

Abstract
Chronic kidney disease (CKD) is characterized by accumulation of proinflammatory cytokines, mainly tumor necrosis factor alpha (TNF-α). Single nucleotide polymorphisms (SNPs) of TNFA gene, including -238 G/A and -308 G/A, have been associated with alteration in the soluble TNF-α (sTNF-α) expression. The aim was to investigate the association of -238 y -308 TNFA gene SNPs with sTNF-α levels in CKD patients. We included 150 CKD patients and 192 control subjects (CS). Both SNPs were genotyped with polymerase chain reaction-restriction fragment length polymorphism technique and sTNF-α levels were measured by enzyme-linked immunosorbent assay. The genotypic distribution of -238 and -308 SNPs was not significantly different between CKD patients and CS (p > 0.001). However, the sTNF-α levels were higher in CKD, compared to CS (p < 0.001). Also, sTNF-α correlated with creatinine (r = 0.279, p = 0.004), urea (r = 0.325, p = 0.001), phosphorus (r = 0.479, p = 0.001), glomerular filtration rate (r = -0.236, p = 0.019) and monocyte count (r = 0.276, p = 0.010). In conclusion, elevated sTNF-α levels are associated with CKD. However, the -238 and -308 TNFA gene SNPs were not associated with susceptibility to CKD and sTNF-α levels in a Mexican population.
Keywords: TNFA polymorphisms, TNF-α levels, CKD
Introduction
Chronic kidney disease (CKD), characterized by a progressive decline in the glomerular filtration rate (GFR) [1], has been recognized as a global public health problem [2]. The prevalence of CKD increases with age and reaches around 17% of individuals over 60 years. Diabetes is the leading risk factor of CKD [3] and inflammation is a key factor as it is activated by the metabolic, biochemical, and hemodynamic derangements known to exist in the diabetic kidney [4,5]. Therefore, overproduction of proinflammatory cytokines may play an important role in inflammation and renal injury [6].
Patients with CKD have markedly elevated levels of cytokines such as tumor necrosis factor-α (TNF-α) [4,7]. TNF-α is mainly produced by monocytes, macrophages, and T cells. However, resident renal cells are also able to produce TNF-α, including mesangial, glomerular, endothelial, dendritic, and renal tubular cells. The effects of TNF-α include promotion of local reactive oxygen species (ROS), the increase of albumin permeability, and the induction of cytotoxicity, apoptosis, and necrosis. TNF-α is implicated in the recruitment of monocyte-macrophages and the reduction of GFR by hemodynamic changes, as well as in altering endothelial permeability. According to experimental data, patients with type 2 diabetes have 3-4 times greater serum levels of TNF-α compared to nondiabetic patients, and these levels are higher in diabetic patients with microalbuminuria compared with those that have normo albuminuria [4,8,9].
The TNF-α gene (TNFA) is located on chromosome 6p21.3, within the highly polymorphic major histocompatibility complex (MHC) region of the human genome. Many single-nucleotide polymorphisms (SNPs) and microsatellites have been identified in the TNF locus [10], and the ones in the promoter region of TNFA are thought to influence TNFA transcription rate and likely have direct functional significance in regulating TNF-α production [11]. In the promoter region of TNFA, two SNPs at positions -238 G/A (rs361525) and -308 G/A (rs1800629), relative to the transcription start site, have been well characterized. They have been shown to influence gene expression [12] and have been linked to several infectious and autoimmune diseases [13], although a lack of association has also been reported [14,15]. Based on this knowledge, the aim of this study was to investigate the possible association of the -238 and -308 TNFA gene polymorphisms with soluble TNF-α (sTNF-α) levels and clinical parameters in CKD patients.
Materials and methods
Patients and controls
We recruited 150 CKD patients from the Nephrology Service of the Clinica No. 46 from the Instituto Mexicano del Seguro Social in Guadalajara, Jalisco, México. Diagnosis of CKD was based on Kidney Disease Outcomes Quality Initiative (K/DOQI), clinical practice guidelines for chronic kidney disease, established by the National Kidney Foundation [16]. The demographic and clinical characteristics of CKD patients are shown in Table 1. In addition, 192 control subjects (CS) without clinical evidence of any autoimmune, inflammatory or infectious disease were enrolled in this study (135 women and 57 men with a mean age of 37 ± 14 years). All subjects were unrelated individuals from a Mexican Mestizo population as defined by the National Institute of Anthropology [17]; and a written informed consent was obtained from all subjects before inclusion in the study, according to the ethical guidelines of the 2013 Declaration of Helsinki.
Table 1.
Demographic and clinical characteristics of CKD patients
| Characteristics | CKD (n = 150) |
|---|---|
| Demographic | |
| Age (years) | 64 ± 11 (25-88) |
| Sex (women/men) | 64/86 |
| Clinical | |
| DM duration (years) | 18 ± 9 (1-43) |
| Serum creatinine (mg/dl) | 3.7 ± 2.7 (0.6-13.8) |
| GFR (ml/min/1.73 m2) | 30.1 ± 2.7 (7-90) |
| Urinary protein (g/24 hrs) | 3.8 ± 5.5 (0.08-24) |
| Glucose (mg/dl) | 122.1 ± 59.3 (34-356) |
| Urea (mg/dl) | 95.5 ± 45.3 (19-265) |
| Phosphorus (mg/dl) | 4.7 ± 1.1 (2.9-7.4) |
| Calcium (mg/dl) | 8.9 ± 0.9 (2.1-10.3) |
| Potassium (mmol/l) | 5.1 ± 0.8 (3.1-8.6) |
| Sodium (mmol/l) | 139.9 ± 3.9 (127-148) |
| Leukocytes (κ/µl) | 7.2 ± 2.1 (2.7-14.9) |
| Erythrocytes (κ/µl) | 4.0 ± 0.7 (2.2-8.0) |
| Platelets (κ/µl) | 252.5 ± 84.9 (72-535) |
| Hemoglobin (g/dl) | 11.6 ± 1.9 (6.9-16.8) |
| Hematocrit (%) | 35.8 ± 5.4 (19.8-50.3) |
| Total cholesterol (mg/dl) | 174.2 ± 43.3 (91.4-296.0) |
| Triglycerides (mg/dl) | 170.4 ± 87.2 (44.2-481.0) |
| SBP (mmHg) | 136.1 ± 27.9 (80-206) |
| DBP (mmHg) | 73.3 ± 14.0 (37-104) |
The data are presented as the mean ± standard deviation, minimum and maximum scores. CKD, chronic kidney disease; DM, diabetes mellitus; GFR, glomerular filtration rate; SBP, systolic blood pressure; DBP, diastolic blood pressure.
Laboratory assessment
White blood cell count, red blood cell count, platelet count, hemoglobin, hematocrit, and levels of creatinine, urinary protein, glucose, urea, phosphorus, calcium, potassium, sodium, total serum cholesterol and triglycerides were assayed using routine biochemical methods in all CKD patients, who had a 12-hour fast.
DNA isolation and TNFA gene polymorphisms genotyping
For genotyping, venous blood samples of patients and CS were collected into EDTA-containing tubes, and the genomic DNA (gDNA) was extracted from peripheral blood leukocytes according to the Miller’s salting-out method [18]. The TNFA gene polymorphisms were identified by the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) technique [19]. Amplification of both -238 and -308 TNFA gene polymorphic regions was performed in a Thermal Cycler, Esco Swift MaxPro, using the following primers (Invitrogen Life Technologies, Carlsbad, CA, USA): for the -238 polymorphism, 5’-AGA AGA CCC CCC TCG GAA CC-3’ (Forward) and 5’-ATC TGG AGG AAG CGG TAG TG-3’ (Reverse); and for the -308 polymorphism, 5’-AGG CAA TAG GTT TTG AGG GCC AT-3’ (Forward) and 5’-TCC TCC CTG CTC CGA TTC CG-3’ (Reverse) [19]. Both forward primers for each TNFA gene polymorphisms containing a single base-pair mismatch adjacent to the polymorphic site to introduce a restriction site into the wild-type nucleotide sequence during the amplification reaction.
The PCR was carried out in a final volume of 12.5 μl containing 0.1 μg/μl of DNA, 3 mM of each primer, 0.025 U/μl of Taq DNA polymerase, 1.25 μl of supplied buffer enzyme 1X, 2.5 mM of MgCl2, and 2.5 mM of each dNTP (Invitrogen Life Technologies, Carlsbad, CA, USA). Amplification conditions were as follows: initial denaturation at 94°C for 4 min, 33 cycles of 94°C for 30 s, 60°C for 30 s and 72°C for 30 s, followed by 72°C for 2 min for ending extension; resulting in fragments of 152 bp for the -238 SNP and 107 bp for the -308 SNP, which were analyzed on a 6% polyacrylamide gel stained with silver nitrate (Invitrogen Life Technologies, Carlsbad, CA, USA). The amplified fragments of both -238 and -308 TNFA gene SNPs were incubated with 3 U of MspI and NcoI restriction enzymes (New England BioLabs, Beverly, MA, USA), respectively, for 1 h at 37°C. Finally, the digested PCR products were electrophoresed on 6% polyacrylamide gels stained with silver nitrate for genotype identification. For the -238 SNP, fragments of 133 and 19 bp represent the wild-type genotype (G/G), fragments of 152, 133 and 19 bp represent the heterozygote genotype (G/A), and a unique fragment of 152 bp represents the homozygote genotype (A/A); and for the -308 SNP, fragments of 87 and 20 bp represent the wild-type genotype (G/G), fragments of 107, 87 and 20 bp represent the heterozygote genotype (G/A), and a only one fragment of 107 bp represents the homozygote genotype (A/A).
Soluble TNF-α serum levels quantification
Soluble TNF-α (sTNF-α) levels were determined in serum samples from CKD patients and CS using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Human sTNF-α ELISA, KHC3011, Invitrogen, Flynn Road, Camarillo, CA). The assay sensitivity was 1.7 pg/ml and it was carried out accordingly to the manufacturer’s instructions.
Statistical analysis
Demographic and clinical characteristics were given as mean values, standard deviation (SD), minimum and maximum scores. Frequencies of genotypes and alleles were estimated and deviation from Hardy-Weinberg expected frequencies (HWE) tested using the Chi squared (c2) test. Association between TNFA genotypes and CKD was measured using Odds ratios (OR) that were tested using the normal approximation to the logarithm of the OR and 95% confidence intervals estimated (Epi Info statistical software 7, Atlanta Georgia). Haplotype frequencies were inferred using HEMHAPFRE software [20]. Linkage disequilibrium (LD) was expressed as Lewontin’s corrected LD coefficient (D’) [21]. Comparing TNFA genotype distributions between CKD and CS used c2 test. Comparing sTNF-α levels between groups used nonparametric Mann-Whitney U tests or Kruskal–Wallis tests. Correlation between sTNF-α levels and other clinical measures used Spearman’s correlation tests for non-parametric variables and Pearson’s correlation for parametric variable (SPSS statistical software 18.0, Chicago Illinois). Differences were considered as significant at p < 0.05.
Results
Frequency of TNFA gene polymorphisms and haplotype analysis in both study groups
Genotype and allele frequencies of -238 G/A and -308 G/A TNFA gene SNPs in CKD patients and CS are shown in Table 2. Both TNFA gene SNPs were in HWE in the CS group (p > 0.05; data not shown). The distribution of genotypic and allelic frequencies of both TNFA gene SNPs was similar between CKD patients and CS (p > 0.05). Therefore, the -238 G/A and -308 G/A TNFA gene SNPs did not affect the risk for CKD.
Table 2.
Genotypic and allelic frequencies of TNFA gene polymorphisms in CKD patients and CS
| SNP | CKD (n = 150) % (n) | CS (n = 192) % (n) | Global p | OR (95% CI); p value |
|---|---|---|---|---|
| -238 | ||||
| Genotypes | ||||
| G/G* | 90 (135) | 89.58 (172) | 1.0 | |
| G/A | 9.33 (14) | 10.42 (20) | 0.50 | 0.76 (0.55-2.30); 0.76 |
| A/A | 0.67 (1) | 0 (0) | 0.26 (0.01-6.48); 0.41 | |
| Alleles | ||||
| G* | 94.67 (284) | 94.80 (364) | 0.94 | 1.0 |
| A | 5.33 (16) | 5.20 (20) | 0.98 (0.50-1.92); 0.94 | |
| -308 | ||||
| Genotypes | ||||
| G/G* | 86.67 (130) | 85.94 (165) | 1.0 | |
| G/A | 12.67 (19) | 14.06 (27) | 0.49 | 1.12 (0.60-2.10); 0.73 |
| A/A | 0.66 (1) | 0 (0) | 0.26 (0.01-6.51); 0.41 | |
| Alleles | ||||
| G* | 93 (279) | 92.97 (357) | 0.99 | 1.0 |
| A | 7 (21) | 7.03 (27) | 0.99 (0.56-1.81); 0.99 |
The values are presented as frequency in percentage (%) and genotypes and alleles number (n). The frequencies comparison between groups was analyzed using Chi square test (x2) and Fisher’s exact test when applicable. Both, -238 and -308 TNFA gene SNPs were in HWE in the CS group (p = 1.00 and p = 0.60, respectively). TNFA, tumor necrosis factor alpha; CKD, chronic kidney disease; CS, control subjects; SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval;
Reference category;
HWE, Hardy-Weinberg equilibrium.
On the other hand, we analyzed the linkage disequilibrium (LD) pattern and haplotypes distribution, combining the -238 and -308 TNFA gene polymorphic sites. The LD analysis showed no significant differences in all possible combinations in the CS group (p > 0.05) (Table 3). Likewise, there was no significant difference in the haplotype frequency distribution between CKD patients and CS (p > 0.05) (Table 4). Therefore, we did not identify a risk haplotype for CKD.
Table 3.
Haplotypes frequencies and LD analysis of TNFA gene polymorphisms in CS
| Haplotype | % | D’ | p value |
|---|---|---|---|
| -308/-238 | |||
| GG | 88.48 | 0.04 | 0.64 |
| GA | 4.49 | -0.04 | 0.98 |
| AG | 6.31 | -0.08 | 0.97 |
| AA | 0.72 | 0.08 | 1.00 |
The linkage disequilibrium (LD) is expressed as Lewontin’s corrected LD coefficient (D’) [21]. The comparison data was evaluated by Chi-square test (x2) and Fisher’s exact test when applicable. TNFA, tumor necrosis factor alpha; CS, control subjects.
Table 4.
Haplotypes frequencies comparison of TNFA gene polymorphisms between CKD patients and CS
| Haplotype | CKD % (n) | CS % (n) | OR (95% CI); p value |
|---|---|---|---|
| -308/-238 | |||
| GG* | 89.46 (268) | 88.48 (340) | 1.0 |
| GA | 3.54 (11) | 4.49 (17) | 0.75 (0.27-2.06); 0.57 |
| AG | 5.21 (16) | 6.31 (24) | 0.85 (0.37-1.93); 0.69 |
| AA | 1.79 (5) | 0.72 (3) | 2.54 (0.51-12.73); 0.26 |
The values are presented as frequency in percentage (%) and haplotypes number (n). The haplotype frequencies were inferred using the HEMHAPFRE software [20]. The comparison data was evaluated by Chi-square test (x2) and Fisher’s exact test when applicable. TNFA, tumor necrosis factor alpha; CKD, chronic kidney disease; CS, control subjects; OR, odds ratio; CI, confidence interval;
Reference haplotype.
Soluble TNF-α levels, clinical correlation in CKD patients and their relationship with TNFA gene polymorphisms in both study groups
The sTNF-α levels were significantly associated with CKD, since CKD patients showed higher sTNF-α levels than CS (p < 0.001) (Figure 1). Likewise, in CKD patients the sTNF-α levels were positively correlated with the creatinine (r = 0.279; p = 0.004), urea (r = 0.325; p = 0.001) and phosphorus levels (r = 0.479; p = 0.001), as well as monocyte count (r = 0.276; p = 0.010); and were negatively correlated with the GFR (r = -0.236; p = 0.019) (Figure 2). Finally, we analyzed sTNF-α levels according to both TNFA gene polymorphisms in CKD patients and CS. However, we did not observe an association between the different genotypes and sTNF-α levels (p > 0.05) (Figure 3).
Figure 1.
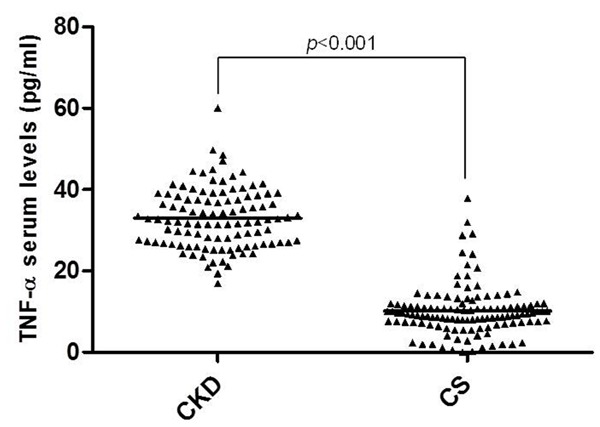
Soluble TNF-α serum levels in CKD patients and CS. The mean difference between CKD patients (n = 106, 32.96 ± 7.40 pg/ml) and CS (n = 120, 10.13 ± 6.14 pg/ml) was analyzed using Mann-Whitney U test. TNF-α, tumor necrosis factor alpha; CKD, chronic kidney disease; CS, control subjects.
Figure 2.
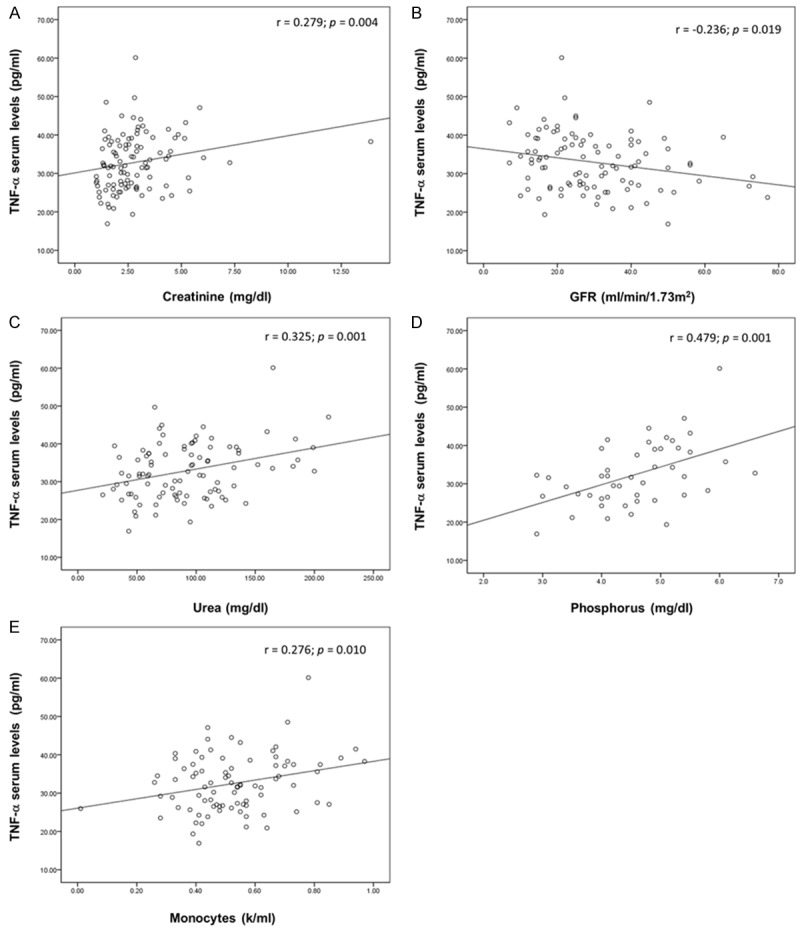
Correlation between soluble TNF-α serum levels and clinical parameters in CKD patients. The analysis was performed using Spearman correlation test (A, n = 104) and Pearson correlation test (B, n = 99; C, n = 94; D, n = 46; E, n = 119). TNF-α, tumor necrosis factor alpha; GFR, glomerular filtration rate; CKD, chronic kidney disease.
Figure 3.
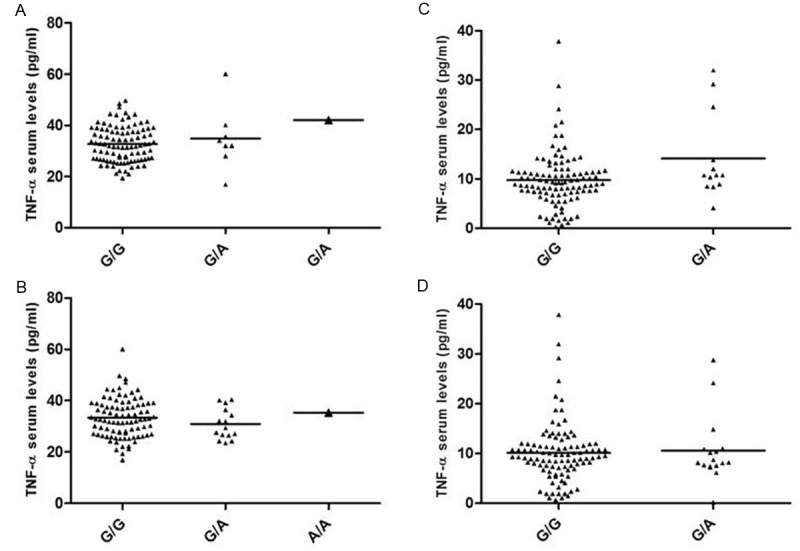
Soluble TNF-α serum levels observed in CKD patients (A and B) and CS (C and D) according to the TNFA gene polymorphisms. (A) TNF-α levels in CKD patients according to the -238 G/A TNFA gene SNP [G/G (32.71 ± 6.91 pg/ml), G/A (34.85 ± 12.26 pg/ml) and A/A (42.07 pg/ml) (p = 0.344)]. (B) TNF-α levels in CKD patients according to the -308 G/A TNFA gene SNP [G/G (33.27 ± 7.63 pg/ml), G/A (30.91 ± 5.95 pg/ml) and A/A (35.36 pg/ml) (p = 0.501)]. (C) TNF-α levels in CS according to the -238 G/A TNFA gene SNP [G/G (9.74 ± 5.57 pg/ml) and G/A (13.14 ± 9.13 pg/ml) (p = 0.158)]. (D) TNF-α levels in CS according to the -308 G/A TNFA gene SNP [G/G (10.06 ± 6.07 pg/ml) and G/A (10.57 ± 6.71 pg/ml) (p = 0.715)]. The mean difference between groups was analyzed using Kruskal-Wallis test. TNF-α, tumor necrosis factor alpha; CKD, chronic kidney disease; CS, control subjects; SNP, single nucleotide polymorphism.
Discussion
Diabetic nephropathy, especially in the context of type 2 diabetes, has become the principal cause of renal failure, which in turn is the main cause of morbidity and mortality in the diabetic population. The classical view of renal injury as a consequence of metabolic and hemodynamic alterations has been transformed significantly. Existing evidence indicates that activation of the innate immunity and the subsequent development of a low-grade chronic inflammatory reaction is an important phenomenon in the pathogenesis and progression of diabetic nephropathy [22]. In this context, inflammatory cytokines have a key role, including mainly TNF-α, a cytokine expressed, synthesized and released by blood cells (mainly monocytes and macrophages), as well as by diverse intrinsic renal cells (endothelial, mesangial, dendritic and tubular epithelial cells) [22,23]; and two TNFA gene SNPs at positions -238 G/A and -308 G/A, relative to the transcription start site, have been shown to influence gene expression [12] and have been linked to several infectious and autoimmune diseases [13]. Therefore, in the present study we analyzed the association of the -238 and -308 TNFA gene SNPs with sTNF-α levels in CKD patients in a Mexican population.
We did not find an association between both -238 G/A and -308 G/A TNFA gene SNPs and CKD. However, to our knowledge, this is the first study that investigates the association between the -238 G/A SNP and CKD. On the other hand, in contrast with our results, the -308 G/A SNP has been previously associated as a strong predisposing risk factor for CKD in north India [6] and Chinese [24] populations. Nevertheless, in accordance with a previous report of this SNP in Asian Indians [25], our results can also be explained by the very low frequency of the polymorphic A/A genotype observed in our study groups. Likewise, we did not observe an association between any haplotype combination of these SNPs and CKD, suggesting that it is unlikely that the -238 G/A and -308 G/A TNFA gene SNPs have an important effect on susceptibility to CKD, although the contribution of other variants located within the promoter region of the TNFA gene in susceptibility to CKD cannot be discarded.
Regarding sTNF-α levels, we found an association of elevated sTNF-α levels with CKD, since CKD patients showed significantly higher sTNF-α levels compared to CS. This is consistent with those observed by Navarro-González et al., Fernández-Real et al. and Shen et al., who also found elevated sTNF-α levels in CKD patients with chronic low-grade inflammation in Spanish and Chinese populations [26-28]. In addition, it has been reported that patients with type 2 diabetes have 3-4 times greater TNF-α serum levels compared to nondiabetic subjects, and these levels are higher in diabetic patients with microalbuminuria compared with those that have normo albuminuria [4]. Based on the above, our results support the cytotoxic role of TNF-α in the glomerular injury, which in turn leads to CKD development.
On the other hand, we also observed a positive correlation between sTNF-α levels with creatinine, urea and phosphorus levels, as well as with the monocyte count, and a negative correlation with the GFR. It has been reported that hemodynamic changes and an alteration of the endothelial permeability reduce the GFR [4,9], which in consequence leads to accumulation of metabolic waste products including creatinine, urea and albumin. In addition, the activation of the TNF-α system [inferred from circulating concentrations of the soluble fraction of TNF-α receptors (sTNFRs) may potentially exert independent effects on urinary albumin, creatinine ratio and GFR in type 2 diabetes [26].
In regards to the correlation between sTNF-α levels and urea, the primary nitrogenous end product of amino acid catabolism, essential for the excretion of nitrogenous waste, it has long been known that the kidney’s permeability to the highly polar molecule urea is greatly increased by the insertion of five urea transporters that allow facilitated urea transport for an adequate renal tubular function, including the ability to potently concentrate urine [29]. In this context, a previous study reported that a bolus infusion of TNF-α at a dose of 1 mg/kg decreased fractional urea excretion, inner medulla urea concentration and osmolality, tubular urea reabsorption and urinary urea whereas it increased plasma osmolality and urea; such findings were attributed to reduced urea transporters [30]. Therefore, we hypothesized that the increased sTNF-α levels observed in our patients may be related to a decrease in the expression of urea transporters expression, favoring urea accumulation. However, this speculation will require more investigation in future studies.
Further, the correlation between the sTNF-α levels and increased phosphorus levels may be explained since TNF-α has been previously reported as a critical mediator of vascular calcification, which in turn is a common feature in CKD patients [31,32]. However, the mechanisms underlying the molecular pathogenesis of vascular calcification in CKD are extremely complex and are influenced by the interaction of several inflammatory factors, lipids, minerals, and oxidative stress [31-33]. Also, the molecular mechanism by which TNF-α promotes CKD-dependent vascular calcification remains uncertain.
Masuda et al. [31], found an important link between TNF-α signaling and one of the endoplasmic reticulum stress signaling pathways (PERK-eIF2a-ATF4-CHOP axis) in the regulation of CKD-dependent vascular calcification. The precise mechanism by which the PERK-eIF2a-ATF4-CHOP signaling pathway augments TNF-α-induced vascular calcification remains unclear, however this is likely related to increased phosphate uptake [31]. Furthermore, their results suggest that Pit-1, a major type III sodium-dependent phosphate transporter, is a crucial regulator in the vascular calcification, since Pit-1 is a major target of the PERK-eIF2a-ATF4-CHOP signaling pathway. In addition, they observed that CHOP (C/EBP homologous protein) significantly increased phosphate uptake by increasing the Pit-1 expression [31].
On the other hand, in addition to inflammation, in advanced stages of CKD, the total renal capacity to eliminate phosphorus is sufficiently reduced such that serum phosphorus concentrations begin to increase [34], which together with increased calcium and calcium-phosphate product, has been attributed to development of vascular calcification in these patients [35]. However, the relationship between sTNF-α levels and increased phosphorus levels will require more investigation in future studies.
With respect to the correlation between the sTNF-α levels and elevated monocyte count, it has been known that monocytes are very important secretors of TNF-α [23,36], which in turn induce leukocyte emigration through the adhesion molecule expression in the vascular endothelium, favoring the activation and differentiation of monocytes and macrophages in local inflammation sites [37].
Finally, we did not observe an association between the sTNF-α levels and the -238 G/A and -308 G/A TNFA gene SNPs in both study groups. This is in accordance with Mekinian et al., who also showed no association between TNF-α level expression and these SNPs in healthy subjects [38]. Therefore, our results might suggest the absence of any functional relationship of these TNFA gene SNPs with sTNF-α levels in CKD patients. However, this is largely speculative since circulating TNF-α levels are also regulated at different stages including transcriptional level, post-transcriptional control of mRNA stability and half-life, post-translational cleavage of the receptor bound to the soluble form and regulation by metabolic parameters including the expression of its receptors [23]. Also, we cannot discard the contribution of other variants located within the promoter region of the TNFA gene in the sTNF-α expression.
In conclusion, elevated sTNF-α levels are associated with CKD in Mestizo Mexican population, confirming the involvement of TNF-α in kidney damage development. However, the -238 G/A and -308 G/A TNFA gene SNPs are not genetic markers of susceptibility to CKD in our population. Likewise, these SNPs did not show an effect on sTNF-α production in CKD patients and CS. However, further studies are required to evaluate the role of other genetic variants within the TNFA gene with CKD and expression levels of TNF-α, as well as in longitudinal studies.
Disclosure of conflict of interest
None.
References
- 1.Abboud H, Henrich WL. Clinical practice. Stage IV chronic kidney disease. N Engl J Med. 2010;362:56–65. doi: 10.1056/NEJMcp0906797. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida T, Kato K, Yokoi K, Oguri M, Watanabe S, Metoki N, Yoshida H, Satoh K, Aoyagi Y, Nishigaki Y, Nozawa Y, Yamada Y. Association of gene polymorphisms with chronic kidney disease in high- or low-risk subjects defined by conventional risk factors. Int J Mol Med. 2009;23:785–792. doi: 10.3892/ijmm_00000193. [DOI] [PubMed] [Google Scholar]
- 3.Shiraishi FG, Stringuetta Belik F, Oliveira E Silva VR, Martin LC, Hueb JC, Goncalves Rde S, Caramori JC, Barreti P, Franco RJ. Inflammation, diabetes, and chronic kidney disease: role of aerobic capacity. Exp Diabetes Res. 2012;2012:750286. doi: 10.1155/2012/750286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012;2012:146154. doi: 10.1155/2012/146154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borgeson E, Godson C. Resolution of inflammation: therapeutic potential of pro-resolving lipids in type 2 diabetes mellitus and associated renal complications. Front Immunol. 2012;3:318. doi: 10.3389/fimmu.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manchanda PK, Kumar A, Kaul A, Mittal RD. Correlation between a gene polymorphism of tumor necrosis factor-alpha (G/A) and end-stage renal disease: a pilot study from north India. Clin Chim Acta. 2006;370:152–157. doi: 10.1016/j.cca.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Rao M, Wong C, Kanetsky P, Girndt M, Stenvinkel P, Reilly M, Raj DS. Cytokine gene polymorphism and progression of renal and cardiovascular diseases. Kidney Int. 2007;72:549–556. doi: 10.1038/sj.ki.5002391. [DOI] [PubMed] [Google Scholar]
- 8.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 9.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 10.Aguillón GJC, Cruzat CA, Cuenca MJ, Cuchacovich TM. El polimorfismo genético del factor de necrosis tumoral alfa como factor de riesgo en patología. Revista médica de Chile. 2002;130:1043–1050. [PubMed] [Google Scholar]
- 11.Jaber BL, Liangos O, Pereira BJ, Balakrishnan VS. Polymorphism of immunomodulatory cytokine genes: implications in acute renal failure. Blood Purif. 2004;22:101–111. doi: 10.1159/000074930. [DOI] [PubMed] [Google Scholar]
- 12.Hajeer AH, Hutchinson IV. Influence of TNFalpha gene polymorphisms on TNFalpha production and disease. Hum Immunol. 2001;62:1191–1199. doi: 10.1016/s0198-8859(01)00322-6. [DOI] [PubMed] [Google Scholar]
- 13.Hollegaard MV, Bidwell JL. Cytokine gene polymorphism in human disease: on-line databases, Supplement 3. Genes Immun. 2006;7:269–276. doi: 10.1038/sj.gene.6364301. [DOI] [PubMed] [Google Scholar]
- 14.Keso T, Perola M, Laippala P, Ilveskoski E, Kunnas TA, Mikkelsson J, Penttila A, Hurme M, Karhunen PJ. Polymorphisms within the tumor necrosis factor locus and prevalence of coronary artery disease in middle-aged men. Atherosclerosis. 2001;154:691–697. doi: 10.1016/s0021-9150(00)00602-x. [DOI] [PubMed] [Google Scholar]
- 15.Zeggini E, Groves CJ, Parkinson JR, Halford S, Owen KR, Frayling TM, Walker M, Hitman GA, Levy JC, O’Rahilly S, Hattersley AT, McCarthy MI. Large-scale studies of the association between variation at the TNF/LTA locus and susceptibility to type 2 diabetes. Diabetologia. 2005;48:2013–2017. doi: 10.1007/s00125-005-1902-4. [DOI] [PubMed] [Google Scholar]
- 16.National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–266. [PubMed] [Google Scholar]
- 17.Gorodezky C, Alaez C, Vazquez-Garcia MN, de la Rosa G, Infante E, Balladares S, Toribio R, Perez-Luque E, Munoz L. The genetic structure of Mexican Mestizos of different locations: tracking back their origins through MHC genes, blood group systems, and microsatellites. Hum Immunol. 2001;62:979–991. doi: 10.1016/s0198-8859(01)00296-8. [DOI] [PubMed] [Google Scholar]
- 18.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oregon-Romero E, Vazquez-Del Mercado M, Ruiz-Quezada SL, Navarro-Hernandez RE, Rangel-Villalobos H, Martinez-Bonilla G, Bernard-Medina AG, Armendariz-Borunda J, Garcia-Banuelos J, Munoz-Valle JF. Tumor necrosis factor alpha-308 and -238 polymorphisms in rheumatoid arthritis. Association with messenger RNA expression and sTNF-alpha. J Investig Med. 2008;56:937–943. doi: 10.2310/JIM.0b013e318189152b. [DOI] [PubMed] [Google Scholar]
- 20.Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- 21.Lewontin RC. The Interaction of Selection and Linkage. I. General Considerations; Heterotic Models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarro-Gonzalez JF, Jarque A, Muros M, Mora C, Garcia J. Tumor necrosis factor-alpha as a therapeutic target for diabetic nephropathy. Cytokine Growth Factor Rev. 2009;20:165–173. doi: 10.1016/j.cytogfr.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Schulz S, Schagdarsurengin U, Suss T, Muller-Werdan U, Werdan K, Glaser C. Relation between the tumor necrosis factor-alpha (TNF-alpha) gene and protein expression, and clinical, biochemical, and genetic markers: age, body mass index and uric acid are independent predictors for an elevated TNF-alpha plasma level in a complex risk model. Eur Cytokine Netw. 2004;15:105–111. [PubMed] [Google Scholar]
- 24.Wang Y, Ng MC, So WY, Ma R, Ko GT, Tong PC, Chan JC. Association between tumour necrosis factor-alpha G-308A polymorphism and risk of nephropathy in obese Chinese type 2 diabetic patients. Nephrol Dial Transplant. 2005;20:2733–2738. doi: 10.1093/ndt/gfi101. [DOI] [PubMed] [Google Scholar]
- 25.Prasad P, Tiwari AK, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK. Association of TGFbeta1, TNFalpha, CCR2 and CCR5 gene polymorphisms in type-2 diabetes and renal insufficiency among Asian Indians. BMC Med Genet. 2007;8:20. doi: 10.1186/1471-2350-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez-Real JM, Vendrell J, Garcia I, Ricart W, Valles M. Structural damage in diabetic nephropathy is associated with TNF-alpha system activity. Acta Diabetol. 2012;49:301–305. doi: 10.1007/s00592-011-0349-y. [DOI] [PubMed] [Google Scholar]
- 27.Navarro JF, Mora C, Muros M, Garcia J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant. 2006;21:3428–3434. doi: 10.1093/ndt/gfl469. [DOI] [PubMed] [Google Scholar]
- 28.Shen L, Lu G, Dong N, Jiang L, Ma Z, Ruan C. Von Willebrand factor, ADAMTS13 activity, TNF-alpha and their relationships in patients with chronic kidney disease. Exp Ther Med. 2012;3:530–534. doi: 10.3892/etm.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt C, Hocherl K, Bucher M. Cytokine-mediated regulation of urea transporters during experimental endotoxemia. Am J Physiol Renal Physiol. 2007;292:F1479–1489. doi: 10.1152/ajprenal.00460.2006. [DOI] [PubMed] [Google Scholar]
- 30.Ramseyer VD, Garvin JL. Tumor necrosis factor-alpha: regulation of renal function and blood pressure. Am J Physiol Renal Physiol. 2013;304:F1231–1242. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2alpha-ATF4-CHOP signaling contributes to TNFalpha-induced vascular calcification. J Am Heart Assoc. 2013;2:e000238. doi: 10.1161/JAHA.113.000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, Heimburger O, Cederholm T, Girndt M. IL-10, IL-6, and TNF-alpha: central factors in the altered cytokine network of uremia--the good, the bad, and the ugly. Kidney Int. 2005;67:1216–1233. doi: 10.1111/j.1523-1755.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- 33.Kendrick J, Chonchol M. The role of phosphorus in the development and progression of vascular calcification. Am J Kidney Dis. 2011;58:826–834. doi: 10.1053/j.ajkd.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menon MC, Ix JH. Dietary phosphorus, serum phosphorus, and cardiovascular disease. Ann N Y Acad Sci. 2013;1301:21–6. doi: 10.1111/nyas.12283. [DOI] [PubMed] [Google Scholar]
- 35.Ossareh S. Vascular calcification in chronic kidney disease: mechanisms and clinical implications. Iran J Kidney Dis. 2011;5:285–299. [PubMed] [Google Scholar]
- 36.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 37.Makhatadze NJ. Tumor necrosis factor locus: genetic organisation and biological implications. Hum Immunol. 1998;59:571–579. doi: 10.1016/s0198-8859(98)00056-1. [DOI] [PubMed] [Google Scholar]
- 38.Mekinian A, Tamouza R, Pavy S, Gestermann N, Ittah M, Mariette X, Miceli-Richard C. Functional study of TNF-alpha promoter polymorphisms: literature review and meta-analysis. Eur Cytokine Netw. 2011;22:88–102. doi: 10.1684/ecn.2011.0285. [DOI] [PubMed] [Google Scholar]