derivatization reagent significantly increased the peptide recovery after desalting with STAGE tips.2 Therefore, our synthetic peptide standards can be very beneficial for accurate quantification of histone H3 K4me2/3, which are arguably the most important histone marks on active gene promoters.
Among the 14 families of histone peptides, we observed very different relative variations, as measured by relative standard deviation (Figs. 4 to 6). The RSD values ranged from 13.1% to 173.5%. These results suggest that PTMs, and even the same PTM, have diverse effects on different peptides. Overall, lysine methylation seemed to have larger effects (both positive and negative) than lysine acetylation. Surprisingly, despite the general believe that serine/threonine phosphorylation has strong deleterious effects on ionization efficiencies, phosphorylation on histone H3T3, S10, or S28 alone was never the most negatively affecting PTM (Figs. 5C, 6B, and 6C) on each peptide. Naturally occurring phosphorylations in biological samples are typically very low in abundance and are quite dynamic. In contrast, lysine methylations and acetylations are more abundant and stable, especially on histones (2, 3, 50). Our results suggest that the difficulties of phosphorylation detection are probably due to the naturally low amount of this mark and possibly are not solely a result of technical limitations, of course based on our detection of these histone peptides examined here. Consistently, a recent study also showed that phosphorylated peptides sometimes also had higher detection efficiencies than their unmodified counterparts (20), thus complicating the view of the effect that these marks have on MS detection efficiency.
The propionyl groups introduced onto histone peptides during our chemical derivatization protocol can also be considered as another form of modification. To assess the impact of propionylation on detection efficiencies, we subjected the histone peptide library to our MS pipeline without the chemical derivatization. However, the unpropionylated peptides were retained so poorly that we could only reliably detect 56 out of 93 peptides (data not shown). Out of the 56, 7 peptides were unmodified (supplemental Fig. S5). We managed to analyze two complete families, the histone H3 54–63 and H3 73–83 peptide families (supplemental Fig. S6). On the histone H3 54–63 peptide, K56ac showed a more consistent increase in detection efficiency than the Unmod counterpart (supplemental Fig. S6A). In contrast, the unpropionylated forms on the histone H3 73–83 peptide (Unmod; K79me1, -2, and -3) were more similar to each other (RSD = 11.4%), whereas K79me2 and -me3 on the propionylated peptides were much higher than their Unmod and K79me1 counterparts (RSD = 86.5%) (Fig. 5 and supplemental Fig. S6B). We concluded that, similar to lysine acetylation and methylation, lysine propionylation has variable effects on detection efficiency when added onto different peptide backbones.
Synthetic heavy-isotope-labeled peptides are usually used as internal controls (absolute quantification), mixed with biological samples and analyzed together via nano-LC-MS/MS. Alternatively, they may also be analyzed separately from the endogenous samples, serving as external controls for detection efficiency (14, 51). We applied both calibration methods and found very similar results between external and internal controls spiked into histones extracted from mouse trophoblast stem and differentiated cells (Fig. 7). For the histone H2A variants, we observed no difference between Int_corr data and Ext_corr data (Fig. 7A, supplemental Table S14). For the histone H4 20–23 peptide family, there was some difference between the two sets of corrected data (Fig. 7B, supplemental Table S15). However, both corrections showed the same trend and were more similar to each other than the original data. Furthermore, the corrected data may change the interpretation of some biological results. In the example shown in Fig. 7, both Int_corr and Ext_corr data show a decrease in marcoH2A level after TS cell differentiation, different from the original data. In fact, the 16 runs presented here were conducted over a period of more than five months on three different instruments, suggesting that the detection efficiencies of these peptides remain stable over time. Therefore, the external corrections may still be used to control for the bias in the detection efficiencies of different peptides, which is something that all researchers in this epigenetics proteomics field should pay closer attention to.
As demonstrated above, the synthetic peptide library presented here can serve as a very useful tool in the quantification of histone PTMs and variants in biological samples. However, some challenges still remain in analyzing the endogenous histone samples. For example, the H4 4–17 peptide family has three isobaric groups within the family: the one-ac, two-ac, and three-ac peptides (Fig. 1). Although we routinely target their m/z (MH2+), it remains difficult to determine the relative amount of each form because some of them do not have unique b or y ions. The synthetic library does provide useful information about retention time differences of some isobaric peptides (Fig. 1C). However, for correct identification and quantification of these peptides, we still have to rely on optimization at the mass spectrometer to obtain full coverage in the MS/MS spectrum.
In conclusion, we have provided a systematic evaluation and quantification of the detection efficiencies of our 93-histone peptide library. We found that there were widespread detection efficiency biases that were both sequence and PTM specific, although no discernible trends could be broadly generalized across all the peptides. Nevertheless, we believe these data will be of great benefit in measuring important histone variants and PTMs and will promote more accurate use of mass-spectrometry-based technologies in the epigenetics field. Our data also serve as a cautionary warning to those wanting to measure histone PTM abundances, as correction factors (internal or external) must be taken into consideration before one makes strong claims describing histone PTM/variant stoichiometry. Furthermore, these data may serve as the first step toward a more comprehensive understanding of peptide behaviors in mass spectrometry, as they represent many different types of protein modifications. Lastly, we further hope this library can be used and mined to extrapolate new parameters or characteristics that could be used to explain the observations made herein and in future studies.
Supplementary Material
Acknowledgments
We thank Dr. Marisa Bartolomei (University of Pennsylvania, Perelman School of Medicine) for trophoblast stem cells, Dr. Xu Han (Temple University, Fox School of Business) for suggestions on statistical analyses, and Rosalynn C. Molden (the Garcia Lab) for helpful discussion.
Footnotes
Author contributions: S.L., J.R., J.R.L., D.A., and B.A.G. designed research; S.L., L.A., T.M., and B.A.G. performed research; G.L.O. and J.R. contributed new reagents or analytic tools; S.L., S.W., M.G., G.L.O., and Z.Y. analyzed data; S.L., T.M., S.L.B., and B.A.G. wrote the paper.
* B.A.G. gratefully acknowledges funding from the National Science Foundation (NSF) Early Faculty CAREER award and an NIH Innovator grant (DP2OD007447) from the Office of the Director.
 This article contains supplemental material.
This article contains supplemental material.
2 D. Arnott and T. Maile, in preparation.
1 The abbreviations used are:
- PTM
- post-translational modification
- ac
- acetylation
- GRAVY
- grand average of hydropathy
- me1
- mono-methylation
- me2
- di-methylation
- me3
- tri-methylation
- ph
- phosphorylation
- RP
- reverse phase
- RSD
- relative standard deviation
- TS cell
- trophoblast stem cell
- Unmod
- unmodified.
REFERENCES
- 1. Goldberg A. D., Allis C. D., Bernstein E. (2007) Epigenetics: a landscape takes shape. Cell 128, 635–638 [DOI] [PubMed] [Google Scholar]
- 2. Zee B. M., Levin R. S., Dimaggio P. A., Garcia B. A. (2010) Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Young N. L., Dimaggio P. A., Garcia B. A. (2010) The significance, development and progress of high-throughput combinatorial histone code analysis. Cell. Mol. Life Sci. 67, 3983–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garcia B. A., Mollah S., Ueberheide B. M., Busby S. A., Muratore T. L., Shabanowitz J., Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin S., Garcia B. A. (2012) Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mandava V., Fernandez J. P., Deng H., Janzen C. J., Hake S. B., Cross G. A. M. (2007) Histone modifications in Trypanosoma brucei. Mol. Biochem. Parasitol. 156, 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ouvry-Patat S. A., Schey K. L. (2007) Characterization of antimicrobial histone sequences and posttranslational modifications by mass spectrometry. J. Mass Spectrom. 42, 664–674 [DOI] [PubMed] [Google Scholar]
- 8. Robin P., Fritsch L., Philipot O., Svinarchuk F., Ait-Si-Ali S. (2007) Post-translational modifications of histones H3 and H4 associated with the histone methyltransferases Suv39h1 and G9a. Genome Biol. 8, R270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drogaris P., Wurtele H., Masumoto H., Verreault A., Thibault P. (2008) Comprehensive profiling of histone modifications using a label-free approach and its applications in determining structure-function relationships. Anal. Chem. 80, 6698–6707 [DOI] [PubMed] [Google Scholar]
- 10. Anderson D. C., Green G. R., Smith K., Selker E. U. (2010) Extensive and varied modifications in histone H2B of wild-type and histone deacetylase 1 mutant Neurospora crassa. Biochemistry 49, 5244–5257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darwanto A., Curtis M. P., Schrag M., Kirsch W., Liu P., Xu G., Neidigh J. W., Zhang K. (2010) A modified “cross-talk” between histone H2B Lys-120 ubiquitination and H3 Lys-79 methylation. J. Biol. Chem. 285, 21868–21876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilczek C., Chitta R., Woo E., Shabanowitz J., Chait B. T., Hunt D. F., Shechter D. (2011) Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 286, 42221–42231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuan W., Xu M., Huang C., Liu N., Chen S., Zhu B. (2011) H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286, 7983–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peach S. E., Rudomin E. L., Udeshi N. D., Carr S. A., Jaffe J. D. (2012) Quantitative assessment of chromatin immunoprecipitation grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol. Cell. Proteomics 11, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sidoli S., Cheng L., Jensen O. N. (2012) Proteomics in chromatin biology and epigenetics: elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteomics 75, 3419–3433 [DOI] [PubMed] [Google Scholar]
- 16. Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., Boeke J. D., Zhao Y. (2012) Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics 11, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng Y., Sweet S. M. M., Popovic R., Martinez-Garcia E., Tipton J. D., Thomas P. M., Licht J. D., Kelleher N. L. (2012) Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc. Natl. Acad. Sci. U.S.A. 109, 13549–13554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Lorenzo A., Bedford M. T. (2011) Histone arginine methylation. FEBS Lett. 585, 2024–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis P. W., Müller M. M., Koletsky M. S., Cordero F., Lin S., Banaszynski L. A., Garcia B. A., Muir T. W., Becher O. J., Allis C. D. (2013) Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marx H., Lemeer S., Schliep J. E., Matheron L., Mohammed S., Cox J., Mann M., Heck A. J. R., Kuster B. (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat. Biotechnol. 31, 557–564 [DOI] [PubMed] [Google Scholar]
- 21. Stemmann O., Zou H., Gerber S. A., Gygi S. P., Kirschner M. W. (2001) Dual inhibition of sister chromatid separation at metaphase. Cell 107, 715–726 [DOI] [PubMed] [Google Scholar]
- 22. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gerber S. A., Kettenbach A. N., Rush J., Gygi S. P. (2007) The absolute quantification strategy: application to phosphorylation profiling of human separase serine 1126. Methods Mol. Biol. 359, 71–86 [DOI] [PubMed] [Google Scholar]
- 24. Craig R., Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 25. Deutsch E. W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J. K., Martin D. B., Nesvizhskii A. I., Aebersold R. (2010) A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.lorikeet—A Javascript MS/MS Spectrum Viewer. https://code.google.com/p/lorikeet/ [Google Scholar]
- 27. Rockwood A. L., Haimi P. (2006) Efficient calculation of accurate masses of isotopic peaks. J. Am. Soc. Mass Spectrom. 17, 415–419 [DOI] [PubMed] [Google Scholar]
- 28. Hedenfalk I., Duggan D., Chen Y., Radmacher M., Bittner M., Simon R., Meltzer P., Gusterson B., Esteller M., Kallioniemi O. P., Wilfond B., Borg A., Trent J., Raffeld M., Yakhini Z., Ben-Dor A., Dougherty E., Kononen J., Bubendorf L., Fehrle W., Pittaluga S., Gruvberger S., Loman N., Johannsson O., Olsson H., Sauter G. (2001) Gene-expression profiles in hereditary breast cancer. N. Engl. J. Med. 344, 539–548 [DOI] [PubMed] [Google Scholar]
- 29. Storey J. D., Tibshirani R. (2003) Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 31. Artimo P., Jonnalagedda M., Arnold K., Baratin D., Csardi G., de Castro E., Duvaud S., Flegel V., Fortier A., Gasteiger E., Grosdidier A., Hernandez C., Ioannidis V., Kuznetsov D., Liechti R., Moretti S., Mostaguir K., Redaschi N., Rossier G., Xenarios I., Stockinger H. (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Golding M. C., Zhang L., Mann M. R. W. (2010) Multiple epigenetic modifiers induce aggressive viral extinction in extraembryonic endoderm stem cells. Cell Stem Cell 6, 457–467 [DOI] [PubMed] [Google Scholar]
- 33. Rothbart S. B., Lin S., Britton L.-M., Krajewski K., Keogh M.-C., Garcia B. A., Strahl B. D. (2012) Poly-acetylated chromatin signatures are preferred epitopes for site-specific histone H4 acetyl antibodies. Sci. Rep. 2, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zentner G. E., Henikoff S. (2013) Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266 [DOI] [PubMed] [Google Scholar]
- 35. Bohrer B. C., Li Y. F., Reilly J. P., Clemmer D. E., DiMarchi R. D., Radivojac P., Tang H., Arnold R. J. (2010) Combinatorial libraries of synthetic peptides as a model for shotgun proteomics. Anal. Chem. 82, 6559–6568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ozdemir A., Masumoto H., Fitzjohn P., Verreault A., Logie C. (2006) Histone H3 lysine 56 acetylation: a new twist in the chromosome cycle. Cell Cycle 5, 2602–2608 [DOI] [PubMed] [Google Scholar]
- 37. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., Jensen O. N. (2006) Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics 5, 1314–1325 [DOI] [PubMed] [Google Scholar]
- 38. Bonenfant D., Coulot M., Towbin H., Schindler P., van Oostrum J. (2006) Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 5, 541–552 [DOI] [PubMed] [Google Scholar]
- 39. Pesavento J. J., Mizzen C. A., Kelleher N. L. (2006) Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal. Chem. 78, 4271–4280 [DOI] [PubMed] [Google Scholar]
- 40. Greer E. L., Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bedford M. T. (2007) Arginine methylation at a glance. J. Cell Sci. 120, 4243–4246 [DOI] [PubMed] [Google Scholar]
- 42. Steen H., Jebanathirajah J. A., Rush J., Morrice N., Kirschner M. W. (2006) Phosphorylation analysis by mass spectrometry: myths, facts, and the consequences for qualitative and quantitative measurements. Mol. Cell. Proteomics 5, 172–181 [DOI] [PubMed] [Google Scholar]
- 43. Young N. L., DiMaggio P. A., Plazas-Mayorca M. D., Baliban R. C., Floudas C. A., Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8, 2266–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin C., Zhang Y. (2005) The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 [DOI] [PubMed] [Google Scholar]
- 45. Pérez-Cadahía B., Drobic B., Davie J. R. (2009) H3 phosphorylation: dual role in mitosis and interphase. Biochem. Cell Biol. Biochim. Biol. Cell 87, 695–709 [DOI] [PubMed] [Google Scholar]
- 46. Rossant J. (2008) Stem cells and early lineage development. Cell 132, 527–531 [DOI] [PubMed] [Google Scholar]
- 47. Simmons D. G., Cross J. C. (2005) Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev. Biol. 284, 12–24 [DOI] [PubMed] [Google Scholar]
- 48. Pesavento J. J., Yang H., Kelleher N. L., Mizzen C. A. (2008) Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 28, 468–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaffe J. D., Wang Y., Chan H. M., Zhang J., Huether R., Kryukov G. V., Bhang H.-E. C., Taylor J. E., Hu M., Englund N. P., Yan F., Wang Z., Robert McDonald E., 3rd, Wei L., Ma J., Easton J., Yu Z., Debeaumount R., Gibaja V., Venkatesan K., Schlegel R., Sellers W. R., Keen N., Liu J., Caponigro G., Barretina J., Cooke V. G., Mullighan C., Carr S. A., Downing J. R., Garraway L. A., Stegmeier F. (2013) Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 45, 1386–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Plazas-Mayorca M. D., Zee B. M., Young N. L., Fingerman I. M., LeRoy G., Briggs S. D., Garcia B. A. (2009) One-pot shotgun quantitative mass spectrometry characterization of histones. J. Proteome Res. 8, 5367–5374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao Y., Wang Y. (2007) A method to determine the ionization efficiency change of peptides caused by phosphorylation. J. Am. Soc. Mass Spectrom. 18, 1973–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.