

ABSTRACT
Microbial communities typically contain many rare taxa that make up the majority of the observed membership, yet the contribution of this microbial “rare biosphere” to community dynamics is unclear. Using 16S rRNA amplicon sequencing of 3,237 samples from 42 time series of microbial communities from nine different ecosystems (air; marine; lake; stream; adult human skin, tongue, and gut; infant gut; and brewery wastewater treatment), we introduce a new method to detect typically rare microbial taxa that occasionally become very abundant (conditionally rare taxa [CRT]) and then quantify their contributions to temporal shifts in community structure. We discovered that CRT made up 1.5 to 28% of the community membership, represented a broad diversity of bacterial and archaeal lineages, and explained large amounts of temporal community dissimilarity (i.e., up to 97% of Bray-Curtis dissimilarity). Most of the CRT were detected at multiple time points, though we also identified “one-hit wonder” CRT that were observed at only one time point. Using a case study from a temperate lake, we gained additional insights into the ecology of CRT by comparing routine community time series to large disturbance events. Our results reveal that many rare taxa contribute a greater amount to microbial community dynamics than is apparent from their low proportional abundances. This observation was true across a wide range of ecosystems, indicating that these rare taxa are essential for understanding community changes over time.
IMPORTANCE
Microbial communities and their processes are the foundations of ecosystems. The ecological roles of rare microorganisms are largely unknown, but it is thought that they contribute to community stability by acting as a reservoir that can rapidly respond to environmental changes. We investigated the occurrence of typically rare taxa that very occasionally become more prominent in their communities (“conditionally rare”). We quantified conditionally rare taxa in time series from a wide variety of ecosystems and discovered that not only were conditionally rare taxa present in all of the examples, but they also contributed disproportionately to temporal changes in diversity when they were most abundant. This result indicates an important and general role for rare microbial taxa within their communities.
INTRODUCTION
Microbial communities predominate Earth’s diverse ecosystems, contributing immense biomass and underpinning integral biogeochemical processes. They sustain the bases of food webs, provide key natural products that support human health and energy needs, and recycle carbon and nutrients that would otherwise stagnate. Despite the central role of microbial communities in biological systems, we are just beginning to understand the intricate interactions between their members and how these interactions contribute to ecosystem functions. Of particular interest is the role of rare microorganisms within a community, which make up the majority of the observed membership at any given time (1–5) (see Fig. S1 in the supplemental material). Determining whether these taxa remain rare or periodically bloom to abundance will change our understanding of each organism’s role in microbially mediated ecosystem functions and, importantly, in the stability of ecosystems in general.
Rare microbial community members encompass an immense diversity (the “rare biosphere”) (6–9). Still, the ecological roles of the vast majority of rare microorganisms remain unclear. Some rare microorganisms are likely on their way to local extinction (8) or are transient taxa that are “passing through” an environment (10–13). Some rare taxa may even be active, providing important functions that are disproportionate to their abundance or growth rate (14–16), and others may be dormant or inactive, awaiting favorable environmental conditions to grow (17, 18). An increase in the abundance of rare microorganisms that “wait” for favorable environmental conditions could be attributed to growth from low abundance, to awakening from dormancy, or to differential survival (i.e., escape from predation). Though there are a variety of ecological explanations for rare-to-prevalent dynamics, we still lack general empirical documentation of the phenomenon among microbial communities, and so their general incidence remains uncertain.
Because rare microbial taxa are difficult to observe, even less is known about their dynamics than is known about their ecological roles. A key unknown is how often rare taxa become abundant and hence play a potentially greater role in the ecology of a given system. However, there are a small but growing number of studies that have documented the dynamics of rare microbial taxa and provide some insights. For example, in the Arctic Ocean, rare microorganisms exhibited biogeography, indicating that some rare taxa, like more abundant taxa, have distributions based on their ecological requirements (19). In a sulfide-rich artesian spring, rare taxa exhibited patchiness over 1 mm (20), which also suggests that rare taxa can have clear distributions at fine spatial scales. Additionally, some coastal sand communities have rare members that do not often become abundant, suggesting that these members have a minimal influence on biogeochemical processes (21). Conversely, in other coastal sand communities, rare microbial taxa were shown to be as sensitive as prevalent taxa to environmental changes caused by an off-shore oil spill (22). The discrepancy between the latter two studies highlights our modest knowledge of the potential contributions of rare taxa and especially calls into question whether such conclusions are transferable to other ecosystems. Therefore, to understand the general importance of rare microbial taxa, their contributions to the larger community and their dynamics, we must systematically interrogate microbial communities from a variety of ecosystems by using consistent methods.
The availability of inexpensive, high-throughput sequencing technologies has led to an increased number of temporal studies of microbial communities (23). One of these studies identified a microbial taxon that bloomed to abundance from an apparently persistently rare state (24, 25). The dynamic of rarity to prevalence has also been observed in two other studies of marine bacterioplankton (14, 26). Here, we asked how the pattern of microbial rarity to prevalence is manifested in communities inhabiting very different ecosystems. We refer to microbial taxa that are typically in low abundance in one locality but occasionally become prevalent over time as “conditionally rare.”
Our objective was to understand the incidence of conditionally rare taxa (CRT) and their contribution to changes in microbial communities through time. We introduce a simple method for identifying CRT from temporal studies of diverse microbial communities and apply this method to a suite of time series data sets generated by using 454 pyrosequencing or Illumina sequencing of 16S rRNA gene fragments. Each data set contained a large percentage of very rare taxa, as typical for microbial communities (see Fig. S1 in the supplemental material). These data sets were previously analyzed by using a closed-reference operational taxonomic unit (OTU)-picking protocol (27) for direct comparison of their temporal patterns (see Table S1 in the supplemental material) (28). Because this OTU-picking protocol discards reads that do not match reference sequences at a minimum of 97% identity, it minimizes the rare OTUs arising from sequencing or PCR errors. The closed-reference protocol also avoids the “OTU splitting” that may occur when OTUs are defined by using a de novo protocol. We show that within many ecosystems, CRT contributed to temporal patterns of microbial diversity disproportionately to their relative abundances, suggesting an important role for CRT in structuring microbial communities over time. We also explicitly examine the influences of sampling frequency, study duration, and sequencing depth on the detection of CRT.
RESULTS
A simple method for detecting CRT.
Conditionally rare dynamics are exhibited when a taxon that is usually in low abundance or below the limit of detection occasionally blooms to an abundance appreciable at the community level. Thus, the frequency of a conditionally rare taxon’s abundance over time exhibits a bimodal distribution. The lower mode of the distribution is near zero at the time points when the taxon was rare or undetected, and the upper mode is centered at the taxon’s average abundance during a “bloom.” A statistical method for detecting a bimodal distribution is to compute the coefficient of bimodality, b (29). We used this coefficient to detect CRT. From the distribution of a taxon’s levels of abundance through time, the coefficient of bimodality, b, is calculated as follows:
where skewness is defined as follows:
and kurtosis is defined as follows:
The coefficient, b, ranges from 0 to 1, where 1 indicates the extreme case of the Bernoulli distribution (as in a binary data set; see Fig. S2A in the supplemental material). Thus, we identified bimodal taxon abundance distributions and then set a minimum relative abundance threshold of >0.01 and confirmed that we were able to identify a previously described conditionally rare Vibrio taxon in the western English Channel time series (24, 25) (see Fig. S2B). We also discovered two additional Vibrio taxa that exhibited similar but distinguishable dynamics in the western English Channel (see Fig. S2B) and confirmed that taxa with seasonal or irregular dynamics did not have a b value, >0.90 (see Fig. S2C). Thus, this method identified known and unknown CRT but excluded taxa that did not have rare-to-prevalent dynamics.
As each data set in this analysis had different sequencing efforts, sampling durations (numbers of days), and intensities (numbers of sampling events per unit of time), it was important to determine how these affected the recoverable enumeration of CRT. To address this, we used three of the most comprehensive data sets available in terms of sequencing effort, study duration, and sampling intensity. The first data set was a human-skin-associated community (male M3, right palm, 8,230 taxa) sampled approximately daily for 1 year and sequenced with Illumina technology (rarefied to 5,031 reads per sample). The second data set was a less rich temperate lake community (Trout Bog epilimnion, Wisconsin, 1,816 taxa) sampled periodically over 4 years with more intensive sampling during the ice-free season and sequenced with Illumina (rarefied to 5,134 reads per sample). The third data set comprised a marine surface water site in the western English Channel L4 (2,017 taxa) that was sampled approximately monthly for 6 years and sequenced by 454 pyrosequencing (rarefied to 3,526 reads per sample). We subsampled these time series along a range of sampling intensities and study durations and then calculated the percentages of CRT (see Fig. S3 in the supplemental material). From these analyses, it is clear that sampling intensity has a greater influence on the detection of CRT than the study duration does. Analysis of the impact of the number of samples included in a study revealed the same pattern across ecosystems (see Fig. S3), suggesting that sampling intensity is the most critical factor and should be taken into consideration when designing studies to explore CRT dynamics. Temporal sampling intensity will be an ecosystem-specific parameter that depends on the anticipated rate of community turnover or average life span of microorganisms in the system (30). See the supplemental material for additional considerations and recommendations for detecting CRT.
As expected, the time series with fewer sequence reads per sample had a higher percentage of CRT at a given sequencing depth (see Fig. S3E). This is because CRT made up a larger percentage of an inadequately sequenced community, which is an artifact of undersampling. The more undersampled the community, the larger the contribution of any taxon, including a conditionally rare taxon, will appear. Thus, unless a community is sequenced exhaustively and sampled at an intensity and duration appropriate for the community and range of environmental conditions in an ecosystem, the number of CRT detected will remain a conservative estimate.
CRT are ubiquitous and contribute disproportionately to community changes.
Acknowledging that detection of CRT will be a conservative estimate and will improve with increasing sampling intensity and duration appropriate to the expected community turnover in an ecosystem, we applied our method to the time series spanning nine distinct ecosystems, 42 microbial communities (consortia sampled at a given locality), and 3,237 individual observations. We found that each community included taxa that exhibited rare-to-prevalent dynamics. The incidence of CRT ranged from 1.5 to 28% of the total community membership (Fig. 1A) (b value, >0.90; relative abundance, >0.5%); however, it is important to note that when comparing CRT contributions to different ecosystems, these values should not be interpreted as absolute. To determine the contribution of CRT to the temporal dynamics of the community (temporal community dissimilarity), we calculated the fraction of Bray-Curtis similarity attributable to CRT (Fig. 1B and 2; see Materials and Methods), which ranged from 0 to 97% of the total community dissimilarity between time points (Fig. 2). This is because when CRT were abundant, their dynamics often explained a large fraction of the community dissimilarity. Interestingly, some ecosystems, such as the human gut (Fig. 2C), exhibited relatively more punctuated contributions of CRT to community dissimilarity over time, while other ecosystems, such as air (Fig. 2A), exhibited a more consistent contribution of CRT.
FIG 1 .
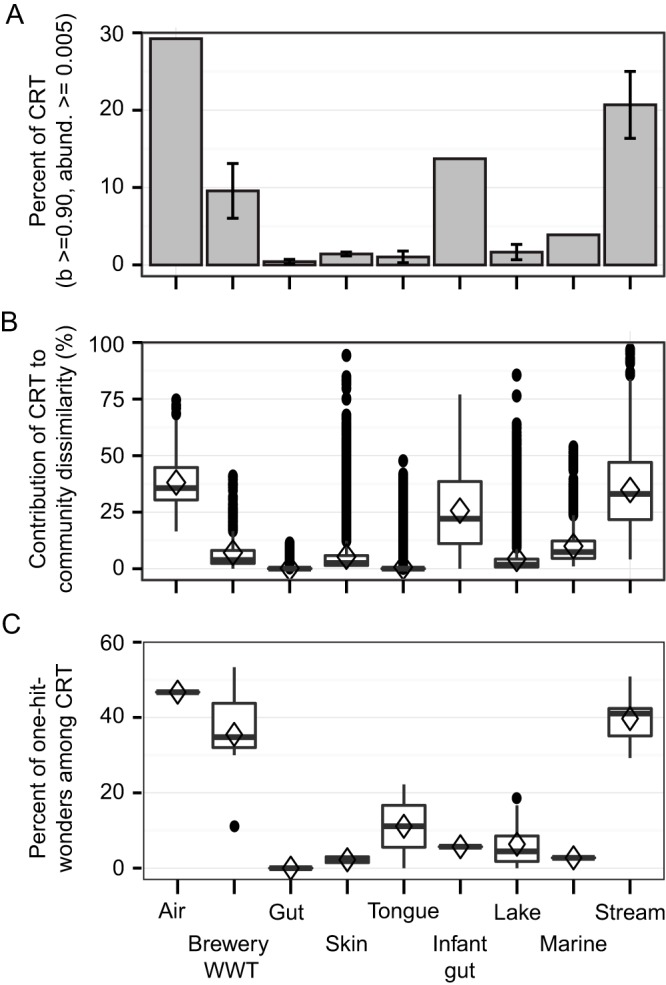
Incidences of CRT and their contributions to community dissimilarity. (A) Incidences of CRT across different ecosystems. Error bars are standard deviations of the means, but none are reported when n = 1 time series. (B) Fraction of temporal community dissimilarity attributed to the dynamics of CRT. Each open diamond is the mean of an ecosystem, whiskers are the lower and upper quartiles, and closed circles show outliers. b value, >0.90; relative abundance, >0.5%. (C) CRT observed only once in a time series, when blooming (one-hit wonders). WWT, wastewater treatment.
FIG 2 .
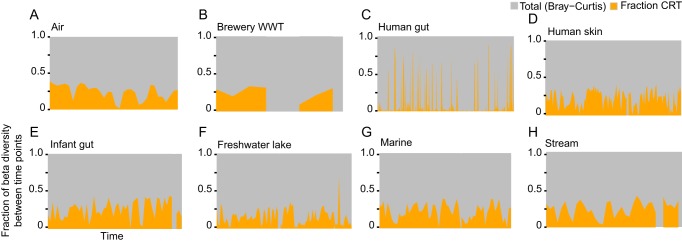
The fraction of consecutive Bray-Curtis dissimilarity attributed to the dynamics of CRT in representative communities. A, air time series, site Sp., 670 days; B, brewery wastewater treatment (WWT), site U4, 305 days; C, human gut, site M3 (male), 442 days; D, human skin, site F4 right palm (female), 185 days; E, infant gut, 834 days; F, freshwater bog lake, site TBE, 1,545 days; G, marine, western English Channel, site L4, 2,156 days; H, freshwater stream, site Orodell, 462 days.
CRT represented a broad range of phylogenetic diversity (see Fig. S4 in the supplemental material), with most environments being dominated by Proteobacteria, except for the infant and adult human guts, which were dominated by Firmicutes, and the human tongue, which had an equal contribution from Cyanobacteria (likely chloroplasts from food matter). There was no evidence that CRT consistently represented certain lineages when different ecosystems were compared (see Fig. S4 in the supplemental material). Additionally, within a community, there were similar lineages represented among CRT and the whole community membership (see SOM in Fig. S5).
Again, because of the differences in sampling and sequencing strategies across data sets (28), we encourage readers to consider the general trends in CRT rather than absolute values. However, despite these nuances, these data show not only that CRT are widespread members of microbial communities but also that CRT contribute to community level temporal changes disproportionately to their relative abundances.
Synchrony among CRT transitions.
To determine whether multiple CRT were synchronized in their transitions from rarity to prevalence, we performed hierarchical cluster analyses. Within each community, we found discrete clusters of CRT that shared the same occurrence patterns over time, as well as some CRT that had occurrence patterns that were independent and did not occur with other CRT (Fig. 3). These results suggest shared environmental drivers or shared sources of dispersal for synchronous CRT (see SOM in Fig. S6 in the supplemental material).
FIG 3 .

CRT clustered by shared occurrence patterns in representative time series. Each taxon was most abundant at the time point colored black. b value, >0.90; relative abundance, >0.5%. WWT, wastewater treatment.
One-hit wonders: can we attribute CRT to large dispersal events?
One mechanism of CRT dynamics could be the immigration and temporary bloom of a foreign taxon. In our data sets, this would be indicated by a taxon that was below the limit of detection, achieved abundance at one time point, and subsequently returned to undetectability; we refer to taxa exhibiting this dynamic as “one-hit wonders.” We wanted to understand how many CRT could be designated one-hit wonders, which would allow us to refine hypotheses about the potential for immigration events to affect community dynamics. We found that while the majority of CRT were detected at multiple time points, a subset of CRT were detected only when they bloomed, possibly because of immigration followed by a bloom and a crash (one-hit wonders: median, 9% of detected CRT; minimum, 0%; maximum, 53%, Fig. 1C). Generally, those communities that had relatively higher levels of temporal variability (i.e., air and stream communities) had more one-hit wonders than communities that were more stable (i.e., lake hypolimnia) (28). An exception to this were the brewery wastewater treatment communities, which were relatively stable but had high percentages of one-hit wonders; however, this time series also had a low sampling intensity, which could contribute to an increase in CRT as discussed above. Another scenario is that a one-hit wonder was always present but below the limit of detection. Because the percentage of one-hit wonders was moderately correlated to the sampling intensity (Pearson’s correlation coefficient, −0.51; P = 0.0005), longer or more intensely sampled time series may reveal multiple occurrences of a conditionally rare taxon that was originally designated a one-hit wonder.
Unraveling CRT ecology by comparing time series to disturbance events.
We propose two classifications of CRT: those that contribute to community dynamics given routine environmental changes (e.g., seasonal changes) (31) and those that contribute after a drastic disturbance. We distinguished these two classifications of CRT in a temperate lake microbial community that was the object of a whole-ecosystem disturbance experiment (32). The community was observed over the ice-free seasons in 2007, 2008, and 2009, and the disturbance experiment was conducted in July 2008. Using the temporal study as a baseline to understand routine dynamics, we could determine CRT that were important for a community response to the disturbance, helping to understand ecological drivers of CRT.
In this study, the epilimnion and hypolimnion thermal layers of a small bog lake (North Sparkling Bog, Wisconsin) were forced to mix at peak summer stratification (July 2008) with two large membranes that oscillated in the water column over the deepest point of the lake for 8 days until thermal homogeneity was achieved. The epilimnion was warm and oxygenated and had high light penetration, while the hypolimnion was cold and anaerobic and had low light penetration. Usually, thermal stratification weakens every spring and autumn as cool air causes the epilimnion temperature to decrease and meet the hypolimnion temperature, initiating seasonal mixing. Previous work showed that the microbial community structure and chemistry recovered to their predisturbance state within 20 days after the forced mixing in summer and that the hypolimnion community was more sensitive to mixing than the epilimnion (32). Therefore, we focused on the response of hypolimnion CRT to the forced mixing in summer.
A total of 24 CRT (b value, >0.90; relative abundance, >0.005; see SOM in the supplemental material) were detected in the hypolimnion of North Sparkling Bog between 2007 and 2009. Changes in the abundance of these 24 CRT could be described by four distinct patterns of increased relative abundance: (i) responding to natural and forced mixing events (Fig. 4A), (ii) responding only to the forced mixing event (Fig. 4B), (iii) responding only to the natural mixing events (Fig. 4C), and (iv) at times that were not defined by any type of mixing (Fig. 4D). The first group of CRT were probably driven by key environmental conditions associated with the phenomenon of mixing (i.e., oxygenation of the hypolimnion, redistribution of nutrients in the water column) and were unaffected by seasonal differences. This dynamic suggests that these CRT were rare but always present. The second group of CRT likely gained a competitive advantage given the novel environmental conditions caused by the forced mixing in summer. For example, OTU 333636, a member of the deltaproteobacterial family Haliangiaceae (Fig. 4B), bloomed after anaerobic conditions had been reestablished in the hypolimnion immediately following the forced mixing event but while the temperature remained elevated above typical seasonal averages (31), suggesting that this CRT thrives in warm and anaerobic waters, which would never have occurred during natural mixing events. The third group of CRT likely had seasonal constraints, and the fourth group of CRT may use multiple strategies to adapt to an increasing biomass, or similar but unmeasured environmental niches were established periodically. Included in the fourth group was a one-hit wonder that did not increase during the forced mixing in summer.
FIG 4 .
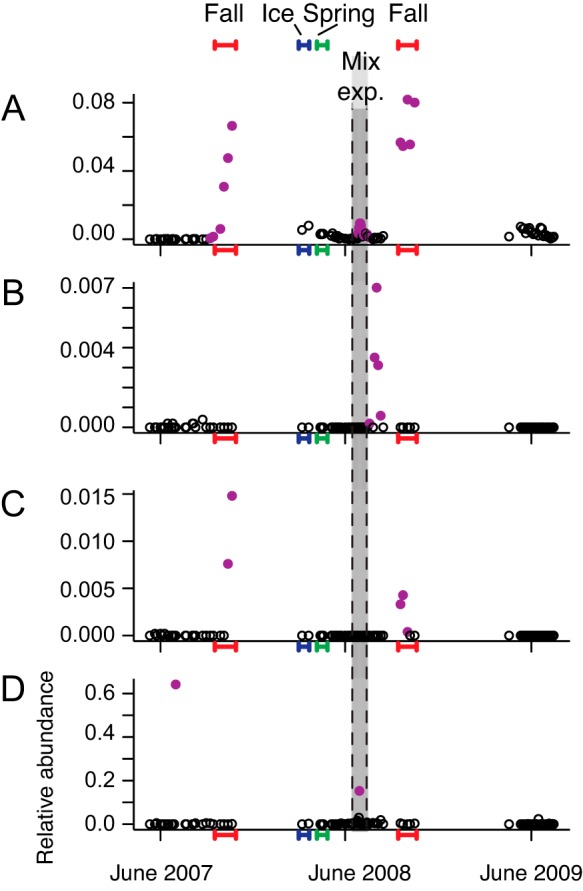
Representative dynamics of CRT from the North Sparkling Bog hypolimnion, observed over three ice-free seasons (2007 to 2009) that included a whole-ecosystem mixing experiment in July 2008 (shaded in gray, between dashed lines). Bloom events are purple points. Samples collected during fall mixing are underlined in red on the x axis, those collected during spring mixing are underlined in green, and those collected under ice are underlined in blue. A, Sphingobacteriales OTU 70346; B, Haliangiaceae OTU 333636; C, Flavobacterium OTU 426108; D, Moraxellaceae OTU 584176. Note the differences in y axis ranges.
DISCUSSION
Our results show that CRT can influence changes in microbial community structure. CRT contributed from 0 to 97% of the variability in the observed temporal community dissimilarity. Though it may seem obvious that CRT would contribute the most to temporal community dissimilarity during their transitions, it was unexpected that they would contribute so disproportionately (i.e., up to 97%) compared with their relative abundance during a “bloom” (mean relative abundance during a bloom, 2.7%; median, 1%). Our previous analysis suggested that the longer a community is observed, the more the perceived magnitude of the changes in community structure is reduced, suggesting very low rates of community change over long-term observations (28). Together, these results indicate that many baseline temporal changes in bacterial and archaeal diversity may be attributed to changes in the relative contributions of taxa that already exist within the community, including CRT transitions.
We provide a simple tool for identifying CRT and suggest that, on the whole, CRT comprise taxa that are always present and that it is less common for these taxa to be introduced by a dispersal event. However, while our strategy identifies taxa that can be targeted for further analysis, it does not explicitly reveal the ecological mechanisms of CRT within a community. These mechanisms are diverse and numerous, and determining the ecological properties of individual taxa is difficult and costly (33–35). However, we provide one example in which we capitalized on a temporal lake study to deduce CRT ecology by contrasting routine dynamics with a disturbance. In doing so, we were able to distinguish CRT that responded to both natural and forced mixing events from those that responded only to a forced event. These methods provide a springboard for hypothesis generation and are useful for understanding the contributions of CRT to different types of ecological dynamics. For example, in the context of human microbial consortia, similar analyses may be done in instances of pathogen invasion or pathobiont formation to understand when, how, and under what environmental conditions a typically rare or invasive member of the human microbiome is able to thrive following such a disturbance.
Though we cannot prove that one-hit-wonder CRT are not artifacts due to PCR (36) or sampling anomalies (37), the fact that the majority of CRT were observed multiple times within a series suggests that this scenario is not common and asserts that CRT would remain important contributors to community dynamics despite occasional misidentification due to artifacts. In reality, one-hit-wonder CRT likely comprise a combination of newly dispersed taxa that fail to thrive long term, rare but persistent taxa that fall below the level of detection when not blooming, CRT that were not observed long enough to detect subsequent blooms (insufficient time series), and artifacts.
There have been two distinct approaches to considering the rare biosphere in microbial ecology: (i) deep sequencing to detect as many rare members as possible (6) and (ii) omission of the entire rare “tail” to clarify overarching community patterns, whether arbitrary (e.g., 50 or fewer sequences) or methodological (e.g., after determining the abundance cutoff at which rare taxa do not contribute substantially to community dissimilarity) (38). Although the ecological roles of many rare taxa are unknown, it has been suggested that rare taxa are not necessarily important for the comparison and interpretation of microbial community patterns (10, 38). As more data from temporal studies of microbial communities are collected, it is likely that the dynamics of CRT will play an increasingly important role in our understanding of both the subtle temporal variability (39) and the disturbance responses of microbial communities. Furthermore, we know that some rare taxa play critical ecological roles in ecosystems, for example, diazotrophs in seawater (40), bacterial and archaeal ammonia oxidizers in soils (41, 42), and methanogens in guts (43). Thus, detection of CRTs will provide clues as to the identities of rare taxa that play previously unknown but equally critical ecological roles. Finally, studies that use unsaturated sequencing efforts to infer community assembly rules may attribute the appearance of new taxa to dispersal, when these taxa may instead already persist in the community in low abundance or in a dormant state (24). Therefore, close inspection of CRT dynamics in sufficiently sequenced communities will provide insights into the different roles of dispersal and blooms in community dynamics.
Given the ubiquity of CRT detected across an array of ecosystems and the large contribution of CRT to community dissimilarity, our results show that rare-to-prevalent dynamics are generally important and that these dynamics are especially critical for the community at the time points of CRT transitions. These data provide evidence that not all of the members of the microbial rare biosphere are always rare but that many contribute to the larger community at key time points. Furthermore, our analysis revealed synchronous dynamics of many CRT within a community and suggests that some CRT may be indicators of environmental changes that are unmeasured, providing clues about the identities of more subtle physical, chemical, or biological drivers of microbial dynamics. Finally, as transient members of the rare biosphere, CRT likely contribute to the high alpha diversity observed in many microbial communities.
MATERIALS AND METHODS
The microbial time series used in this study were previously published as separate studies (12, 25, 44–47), except for the lake data set, which is available from the Earth Microbiome Project (http://www.earthmicrobiome.org) (48). The whole-lake manipulation, including physical and chemical lake conditions, was described previously (32). The descriptions, quality control, and normalization of these data sets also are detailed elsewhere (28). OTUs were defined at 97% sequence identity of the 16S rRNA gene. We chose to include these 42 time series because they had study durations of at least 60 days. Because microbial communities have different degrees of richness, relative abundances were used when comparing community members. The overarching patterns of CRT were robust when different thresholds were used for the coefficient of bimodality and maximum relative abundance (see SOM in Fig. S7 in the supplemental material).
The study duration was the total number of days spanning the time series collection. Sampling intensity was the average number of days between observations. The influences of study duration and sampling intensity on the detection of CRT were investigated by subsampling the human male M3 gut, freshwater lake Trout Bog epilimnion, and marine L4 western English Channel time series by a “moving-window” approach (49). This approach involves the partitioning of a time series into as many window subsets as possible given the number of observations and calculation of the number of CRT detected within each window. For example, a 250-time-point series would first be divided into one 250-point window, two 249-point windows, three 248-point windows, etc. Subsampling of a data set to fewer sequences per sample (rarefaction) was performed by using the multiple_rarefactions.py script in QIIME v. 1.6.0 (50). We also rarefied the observed taxa classified as CRT by generating replicated, subsampled data sets at systematically varied sampling effort (i.e., number of samples). The percentage of CRT was calculated for each subsampled data set as described above. To extrapolate rarefaction curves to a standard sample size, the three parameters of the function
were estimated by maximum likelihood using custom scripts in R.
The R environment for statistical computing v 2.15.2 was used for all other analyses (51). Hierarchical clustering of CRT (to determine synchronous responses) was performed as described previously (10), by using dynamics of CRT standardized for each time series and k-means clustering of common occurrence patterns. To assess whether the subset of CRT represented a composition or structure different from that of the whole community, we used Pearson’s product-moment correlation. Some plots were made in R with the ggplot2 package (52). We calculated Bray-Curtis dissimilarity as a metric of community dissimilarity as follows:
where BC is the Bray-Curtis dissimilarity between communities j and k and X is the relative abundance of taxon i. For each time series, we calculated the Bray-Curtis similarity of all of the samples and then calculated the dissimilarity attributed to the taxa that were identified as conditionally rare (b value, ≥0.90; relative abundance, ≥0.05%). Because the Bray-Curtis dissimilarity is a scaled summation of abundance differences between two communities, we can easily partition Bray-Curtis dissimilarity between two samples attributable to a subset of the community. To do so, we use only CRT when calculating the summation in the numerator of the Bray-Curtis dissimilarity expression but use all of the taxa when calculating the scaling summation in the denominator. In this way, the Bray-Curtis dissimilarity of CRT and non-CRT will sum to the Bray-Curtis dissimilarity of the whole sample. We then divided the Bray-Curtis dissimilarity of CRT by the total community Bray-Curtis dissimilarity to report the fraction of beta diversity attributed to CRT. R scripts for calculation of CRT are freely available on GitHub (53).
SUPPLEMENTAL MATERIAL
Supplemental results and discussion to accompany the text. Download
Taxa ranked by abundance in a representative community from each ecosystem examined in this study. Colors show the relative abundance of the taxon observed over the time series. Similar to many other environmental microbial communities, these communities have a large percentage of low-abundance taxa. Download
Use of the coefficient of bimodality (b) to detect CRT in microbial communities. (A) b values for uniform, Bernoulli, and trimodal distributions. (B) Dynamics of three conditionally rare Vibrio taxa (OTU defined at 97% sequence identity) detected in the western English Channel with a b value of >0.95 and a maximum relative (Max rel.) abundance of ≥0.01. (C) Examples of taxa that did not fit the conditionally rare criteria. Panels B and C include the time series of the taxon’s relative abundance (left) and the distribution of the taxon’s levels of abundance though time (right). Download
Influence of time series duration and sampling intensity on the detection of CRT. In panels A and C, the color gradient shows the percentages (of the total number of community members) of CRT detected. (A) The marine western English Channel (L4 site). (B) A freshwater lake, Trout Bog epilimnion, in northern Wisconsin. (C) A human male right palm skin community. The patchy nature of this distribution is attributed to the frequent disturbances associated with the habitat (e.g., hand washing that removed taxa and contact with a variety of objects that added taxa). (D) Influence of the number of samples, a proxy for sampling intensity, on the CRT detected. (E) Influence of sequencing depth on the CRT detected. b value, >0.90; relative abundance, >0.5%. Note the differences in y axis ranges. Download
Taxonomic distribution of CRT among microbial communities, summarized by ecosystem. Note the differences in x axis ranges. WWT is brewery wastewater treatment. b value, >0.90; relative abundance, >0.5%. Download
Summary of significant (P < 0.05, blue) and not significant (NS, pink) two-sided Pearson’s correlation tests to compare the composition of each whole community and that of its subset of CRT. Community composition was summarized at the phylum (circle), class (triangle), and order (square) levels. Download
CRT clustered by shared occurrence patterns from the western English Channel time series. Each taxon was most abundant at the time point colored black. b value, >0.90; relative abundance, >0.5%. Taxonomic assignments are provided. Some synchronous CRT were from closely related phylogenetic lineages (i.e., blue-highlighted example of cooccurring Pseudoalteromonas taxa), while other synchronous CRT were more diverse (i.e., yellow-highlighted example of Vibrio taxa cooccurring with Tatumella and Pseudoalteromonas taxa), suggesting that patterns of CRT cooccurrences are complex and could be either redundant or modular. Download
Incidence of CRT in each ecosystem with different coefficient-of-bimodality (b) and maximum-abundance (ra) thresholds and the inclusion or omission of singletons. Few CRT were detected with a maximum-abundance threshold of 0.05 (5%), indicating that this cutoff is uninformative because it is too high. This analysis suggests that an abundance threshold between 0.005 and 0.01 is consistent within a data set and bimodality cutoffs of ≥0.90 are consistent. Download
Description of the data sets used in this study.
ACKNOWLEDGMENTS
A Gordon and Betty Moore Foundation postdoctoral fellowship from the Life Sciences Research Foundation supported A.S. This work was supported in part by the U.S. Department of Energy under contract DE-AC02-06CH11357 and by the Howard Hughes Medical Institute.
We thank the anonymous reviewers for thoughtful comments on previous versions of this work.
Footnotes
Citation Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5(4):e01371-14. doi:10.1128/mBio.01371-14.
REFERENCES
- 1. Dunbar J, Barns SM, Ticknor LO, Kuske CR. 2002. Empirical and theoretical bacterial diversity in four Arizona soils. Appl. Environ. Microbiol. 68:3035–3045. 10.1128/AEM.68.6.3035-3045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ. 2001. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67:4399–4406. 10.1128/AEM.67.10.4399-4406.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reeder J, Knight R. 2010. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat. Methods 7:668–669. 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Curtis TP, Sloan WT. 2004. Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Curr. Opin. Microbiol. 7:221–226. 10.1016/j.mib.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 5. Pedrós-Alió C. 2007. Ecology. Dipping into the rare biosphere. Science 315:192–193. 10.1126/science.1135933. [DOI] [PubMed] [Google Scholar]
- 6. Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120. 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fuhrman JA. 2009. Microbial community structure and its functional implications. Nature 459:193–199. 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 8. Pedrós-Alió C. 2012. The rare bacterial biosphere. Ann. Rev. Mar. Sci. 4:449–466. 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- 9. Bent SJ, Forney LJ. 2008. The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J. 2:689–695. 10.1038/ismej.2008.44. [DOI] [PubMed] [Google Scholar]
- 10. Shade A, McManus PS, Handelsman J. 2013. Unexpected diversity during community succession in the apple flower microbiome. mBio 4(2):e00602-12. 10.1128/mBio.00602-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shade A, Read JS, Welkie DG, Kratz TK, Wu CH, McMahon KD. 2011. Resistance, resilience and recovery: aquatic bacterial dynamics after water column disturbance. Environ. Microbiol. 13:2752–2767. 10.1111/j.1462-2920.2011.02546.x. [DOI] [PubMed] [Google Scholar]
- 12. Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, Gordon JI, Knight R. 2011. Moving pictures of the human microbiome. Genome Biol. 12:R50. 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Gast CJ, Walker AW, Stressmann FA, Rogers GB, Scott P, Daniels TW, Carroll MP, Parkhill J, Bruce KD. 2011. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 5:780–791. 10.1038/ismej.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proc. Natl. Acad. Sci. U. S. A. 108:12776–12781. 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones SE, Lennon JT. 2010. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U. S. A. 107:5881–5886. 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shade A, Hogan CS, Klimowicz AK, Linske M, McManus PS, Handelsman J. 2012. Culturing captures members of the soil rare biosphere. Environ. Microbiol. 14:2247–2252. 10.1111/j.1462-2920.2012.02817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9:119–130. 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 18. Epstein SS. 2009. Microbial awakenings. Nature 457:1083. 10.1038/4571083a. [DOI] [PubMed] [Google Scholar]
- 19. Galand PE, Casamayor EO, Kirchman DL, Lovejoy C. 2009. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. U. S. A. 106:22427–22432. 10.1073/pnas.0908284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Youssef NH, Couger MB, Elshahed MS. 2010. Fine-scale bacterial beta diversity within a complex ecosystem (Zodletone Spring, OK, USA): the role of the rare biosphere. PLoS One 5(8):e12414. 10.1371/journal.pone.0012414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gobet A, Böer SI, Huse SM, van Beusekom JE, Quince C, Sogin ML, Boetius A, Ramette A. 2012. Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6:542–553. 10.1038/ismej.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newton RJ, Huse SM, Morrison HG, Peake CS, Sogin ML, McLellan SL. 2013. Shifts in the microbial community composition of Gulf Coast beaches following beach oiling. PLoS One 8(9):e74265. 10.1371/journal.pone.0074265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, Meyer F, Stevens R, Bailey M, Gordon J, Kowalchuk G, Gilbert JA. 2012. Unlocking the potential of metagenomics through replicated experimental design. Nat. Biotechnol. 30:513–520. 10.1038/nbt.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caporaso JG, Paszkiewicz K, Field D, Knight R, Gilbert JA. 2012. The western English Channel contains a persistent microbial seed bank. ISME J. 6:1089–1093. 10.1038/ismej.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B, Huse S, McHardy AC, Knight R, Joint I, Somerfield P, Fuhrman JA, Field D. 2012. Defining seasonal marine microbial community dynamics. ISME J. 6:298–308. 10.1038/ismej.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vergin KL, Done B, Carlson CA, Giovannoni SJ. 2013. Spatiotemporal distributions of rare bacterioplankton populations indicate adaptive strategies in the oligotrophic ocean. Aquat. Microb. Ecol. 71:1–13. 10.3354/ame01661. [DOI] [Google Scholar]
- 27. McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610–618. 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N. 2013. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J. 7:1493–1506. 10.1038/ismej.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ellison AM. 1987. Effect of seed dimorphism on the density-dependent dynamics of experimental populations of Atriplex triangularis (Chenopodiaceae). Am. J. Bot. 74:1280–1288. 10.2307/2444163. [DOI] [Google Scholar]
- 30. Shade A, Peter H, Allison SD, Baho DL, Berga M, Bürgmann H, Huber DH, Langenheder S, Lennon JT, Martiny JB. 2012. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 3:417. 10.3389/fmicb.2012.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimadzu H, Dornelas M, Henderson PA, Magurran AE. 2013. Diversity is maintained by seasonal variation in species abundance. BMC Biol. 11:98. 10.1186/1741-7007-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shade A, Read JS, Youngblut ND, Fierer N, Knight R, Kratz TK, Lottig NR, Roden EE, Stanley EH, Stombaugh J, Whitaker RJ, Wu CH, McMahon KD. 2012. Lake microbial communities are resilient after a whole-ecosystem disturbance. ISME J. 6:2153–2167. 10.1038/ismej.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Woyke T, Xie G, Copeland A, González JM, Han C, Kiss H, Saw JH, Senin P, Yang C, Chatterji S, Cheng JF, Eisen JA, Sieracki ME, Stepanauskas R. 2009. Assembling the marine metagenome, one cell at a time. PLoS One 4(4):e5299. 10.1371/journal.pone.0005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishoey T, Woyke T, Stepanauskas R, Novotny M, Lasken RS. 2008. Genomic sequencing of single microbial cells from environmental samples. Curr. Opin. Microbiol. 11:198–204. 10.1016/j.mib.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sangwan N, Verma H, Kumar R, Negi V, Lax S, Khurana P, Khurana JP, Gilbert JA, Lal R. 2014. Reconstructing an ancestral genotype of two hexachlorocyclohexane-degrading Sphingobium species using metagenomic sequence data. ISME J. 8:398–408. 10.1038/ismej.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pinto AJ, Raskin L. 2012. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7(8):e43093. 10.1371/journal.pone.0043093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou J, Jiang YH, Deng Y, Shi Z, Zhou BY, Xue K, Wu L, He Z, Yang Y. 2013. Random sampling process leads to overestimation of β-diversity of microbial communities. mBio 4(3):e00324-13. 10.1128/mBio.00324-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gobet A, Quince C, Ramette A. 2010. Multivariate cutoff level analysis (MultiCoLA) of large community data sets. Nucleic Acids Res. 38(15):e155. 10.1093/nar/gkp784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jones SE, Cadkin TA, Newton RJ, McMahon KD. 2012. Spatial and temporal scales of aquatic bacterial beta diversity. Front. Microbiol. 3:318. 10.3389/fmicb.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. LaRoche J, Breitbarth E. 2005. Importance of the diazotrophs as a source of new nitrogen in the ocean. J. Sea Res. 53:67–91. 10.1016/j.seares.2004.05.005. [DOI] [Google Scholar]
- 41. Hermansson A, Lindgren PE. 2001. Quantification of ammonia-oxidizing bacteria in arable soil by real-time PCR. Appl. Environ. Microbiol. 67:972–976. 10.1128/AEM.67.2.972-976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, Prosser JI, Schuster SC, Schleper C. 2006. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806–809. 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 43. Horz HP, Conrads G. 2010. The discussion goes on: what is the role of Euryarchaeota in humans? Archaea 2010: 967271. 10.1155/2010/967271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bowers RM, McCubbin IB, Hallar AG, Fierer N. 2012. Seasonal variability in airborne bacterial communities at a high-elevation site. Atmos. Environ. 50:41–49. 10.1016/j.atmosenv.2012.01.005. [DOI] [Google Scholar]
- 45. Werner JJ, Knights D, Garcia ML, Scalfone NB, Smith S, Yarasheski K, Cummings TA, Beers AR, Knight R, Angenent LT. 2011. Bacterial community structures are unique and resilient in full-scale bioenergy systems. Proc. Natl. Acad. Sci. U. S. A. 108:4158–4163. 10.1073/pnas.1015676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. 2011. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U. S. A. 108(Suppl 1):4578–4585. 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Portillo MC, Anderson SP, Fierer N. 2012. Temporal variability in the diversity and composition of stream bacterioplankton communities. Environ. Microbiol. 14:2417–2428. 10.1111/j.1462-2920.2012.02785.x. [DOI] [PubMed] [Google Scholar]
- 48. Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Desai N, Eisen JA, Evers D, Field D, Feng W, Huson D, Jansson J, Knight R, Knight J, Kolker E, Konstantindis K, Kostka J, Kyrpides N, Mackelprang R, McHardy A, Quince C, Raes J, Sczyrba A, Shade A, Stevens R. 2010. Meeting report: the Terabase Metagenomics Workshop and the Vision of an Earth Microbiome Project. Stand. Genomic Sci 3:243–248. 10.4056/sigs.1433550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. White EP, Adler PB, Lauenroth WK, Gill RA, Greenberg D, Kaufman DM, Rassweiler A, Rusak JA, Smith MD, Steinbeck JR. 2006. A comparison of the species-time relationship across ecosystems and taxonomic groups. Oikos 112:185–195. 10.1111/j.0030-1299.2006.14223.x. [DOI] [Google Scholar]
- 50. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Core R. Team; 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 52. Wickham H. 2009. Siggplot2: elegant graphics for data analysis. Springer Verlag, New York, NY. [Google Scholar]
- 53. Shade A. 21 May 2014. Conditionally rare taxa detection scripts v1.0. European Organization for Nuclear Research, Geneva, Switzerland. 10.5281/zendo/10040. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental results and discussion to accompany the text. Download
Taxa ranked by abundance in a representative community from each ecosystem examined in this study. Colors show the relative abundance of the taxon observed over the time series. Similar to many other environmental microbial communities, these communities have a large percentage of low-abundance taxa. Download
Use of the coefficient of bimodality (b) to detect CRT in microbial communities. (A) b values for uniform, Bernoulli, and trimodal distributions. (B) Dynamics of three conditionally rare Vibrio taxa (OTU defined at 97% sequence identity) detected in the western English Channel with a b value of >0.95 and a maximum relative (Max rel.) abundance of ≥0.01. (C) Examples of taxa that did not fit the conditionally rare criteria. Panels B and C include the time series of the taxon’s relative abundance (left) and the distribution of the taxon’s levels of abundance though time (right). Download
Influence of time series duration and sampling intensity on the detection of CRT. In panels A and C, the color gradient shows the percentages (of the total number of community members) of CRT detected. (A) The marine western English Channel (L4 site). (B) A freshwater lake, Trout Bog epilimnion, in northern Wisconsin. (C) A human male right palm skin community. The patchy nature of this distribution is attributed to the frequent disturbances associated with the habitat (e.g., hand washing that removed taxa and contact with a variety of objects that added taxa). (D) Influence of the number of samples, a proxy for sampling intensity, on the CRT detected. (E) Influence of sequencing depth on the CRT detected. b value, >0.90; relative abundance, >0.5%. Note the differences in y axis ranges. Download
Taxonomic distribution of CRT among microbial communities, summarized by ecosystem. Note the differences in x axis ranges. WWT is brewery wastewater treatment. b value, >0.90; relative abundance, >0.5%. Download
Summary of significant (P < 0.05, blue) and not significant (NS, pink) two-sided Pearson’s correlation tests to compare the composition of each whole community and that of its subset of CRT. Community composition was summarized at the phylum (circle), class (triangle), and order (square) levels. Download
CRT clustered by shared occurrence patterns from the western English Channel time series. Each taxon was most abundant at the time point colored black. b value, >0.90; relative abundance, >0.5%. Taxonomic assignments are provided. Some synchronous CRT were from closely related phylogenetic lineages (i.e., blue-highlighted example of cooccurring Pseudoalteromonas taxa), while other synchronous CRT were more diverse (i.e., yellow-highlighted example of Vibrio taxa cooccurring with Tatumella and Pseudoalteromonas taxa), suggesting that patterns of CRT cooccurrences are complex and could be either redundant or modular. Download
Incidence of CRT in each ecosystem with different coefficient-of-bimodality (b) and maximum-abundance (ra) thresholds and the inclusion or omission of singletons. Few CRT were detected with a maximum-abundance threshold of 0.05 (5%), indicating that this cutoff is uninformative because it is too high. This analysis suggests that an abundance threshold between 0.005 and 0.01 is consistent within a data set and bimodality cutoffs of ≥0.90 are consistent. Download
Description of the data sets used in this study.