

Abstract
Chronic inflammation is a well-recognized pathogenic factor in tumor initiation and progression. Mice lacking the pro-oncogenic transcription factor STAT3 were shown to be protected from both colitis-associated and epidermal cancers induced by the AOM/DSS and DMBA/TPA protocols, respectively. However, these murine models did not distinguish between the two STAT3 isoforms, the full-length STAT3α, believed to exert most pro-oncogenic functions attributed to STAT3, and the shorter STAT3β, often referred to as a dominant-negative, but possessing specific transcriptional activities. Here we assessed the contribution of STAT3β to inflammation-driven tumorigenesis making use of mice lacking this isoform, but still expressing STAT3α (STAT3Δβ/Δβ). We show that the lack of STAT3β leads to exacerbated acute responses to both TPA and DSS, thus confirming its anti-inflammatory role. Enhanced inflammation correlates with earlier tumor onset in both the epidermis and the intestine in STAT3Δβ/Δβ mice. In contrast, overall tumor development and final tumor burden were unaffected. These results suggest that STAT3β, by limiting inflammation during the initial phases of tumorigenesis, contributes to tissue homeostasis and counteracts malignant transformation and initial tumor growth. Accordingly, the balance between the two STAT3 isoforms, likely determined by the complex signaling networks shaping the tumor microenvironment and driving tumor transformation and progression, is apparently crucial to determine the initial tumor transformation rates in inflammation-associated cancers.
Keywords: STAT3 isoforms, STAT3β, inflammation, inflammation-driven tumorigenesis, colitis-associated cancer, AOM/DSS protocol, skin tumorigenesis, DMBA/TPA protocol, genetically modified mice
Introduction
Signal Transducer and Activator of Transcription (STAT)3 is an extremely pleiotropic transcription factor that is activated downstream of multiple cytokine and growth factor receptors by tyrosine phosphorylation, and displays a tissue- and signal-specific repertoire of target genes [1-3]. STAT3 is considered as an oncogene, since it is constitutively active in a wide variety of tumors and often required for tumor cell transformation, survival and proliferation [4]. Its activation in tumors mainly occurs downstream of aberrantly activated pro-oncogenic signals, although activating mutations have been recently identified in a subset of hepatocellular adenomas [5]. On the other hand, STAT3 is also able to act as a tumor suppressor under specific conditions, underscoring the biological complexity of its functions. For example, STAT3 gene inactivation was shown to enhance tumorigenesis in a model of thyroid adenocarcinoma [6] and in ApcMin-induced intestinal tumorigenesis [7].
STAT3 exists in two alternatively spliced isoforms, the full length STAT3α and the truncated STAT3β [8]. The latter lacks the C-terminal Transcriptional Activation Domain (TAD) and has long been considered as a dominant negative form. However, others and we have demonstrated that the β isoform can play unique functions, activating both common and specific repertoires of STAT3 target genes, and is able to rescue the embryonal lethality of a null STAT3 mutation [8-11]. STAT3β can play opposite functions in tumorigenesis. When over-expressed, it can oppose the pro-oncogenic effects of STAT3α in several tumor cell lines, acting as a dominant negative form [4]. Moreover, the induction of a splicing switch towards the beta isoform leads to apoptosis and cell-cycle arrest in STAT3-dependent cell lines that show persistent STAT3 tyrosine phosphorylation, via the activation of a unique gene expression signature [12]. In contrast, STAT3β apparently acts as an oncogene by triggering aggressive T cell leukemia when overexpressed in transplanted bone marrow cells [13]. On the other hand, STAT3β appears to act as a suppressor of systemic inflammation. Mice lacking STAT3β are more sensitive to endotoxic shock [9,10], and were recently shown to develop exacerbated atherosclerosis in the absence of ApoE [14]. Accordingly, STAT3β-deficient macrophages display enhanced production of inflammatory cytokines and reduced levels of the anti-inflammatory cytokine IL-10 upon lipopolysaccharide (LPS) treatment [10]. These unique properties of the beta isoform may contribute to the extraordinary functional complexity of STAT3 under both physiological and pathological conditions.
Continuous, non-resolving inflammation is a common feature of the tumor microenvironment and plays a crucial role in both initiation and progression of many malignancies [15,16]. Accordingly, chronic inflammatory conditions such as inflammatory bowel diseases, hepatitis or pancreatitis are well known cancer-predisposing factors. STAT3 is considered as a key player in mediating inflammation-driven tumorigenesis, being constitutively activated by chronically high levels of the pro-inflammatory cytokine IL-6 and participating in a feed-forward loop with IL-6 and the pro-oncogenic transcription factor NF-κB [4,17]. Indeed, both IL-6 and STAT3 have been shown to be essential pathogenic factors in Colitis-Associated Cancer (CAC) [18,19]. This is modeled in the mouse by a single administration of the pro-carcinogen azoxymethane (AOM), whose mutagenic effects are then revealed by chronic inflammation triggered via repeated treatments with the luminal toxin dextran sodium sulphate (DSS) in the drinking water [20]. STAT3 is also required for tumor development in another well accepted tumor model where chronic inflammation plays a prominent role, the DMBA/TPA multi-step skin carcinogenesis protocol [21,22]. This consists of a single topical exposure of the murine skin to the 7, 12-Dimethylbenz(a)anthracene (DMBA) mutagen, followed by repeated applications of the tumor promoting agent 12-O-tetradecanoylphorbol 13-acetate (TPA). In both models, STAT3 plays a prominent role in favoring cell survival upon the mutagenic event, but it is also thought to contribute to tumor progression [18,19,22,23]. However, the specific roles played by the alpha and beta isoforms in the context of inflammation-associated tumors have not been addressed.
Here, we assess the contribution of the STAT3β isoform to inflammation-driven carcinogenesis by exposing mice specifically lacking STAT3β (STAT3Δβ/Δβ) to both the AOM/DSS and the DMBA/TPA carcinogenesis protocols. Consistent with its anti-inflammatory role, in both models STAT3Δβ/Δβ mice display enhanced initial inflammation and earlier onset of tumor that, however, do not correlate with increased final tumor burden, supporting the idea that STAT3β may play an anti-oncogenic role in inflammation-driven cancer mainly acting at the earlier steps of tumor formation.
Materials and methods
Mice and treatments
STAT3Δβ/Δβ mice were generated as described in [10], backcrossed for 5 generation with the Balb/c background, maintained in the transgenic unit of the Molecular Biotechnology Center (University of Turin, Turin, Italy) and given water and food ad libitum. Genetic screening was performed by PCR as previously described [10]. Procedures were conducted in conformity with national and international laws and policies as approved by the University of Turin Ethical Committee.
DMBA/TPA protocol
The two-stage skin carcinogenesis experiments were performed as previously described [22], with some modifications. Briefly, eight-weeks old STAT3Δβ/Δβ and control female littermates were shaved on the back 48h before exposure of the skin to DMBA (Sigma Aldrich, 25 µg in 200 μg of acetone), followed after two weeks by biweekly treatments with TPA (Sigma Aldrich, 4 µg in 200 μg of acetone) for 42 weeks. Mice were monitored for tumor formation weekly. Skin papillomas were counted and measured with a caliper. Papillomas were collected at the end of the study for histopathological evaluation.
For acute responses to TPA, twelve-weeks old female mice were treated with a single dose of TPA (0.1 mM in acetone) or with acetone alone on their shaved backs, and sacrificed after 24 or 72 hours for histopathological analysis. Proliferation and inflammation were evaluated in blind as follows: Proliferation: (-) epidermis 1-2 layers, thin stratum corneum and thin or absent granular layer; (+/-) some areas displaying more than two cell layers in the epidermis; (+) epidermis 2-4 cell layers, thicker stratum corneum; (++) many cell layers, both stratum corneum and granular layer more evident; (+++) thick epidermis, stratum corneum and granular layer very evident along the entire section; Inflammation: (-) none or very few inflammatory cells, dispersed into the dermis; (+) few inflammatory cells in the adipose tissue and many in dermis and epidermis; (++) some detachment areas between epidermis and dermis due to inflammation, few inflammatory cells in the adipose tissue; (+++) numerous detachment areas between epidermis and dermis, aggregates of inflammatory cells in the adipose tissue.
AOM/DSS protocol
The CAC tumorigenesis experiments were performed as previously described [24]. Briefly, six-weeks old STAT3Δβ/Δβ and control male littermates were injected i.p. with AOM (Sigma Aldrich, 12.5 mg/kg in PBS), followed by three five-days cycles of 2.5% DSS in the drinking water (MP Biomedicals, MW 35,000-50,000, first and second cycle) or 2% DSS (third cycle) with 14 days intervals. Mice were sacrificed at day 60 or 80 after AOM injection and their intestine prepared as a “Swiss Roll” for subsequent analysis [25]. Macroscopic tumors were counted and measured (see below).
For acute DSS treatment, mice received water with 3% DSS for five days and were sacrificed at different times after the end of the treatment [26]. Colitis scoring: mice were daily monitored for colitis features (see below) and sacrificed at 0, 5, 7 or 9 days from the beginning of the experiment. The colons were removed and their lengths measured from the anus to the beginning of the caecum, as an indirect marker of inflammation, followed by histopathological analysis as above [27].
The clinical course of the disease was followed daily and represented by a colitis clinical score as previously described [27], with some modifications. The colitis clinical score was obtained as the sum of the scores assigned for body weight loss (0, none; 1, 1-5%; 2, 5-10%; 3, 10-20%; 4, more than 20%), stool consistency (0, well formed pellets; 2, pasty and semiformed stools; 4, liquid stools), and the presence or absence of fecal blood (0, negative; 2, light bleeding; 4, marked bleeding). Accordingly, the score can range from 0 (healthy) to 12 (maximal colitis activity).
Histological analysis and immunohistochemistry
For papillomas or TPA treated skins, back skins from sacrificed mice were fixed in 4% buffered formalin for 24h and embedded in paraffin. 4 µm thick sections were stained with haematoxylin and eosin (H&E) and analyzed in blind by a pathologist.
For the colon, Swiss rolls (see above) were fixed in 4% buffered formaldehyde for 24h and subsequently embedded in paraffin [25]. Sections were stained with H&E and analyzed for tumor evaluation and inflammation.
Immunohistochemical analysis were performed on paraffinembedded sections (4 µm), with the following antibodies: anti-CD3 (Neomarkers, RM9107), anti phospho-STAT3 (Tyr705, #9131, Cell Signaling Technology), anti-PCNA (sc-56, Santa Cruz Biotechnology), anti-CD18 (BMA, BioMedicals, Augst, CH), anti-neutrophil (NIMPR14, ab2557, Abcam). Quantification was performed using the Metamorph software (Universal Imaging Corporation, Downingtown, PA, USA), calculating the ratio between the percentage of staining with respect to total tissue for each field.
Tumors quantitation
The quantitation of colon polyps was performed as previously described [7]. Briefly, median sections from the entire Swiss rolls were stained with H&E and scanned using TissueFaxsTM software (TissueGnostics GmbH, Vienna, Austria; www.tissuegnostics.com), and the quantitation of total colon area, tumor area and tumor number performed using the HistoQuestTM (TissueGnostics) software.
Real-time PCR
Total RNA was extracted from 0.5 cm portions of proximal plus distal colon from DSS treated mice using the PureLink Micro-to-Midi total RNA Purification System (Invitrogen, Carlsbad, CA, USA) and its concentration was determined with a NanoDrop™ 1100 (NanoDrop Technologies, Wilmington, DE, USA). Total RNA was reverse-transcribed with a high capacity cDNA reverse transcription kit (#4368813, Applied BioSystem) according to manufacturer’s instruction and amplified with specific primers. Taqman PCR reactions were performed as previously described [28], using the Universal Probe Library system (Roche Italia, Monza, Italy), and quantified with the Molecular Analyst software (Bio-Rad Laboratories). The 18S rRNA pre-developed TaqMan assay (#4319413, Applied Biosystems) was used as an internal control. Expression of the target genes was calibrated against the values obtained in untreated animals (d0).
Specific primers and probes used: IL-6, 5’GCTACCAAACTGGATATAATCAGGA-3’, 5’-CAGGTAGCTATGGTACTCCAGAA-3’, UPL probe #6; MIP-2, 5’-GAAAATCATCCAAAAGATACTGAACA-3’, 5’CTTTGGTTCTTCCGTTGAGG-3’, UPL probe #26; CCL2, 5’-GATCATCTTGCTGGTGAATGAGT-3’, 5’CATCCACGTGTTGGCTCA3’, UPL probe #62; COX2, 5’-GATGCTCTTCCGAGCTGTG-3’, 5’-GGATTGGAACAGCAAGGATTT-3’, UPL probe #45; SOCS3, 5’-atttcgcttcgggactagc-3’, 5’-aacttgcttgggtgaccat-3’, UPL probe #83.
Statistical analysis
An unpaired t test was used to calculate a P value between the specified groups, using the GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). Statistically significant P values and symbols: P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***).
Results
STAT3Δβ/Δβ mice display enhanced skin inflammation upon TPA acute treatment
We have previously shown that STAT3Δβ/Δβ mice are more susceptible to LPS-induced acute inflammation, displaying enhanced production of inflammatory cytokines such as IL-6 and TNFα and increased tissue damage. In order to investigate acute inflammatory responses in the skin, STAT3Δβ/Δβ mice and control littermates were shaved, treated with TPA and sacrificed after 24 or 72 hours for histological analysis. No morphological differences were detected between untreated mice (Figure 1A, 0h). Inflammation was readily detectable at 24 hours after TPA treatment in mice of both genotypes but particularly marked in the STAT3Δβ/Δβ mice, where inflammatory cells can be appreciated to abundantly infiltrate the dermal-epidermal junction, the dermis and, to a minor extent, the subcutaneous adipose tissue (Figure 1A and 1B). Inflammation was significantly decreased at 72 hours in wild type mice while remaining high in the STAT3Δβ/Δβ skins, in particular in the lower dermis and in the subcutaneous adipose tissue. Increased inflammatory infiltrate was also detected by immunostaining with the pan-leukocyte marker CD18 (Figure 1C). As expected, TPA treatment induced cell proliferation, as judged by the increased number of epidermal keratinocyte layers (see Materials and Methods section), as well as by the increased percentage of PCNA+ cells in both the dermis and the epidermis (Figure 1D). Proliferation was however equivalent in mice of the two genotypes. In agreement with the enhanced inflammation, STAT3Δβ/Δβ mice displayed higher phosphorylation levels of STAT3α, particularly in the dermis (Figure 1E). These data suggest that STAT3Δβ/Δβ mice are more susceptible to TPA-induced acute inflammation than their STAT3WT/WT controls.
Figure 1.
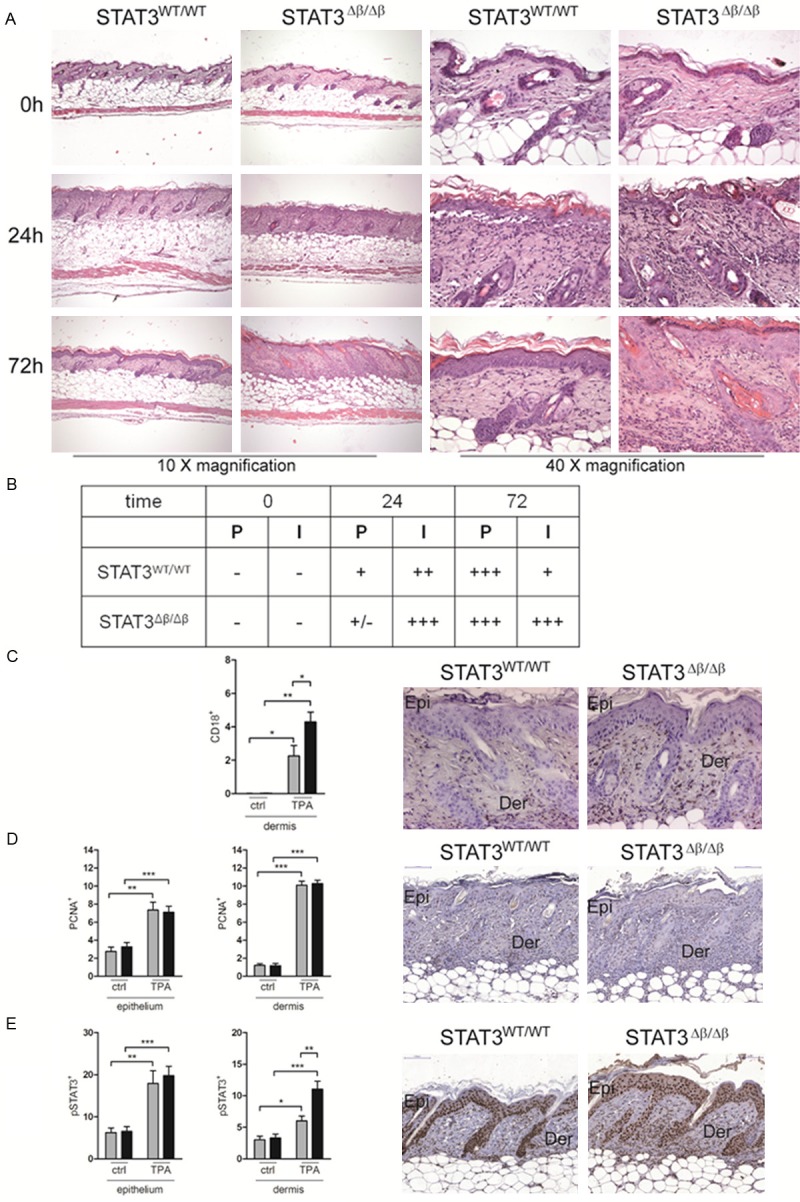
Increased skin inflammation in response to TPA in STAT3Δβ/Δβ mice. The shaved skins of STAT3WT/WT and STAT3Δβ/Δβ mice (0h, n=4; 24h and 72h, n=6 per genotype) topically treated with 0.1 mM TPA were excised at the indicated time points and stained with Haematoxylin and Eosin (H&E) (A). Representative images are shown with the indicated magnifications. (B) The degree of proliferation (P) or inflammation (I) was calculated as described in the Materials and Methods section. Results are representative of two independent experiments each with 3 mice/genotype. (C-E) Representative images (20x) of IHC stainings with anti-CD18 (C), anti-PCNA (D) or anti-phosphoSTAT3 (E) antibodies. Epi, epidermis; Der, dermis. Quantification: the histograms represent the mean±SEM of 3 fields per sample (n=3), where the extent of the positive area over the total area was calculated using the Metamorph software, separately for the epidermis and the dermis. Grey bars, STAT3WT/WT mice; black bars, STAT3Δβ/Δβ mice. The asterisks indicate statistically significant differences between the indicated groups. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Earlier onset of skin papillomas in STAT3Δβ/Δβ mice
To assess whether the enhanced inflammation and the high levels of activation of the STAT3α isoform observed in the STAT3Δβ/Δβ mice in response to TPA correlates with increased susceptibility to inflammation-driven skin cancer, mice were subjected to the DMBA/TPA multi-step carcinogenesis protocol [21]. In the absence of STAT3β, there was an earlier onset of squamous cells papillomas (SCPs) (week 11 versus week 17 after the first TPA exposure). Moreover, while 40% of the STAT3WT/WT mice had not developed SCPs at the end of the experiment (week 42), only 14% of the STAT3Δβ/Δβ mice were still papilloma-free (Figure 2A), suggesting enhanced tumor incidence. However, the number and size of SCPs detected per mouse, although variable, was overall equivalent in mice of the two genotypes (Figure 2B). All lesions displayed the histological features of benign papillomas (Figure 2C), with the exception of one in situ carcinoma detected in one out of 14 STAT3Δβ/Δβ mice analyzed (not shown). These data suggest that although in the ab-sence of STAT3β, tumor formation is accelerated and parallels the enhanced inflammatory response to TPA, the overall tumor progression is apparently not affected.
Figure 2.
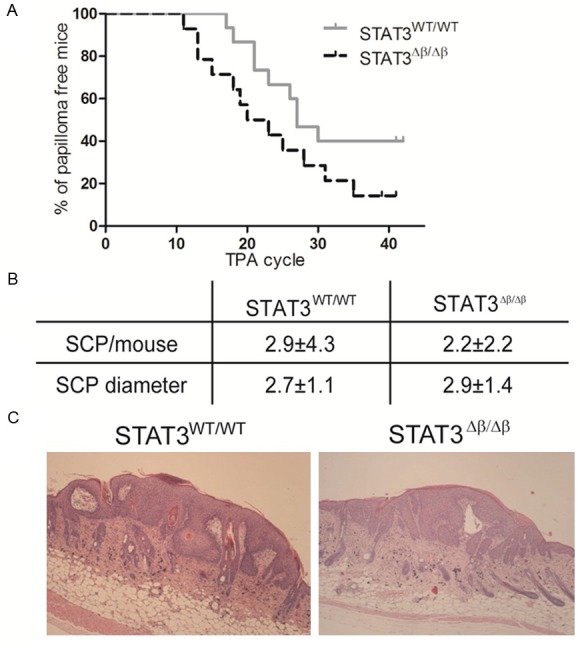
STAT3Δβ/Δβ mice show earlier papilloma onset but unaltered tumor load. STAT3WT/WT and STAT3Δβ/Δβ mice were treated with DMBA/TPA as described and observed for squamous cell papilloma (SCP) development for 42 weeks. A. Shows the percentage of papilloma-free mice. Grey line, STAT3WT/WT mice (n=15); black line, STAT3Δβ/Δβ mice (n=14). B. Shows the number and mean diameter±SD of SCPs per mouse. C. Representative SCPs images from mice of the indicated genotypes.
STAT3Δβ/Δβ mice display exacerbated colitis after DSS treatment
To assess acute responses to DSS-induced colon inflammation, STAT3Δβ/Δβ mice and control littermates were given one five-days cycle of DSS in the drinking water and sacrificed at different time points (day 0, 5, 7 and 9 from the beginning of treatment). STAT3Δβ/Δβ mice were significantly more susceptible to acute DSS-induced inflammation. Shortening of the colon, a typical colitis marker, was significantly more pronounced at days 5 and 7 (Figure 3A), correlating with a significantly higher colitis clinical score, which considers bleeding, weight loss and stool consistency as described in the Materials and Methods section (Figure 3B). Increased inflammation was also confirmed by the higher expression levels of mRNAs encoding pro-inflammatory markers such as IL-6, MIP2, CCL2 and COX2 (Figure 3C). Expression of the classical STAT3 target gene SOCS3, a negative regulator of the JAK-STAT pathway, was also enhanced (Figure 3C). Interestingly, by day 9 both colon length and clinical score improved becoming comparable to those of the wild type controls, suggesting resolution of the inflammation. Immunohistochemical analysis could clearly show significantly enhanced infiltration of both CD3+ T lymphocytes and Gr1+ granulocytes in colons derived from STAT3Δβ/Δβ mice at day 5 of treatment, confirming higher inflammation (Figure 4A and 4B). Interestingly, the number of cells expressing activated STAT3α, initially lower in the STAT3Δβ/Δβ mice, increased sharply upon DSS treatment to become equivalent to those detected in the wild type littermates (Figure 4C, see fold induction). Finally, DSS-induced proliferation was similar in mice of the two genotypes, as shown by equivalent numbers of PCNA+ cells (Figure 4D).
Figure 3.
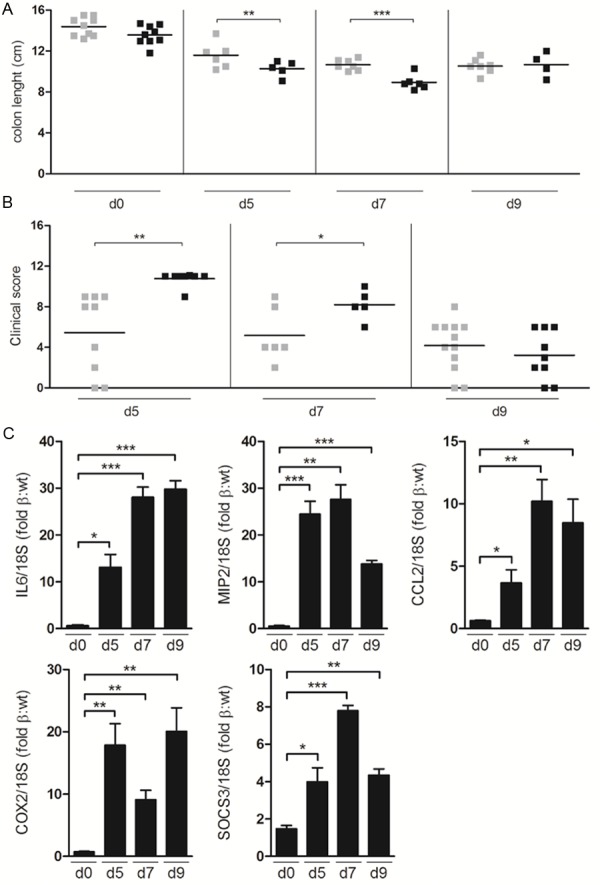
Exacerbated DSS-induced colitis in STAT3Δβ/Δβ mice. Mice treated for five days with 2.5% DSS in the drinking water were sacrificed at different times (day (d) 0, 5, 7, 9). Colon length (A) and clinical score (B) were evaluated as described in the Materials and Methods section. Colon length, STAT3WT/WT mice, n= 9 (d0), n=6 (d5), n=7 (d7 and d9); STAT3Δβ/Δβ mice, n=9 (d0), n=5 (d5), n=6 (d7) and n=4 (d9). Clinical score, STAT3WT/WT mice, n=9 (d5), n=6 (d7) and n=12 (d9); STAT3Δβ/Δβ mice, n=8 (d5), n=5 (d7) and n=9 (d9). (C) Taqman PCR analysis of the expression of IL-6, MIP-2, CCL2, COX2, and SOCS3, normalized to 18S RNA as an internal control, in the DSS-treated colons. Data are expressed as induction fold of STAT3Δβ/Δβ respect to STAT3WT/WT mice, and are the mean±SEM of 3 samples/genotype. Asterisks indicate statistically significant differences between the indicated goups. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 4.
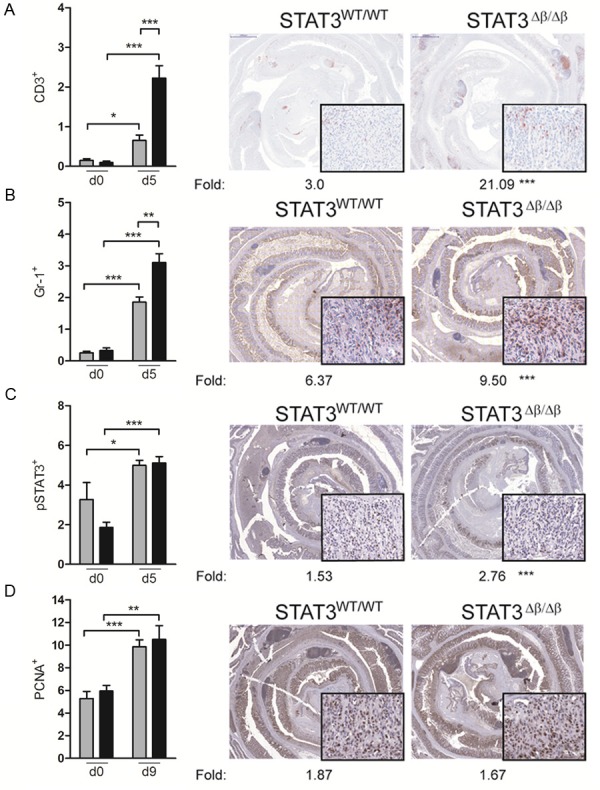
Increased DSS-induced inflammation in STAT3Δβ/Δβ mice. Colon sections from acutely DSS treated mice (see legend to Figure 3) were stained with the indicated antibodies to detect infiltrating CD3+ T lymphocytes (A) Gr1+ granulocytes (B), p-Y-STAT3+ (C) or PCNA+ (D) cells. Representative images are shown at low magnification (2x), with 20x insets. The histograms represent the mean±SEM of 6 fields per sample where the extent of the positive area over the total epidermal area was calculated using the Metamorph software. Grey bars, STAT3WT/WT mice (n=3); black bars, STAT3Δβ/Δβ mice (n=3). The induction fold between the values obtained from treated and untreated mice is indicated below the images. Note that d9 is shown for the PCNA staining because no increase was detected at d5. Asterisks indicate statistically significant differences between the percentages of positive cells (histograms) or the induction folds (pictures) of the indicated groups. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Colitis-driven carcinogenesis in STAT3WT/WT and STAT3Δβ/Δβ mice
In order to assess whether the absence of STAT3β, resulting in the observed increased sensitivity to DSS-induced colon inflammation, could enhance susceptibility to colitis associated cancer (CAC), we took advantage of the AOM/DSS CAC model [20,26], injecting mice of both genotypes with AOM, followed by three five-days cycles of DSS in the drinking water (Figure 5A). In a first round of experiments, mice were sacrificed at day 60 for the analysis of tumor load. Histological analysis showed that at this stage 6 out of 18 (33.4%) STAT3WT/WT mice were still tumor-free, against only 2 out of 12 (16.7%) STAT3Δβ/Δβ mice (Figure 5B). This increased incidence was likely due to an earlier onset of tumor lesions in the absence of STAT3β, since both tumor number and tumor area per mouse were comparable between animals of the two genotypes, suggesting equivalent grown rates and progression (Figure 5C and 5D). Accordingly, the penetrance was 100% in mice of both genotypes when a second round of experiments was performed where mice were sacrificed 20 days later, and again no differences in tumor burden were observed (Figure 5C and 5D). Histological analysis performed on colon samples at day 80 showed no difference in tumor morphology between the two genotypes (Figure 5E and 5F). Thus, the stronger inflammatory response to DSS displayed by the STAT3Δβ/Δβ mice correlated with an earlier tumor onset but did not affect overall tumor progression, similar to what observed in the DMBA/TPA skin carcinogenesis model.
Figure 5.
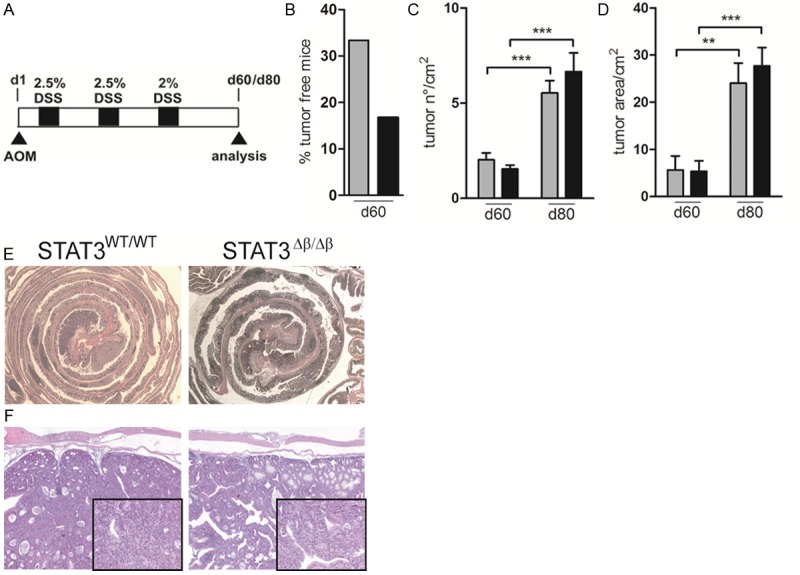
AOM/DSS treatment triggers earlier colonic tumor onset in STAT3Δβ/Δβ mice. (A) Schematic overview of the experimental model. Mice were injected once i.p. with AOM (day 1, 12.5 mg/kg), and received the indicated percentage of DSS in the drinking water for three five-days cycles starting from five days later (black squares), with 2 weeks regular water intervals. Mice were then sacrificed at day 60 or day 80, as indicated. (B) Percentage of tumor free mice of the two genotypes at day 60 of treatment. (C, D) Mean±SEM of the number of tumors per square centimeter (C) and of the tumor area (D) in mice sacrificed at d60 and d80, as indicated. Grey bars, STAT3WT/WT, n=12 (d60), n=13 (d80); black bars, STAT3Δβ/Δβ;, n=13 (d60), n=11 (d80). The asterisks indicate statistically significant differences between the indicated groups. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. (E, F) Representative images of H&E staining on colon samples from mice of the indicated genotypes at a magnification of 2.5X (E) and 20X (F).
Discussion
Tumor-promoting inflammation has been recently recognized as an important enabling characteristic of cancer, contributing to multiple hallmark features of tumor cells by releasing mutagenic reactive oxygen species, as well as by supplying extrinsic factors that help cell survival and growth, thus promoting tumor initiation. On the other hand, the intrinsic inflammatory programs elicited by oncogenic mutations in tumor cells contribute shaping a tumor-promoting microenvironment rich in factors that induce cell survival, proliferation, angiogenesis, motility and invasion, thus stimulating tumor progression [15,16].
Both IL-6 and its main downstream mediator STAT3 have been identified as main players in regulating tumor-associated inflammation and the cross-talk between the microenvironment and tumor cells [4,29,30]. In particular, STAT3 deletion in either keratinocytes or intestinal epithelial cells was shown to protect mice from tumor development in both the DMBA/TPA- and AOM/DSS-mediated carcinogenesis models of the skin and colon [18,19,22,23]. STAT3 acted at multiple levels in both models, mediating both survival to the mutagenic insult (tumor initiation stage) and subsequent proliferation (tumor promotion stage). Although the relative contribution of the two STAT3 isoforms, alpha and beta, to inflammation-driven tumor onset and progression has never been assessed, STAT3β has long been considered as a dominant negative form of the pro-oncogenic activities of the alpha isoform [4]. On the other hand, while STAT3α can act both as a pro- and anti-inflammatory factor, depending on the activating signal, STAT3β functions appear to be prominently anti-inflammatory [9,10,14]. This is confirmed by our observation that mice specifically lacking the STAT3β isoform display exacerbated acute inflammatory responses to both TPA and DSS. Indeed, a single application of TPA was sufficient to induce more abundant and longer lasting inflammatory skin infiltration in STAT3Δβ/Δβ mice, correlating with a higher increase of cells displaying tyrosine-phosphorylated STAT3 in the dermis with respect to wild type mice. Proliferation, which usually follows damage/inflammation to favour healing and is considered as a fundamental step in the tumor-promoting effects of TPA, was however equivalent in both the dermis and epithelium of mice of the two genotypes. Likewise, STAT3Δβ/Δβ mice exhibited enhanced colitis symptoms and significantly more extensive infiltration of both granulocytes and T lymphocytes in the colon mucosa as compared to their wild type littermates upon a single DSS cycle. This is reminiscent of the more dramatic DSS-induced colitis observed in mice lacking STAT3 in the intestinal epithelial cells, which may therefore be at least partly due to the lack of the beta isoform. Under this scenario of increased inflammation and high STAT3α activity, a higher tumor incidence/burden upon application of the carcinogenesis protocols (DMBA/TPA and AOM/DSS) could be expected. Indeed, in both models a trend towards an earlier onset of tumor lesions was observed, likely as a consequence of the higher concentration of inflammatory mediators in the absence of STAT3β. Accordingly, we have recently shown that even low levels of constitutive STAT3α activation in the absence of STAT3β can serve as a first hit in the process of malignant transformation, making the cells exquisitely sensitive to mutagenic insults [31]. However, the initial enhanced inflammation was rapidly normalized, and the overall tumor burden per mouse and the mean tumor size/tumor area were similar in the presence or absence of STAT3β, in line with the equivalent proliferation levels observed. These results suggest that also in cancer STA3β is not a physiological dominant negative factor for STAT3α unless it is overexpressed. Thus, while confirming the potent anti-inflammatory role of STAT3β and highlighting its potential oncosuppressive role in the initiation phases of skin and colon tumorigenesis, our data suggest that the effects of the activation of STAT3α in the absence of the counteracting STAT3β activity are not as dramatic as could be expected, in particular in driving tumor progression. For CAC, this observation may be partly explained by the observation that the colons of DSS-treated STAT3Δβ/Δβ mice displayed higher expression levels of the JAK-STAT negative feedback factor SOCS3 (Figure 3C), a well-recognized specific STAT3α target that is believed to act as an oncosuppressor in CAC [10,32]. Thus, elevated SOCS3 levels in the STAT3Δβ/Δβ mice could be involved in counterbalancing the potentially pro-oncogenic effects of higher inflammation and unchecked STAT3α activity.
Fine tuning of the degree and length of inflammation is fundamental to warrant resolution without promoting tumorigenesis. Our results suggest that the balance between the activated alpha and beta isoforms of STAT3, determined by the interplay between the complex signaling networks activated in the tumor microenvironment, plays an important role in shaping the inflammatory responses and has a major impact on the initial degree of malignant transformation.
Acknowledgements
We wish to thank Dr. Paolo Provero for help with statistical analysis. This work was supported by the Italian Association for Cancer Research (AIRC, IG 13009 to VP, IG-12023 to RC), the Ateneo San Paolo Foundation and the Truus and Gerrit van Riemsdijk Foundation, Liechtenstein (grants to VP), the Piedmont Region, Converging Technologies Platform (EC and VP), the FP7 ERC-2009-StG (Proposal No. 242965- “Lunely” to RC); the International Association for Cancer Research (AICR) (grant 12-0216 to RC), the Telethon Foundation (grant TCP 06001 to EC), and a FWF DK-plus grant IAI, a CCC research grant, the FWF grant P25925-B20 and the FWF grant P24802 to RE.
Disclosure of conflict of interest
None.
Abbreviations
- STAT
Signal Transducer and Activator of Transcription
- DMBA
Dimethylbenzanthracene
- TPA
12-O-tetradecanoylphorbol-13-acetate
- AOM
azoxymethane
- DSS
Dextran Sulfate Sodium
- CAC
Colitis-Associated Cancer
- SOCS
Suppressor of Cytokine Signaling
References
- 1.Avalle GRaVP L. Universal and Specific Functions of STAT3 in Solid Tumours. In: Thomas Decker MM, editor. Jak-Stat Signaling: From Basics to Disease. Vienna: Springer Vienna; 2012. pp. 305–333. [Google Scholar]
- 2.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demaria M, Camporeale A, Poli V. STAT3 and metabolism: How many ways to use a single molecule? Int J Cancer. 2014;135:1997–2003. doi: 10.1002/ijc.28767. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pilati P, Mocellin S, Bertazza L, Galdi F, Briarava M, Mammano E, Tessari E, Zavagno G, Nitti D. Prognostic value of putative circulating cancer stem cells in patients undergoing hepatic resection for colorectal liver metastasis. Ann Surg Oncol. 2012;19:402–408. doi: 10.1245/s10434-011-2132-2. [DOI] [PubMed] [Google Scholar]
- 6.Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simoes M, Lima J, Maximo V, Soares P, Lyden D, Bromberg JF. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:E2361–2370. doi: 10.1073/pnas.1201232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musteanu M, Blaas L, Mair M, Schlederer M, Bilban M, Tauber S, Esterbauer H, Mueller M, Casanova E, Kenner L, Poli V, Eferl R. Stat3 is a negative regulator of intestinal tumor progression in Apc (Min) mice. Gastroenterology. 2010;138:1003–1011. e1001–1005. doi: 10.1053/j.gastro.2009.11.049. [DOI] [PubMed] [Google Scholar]
- 8.Dewilde S, Vercelli A, Chiarle R, Poli V. Of alphas and betas: distinct and overlapping functions of STAT3 isoforms. Front Biosci. 2008;13:6501–6514. doi: 10.2741/3170. [DOI] [PubMed] [Google Scholar]
- 9.Yoo JY, Huso DL, Nathans D, Desiderio S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331–344. doi: 10.1016/s0092-8674(02)00636-0. [DOI] [PubMed] [Google Scholar]
- 10.Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401–409. doi: 10.1038/ni1052. [DOI] [PubMed] [Google Scholar]
- 11.Ng IH, Ng DC, Jans DA, Bogoyevitch MA. Selective STAT3-alpha or -beta expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem J. 2012;447:125–136. doi: 10.1042/BJ20120941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zammarchi F, de Stanchina E, Bournazou E, Supakorndej T, Martires K, Riedel E, Corben AD, Bromberg JF, Cartegni L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U S A. 2011;108:17779–17784. doi: 10.1073/pnas.1108482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ecker A, Simma O, Hoelbl A, Kenner L, Beug H, Moriggl R, Sexl V. The dark and the bright side of Stat3: proto-oncogene and tumor-suppressor. Front Biosci (Landmark Ed) 2009;14:2944–2958. doi: 10.2741/3425. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Baldwin W, Lee C, Desiderio S. Stat3β mitigates development of atherosclerosis in apolipoprotein E-deficient mice. J Mol Med (Berl) 2013;91:965–976. doi: 10.1007/s00109-013-1013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2011;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, Matthews V, Schmid RM, Kirchner T, Arkan MC, Ernst M, Greten FR. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karim BO, Huso DL. Mouse models for colorectal cancer. Am J Cancer Res. 2013;3:240–250. [PMC free article] [PubMed] [Google Scholar]
- 21.Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009;4:1350–1362. doi: 10.1038/nprot.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kataoka K, Kim DJ, Carbajal S, Clifford JL, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis. 2008;29:1108–1114. doi: 10.1093/carcin/bgn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 25.Chae S, Eckmann L, Miyamoto Y, Pothoulakis C, Karin M, Kagnoff MF. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J Immunol. 2006;177:1214–1220. doi: 10.4049/jimmunol.177.2.1214. [DOI] [PubMed] [Google Scholar]
- 26.Fritsch Fredin M, Vidal A, Utkovic H, Gotlind YY, Willen R, Jansson L, Hultgren Hornquist E, Melgar S. The application and relevance of ex vivo culture systems for assessment of IBD treatment in murine models of colitis. Pharmacol Res. 2008;58:222–231. doi: 10.1016/j.phrs.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Siegmund B, Fantuzzi G, Rieder F, Gamboni-Robertson F, Lehr HA, Hartmann G, Dinarello CA, Endres S, Eigler A. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1264–1273. doi: 10.1152/ajpregu.2001.281.4.R1264. [DOI] [PubMed] [Google Scholar]
- 28.Schiavone D, Dewilde S, Vallania F, Turkson J, Di Cunto F, Poli V. The RhoU/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem J. 2009;421:283–292. doi: 10.1042/BJ20090061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 2011;19:429–431. doi: 10.1016/j.ccr.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demaria M, Misale S, Giorgi C, Miano V, Camporeale A, Campisi J, Pinton P, Poli V. STAT3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ. 2012;19:1390–1397. doi: 10.1038/cdd.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833–4841. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]