

Abstract
GATA-1, an X-linked gene, encodes a transcription factor that plays a role in erythropoiesis and megakaryopoiesis. GATA-1 mutations have been associated with various diseases, such as X-linked thrombocytopenia. ALAS2 is an X-linked erythroid-specific isoenzyme expressed during erythropoiesis. Mutations of ALAS2 were associated with X-linked sideroblastic anemia. We report a case of newborn twin boy with anemia and thrombocytopenia at birth. A bone marrow biopsy at 4 months of age showed marked dyserythropoiesis, dysmegakaryopoiesis, and rare ringed sideroblasts. Gene sequencing study showed a previously reported mutation in GATA-1 at c.622G>A location (G208R) and a novel ALAS2 mutation at c.1436G>A location (R479Q).
Keywords: GATA1 mutation, ALAS2 mutation, macrothrombocytopenia, dysmegakaryopoiesis, dyserythropoiesis, ringed sideroblasts
Introduction
GATA1 is an X-linked gene that encodes a transcription factor that regulates the expression of a very large number of genes in many cell types. It contains a DNA binding domain and a transactivator domain. The N-finger in the DNA binding domain plays an important role in GATA1’s activity, both by binding DNA to increase the stability, and by recruiting cofactors, such as Friend of GATA1 (FOG-1), which is important for erythropoiesis and megakaryopoiesis [1-3]. Cases associated with transient myeloproliferative disease and acute megakaryocytic leukemia in children with Down syndrome have somatic mutations in exon 2 of the GATA-1 gene, resulting in the short isoform, GATA-1s [4]. Germline missense mutations in exon 4 of GATA-1 that lead to alterations in various amino acids of the N-terminal zinc-finger domain have been linked to various diseases, such as X-linked thrombocytopenia and anemia and congenital erythropoietic porphyria [5-9].
ALAS, 5-aminolevulinate synthase, catalyzes the formation of 5-aminolevulinate from succinyl CoA and glycine. It is a rate-limiting mitochondrial enzyme. The ALAS2 is an erythroid-specific isoenzyme that is expressed during erythropoiesis [10]. The ALAS2 mutations known are heterogeneous and clustered in the exons that encode the catalytic domain.
A case of a newborn male with anemia and thrombocytopenia with concomitant GATA1 and a novel ALAS2 mutation is reported.
Case presentation
The patient was a male born at 36 weeks gestation as the product of a dichorionic diamniotic twin pregnancy resulting from non-consanguinous parents. At birth, CBC showed anemia and thrombocytopenia with hemoglobin of 10.2 g/dL, reticulocyte count of 7.9%, and platelet count of 22,000/µL. Physical examination was significant for hepatosplenomegaly and “blueberry muffin” rashes. There were no dysmorphic features or skeletal abnormalities. Work-up for infections and genetic diseases was negative. Review of the placenta showed immature placenta without acute or chronic villitis and viral inclusions. There were many nucleated RBCs within the villi. The co-twin placenta was mature and unremarkable.
The patient required several packed red cell and platelet transfusions within the first month of life. The first bone marrow aspirate was done at 2 months of age and showed pauci-spicular bone marrow with marked erythroid hypoplasia and mild dyserythropoiesis. The megakaryopoiesis was unremarkable without obvious dysmegakaryopoiesis. Few early myeloid precursors contained cytoplasmic vacuoles. An iron stain showed rare ringed sideroblasts (Figure 1A). At the time, sideroblastic anemia, especially Pearson syndrome, was considered but ruled out when mitochondrial DNA deletion was not detected. A repeat bone marrow biopsy was performed at 4 months of age due to persistent anemia and thrombocytopenia requiring multiple packed red cell and platelet transfusions (Figure 2). CBC at this time showed anemia and macrothrombocytopenia (Table 1). The RBCs on the peripheral smear showed marked anisopoikilocytosis with coarse basophilic stippling and many large agranular platelets (Figure 1B). The bone marrow aspirate showed marked dyserythropoiesis. The megakaryocytes were dysplastic with small, eccentric crescent shaped nuclei and clear cytoplasm. An X-linked GATA1 gene sequencing study was performed (Prevention Genetics, Marshfield, WI) and showed a missense mutation at c.622 G>A location resulting in a glycine to arginine amino acid change (G208R). In addition, a comprehensive mitochondrial nuclear gene panel was performed (GeneDX, Gaithersburg, MD) and showed a novel ALAS2 mutation at c.1438 G>A resulting in an arginine to glutamine amino acid change (R479Q). This variant has not been published as a mutation, nor has it been reported as a benign polymorphism.
Figure 1.
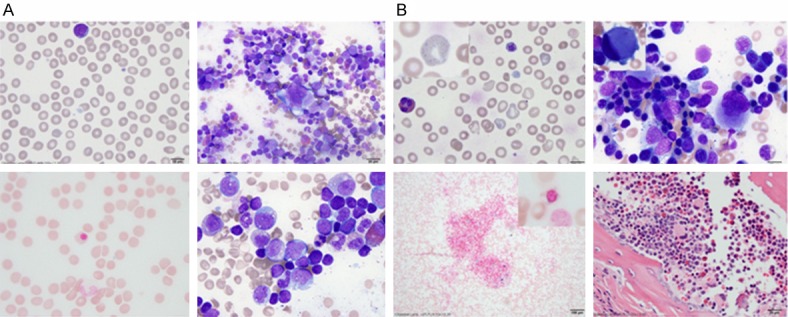
Comparison of peripheral blood and bone marrow morphology at 2 months (A) and 4 months of age (B). In panel A: Top left: Peripheral blood showed normocytic RBCs without anisopoikiocytosis. A very rare agranular large platelet is seen (MPV 10.3). Top right: Bone marrow aspirate showed trilineage hematopoiesis with marked erythroid hypoplasia. The megakaryocytes appeared unremarkable. Bottom left: Iron stain on the bone marrow aspirate showed a rare ringed sideroblast. Bottom right: Occasional early stage myeloid cells contained small cytoplasmic vacuoles. In panel B: Top left: Peripheral blood showed normocytic anemia with marked anisopoikilocytosis. The insert showed the presence of one of the many RBCs with coarse basophilic stippling. There were many giant agranular platelets (MPV 12.9). Top right: The bone marrow aspirate showed moderate dyserythropoiesis with increased monolocated/hypolobated megakaryocytes with eccentrically located nucleus (insert). Bottom left: The bone marrow showed increased iron storage for age with rare ringed sideroblasts (insert). Bottom right: The bone marrow core biopsy showed the presence of the small megakaryocytes with crescent shaped eccentrically located nuclei.
Figure 2.
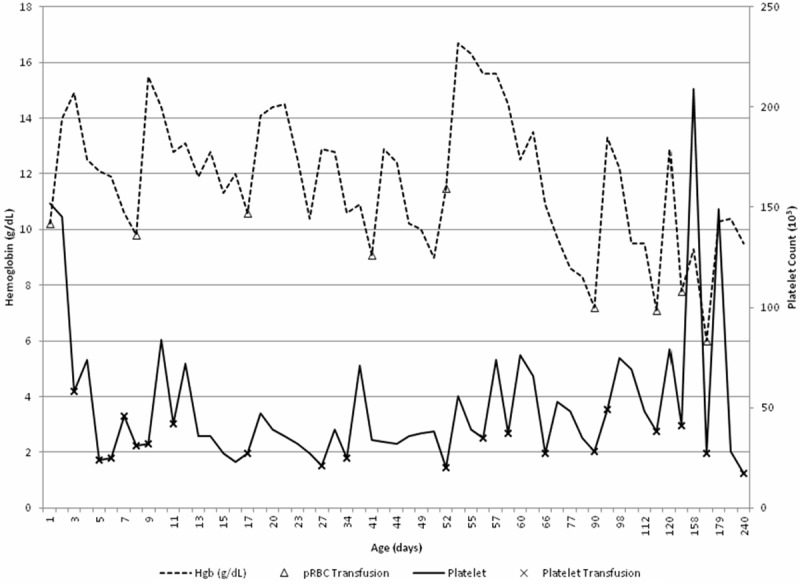
Hemoglobin, platelet count and transfusion requirements over time. The patient’s platelet count (solid line) and hemoglobin concentration (dashed line) over time until the age of 240 days (8 months) are shown. Transfusion of packed red cells (Δ) and platelets (x) over time are also displayed on these curves.
Table 1.
CBC and bone marrow findings at initial and subsequent bone marrow biopsies
| 2 month old (normal range) | 4 month old (normal range) | ||
|---|---|---|---|
| CBC | WBC (×103/µl) | 16.31 (5.0-19.5) | 23.46 (5.0-19.5) |
| Hgb (g/dl) | 10.9 (9.4-13.0) | 7.1 (9.4-13.0) | |
| MCV (fl) | 91.7 (84-106) | 98.5 (84-106) | |
| Platelet (×103/µl) | 27 (150-400) | 38 (150-400) | |
| MPV (fl) | 10.3 (7.0-11.3) | 12.9 (7.0-11.3) | |
| Bone marrow | Erythropoiesis | Decreased; Mild dysplasia | Predominant; Marked Dysplasia |
| Granulopoiesis | Normal | Normal | |
| Megakaryopoiesis | Mostly normal | Dysplasia |
At the age of one, the patient continues to have persistent thrombocytopenia with platelet counts averaging 30,000 and mild anemia (Figure 2). He continues to have significant bleeding episodes requiring regular platelet transfusions. Of note, this patient’s twin lacks the GATA-1 mutation as well as any anemia or thrombocytopenia. The twin’s ALAS2 mutation status is unknown at this time.
Discussion
GATA1, an X-linked gene, is the founding member of the GATA transcription factor family. It is primarily expressed in hematopoietic tissues and plays an important role in erythropoiesis and megakaryopoiesis [12-14]. There seems to be a dose-related effect: erythroid precursors can mature at low levels of GATA1 whereas megakaryocytes need higher levels. In murine studies, deletion of GATA1 has shown a compensatory increase in GATA2, which may explain why these proerythroblasts express some genes at wild type levels such as beta globin H1 and alpha globin [3]. GATA1 deficient megakaryocytes are more numerous, smaller, have larger and segmented nuclei, have a paucity of alpha granules, do not express GPIb and stain weaker with acetylcholinesterase [3,15]. In essence, these megakaryocytes are developmentally arrested and proliferate excessively [16].
Our case demonstrated the presence of previously described missense mutation that led to G208R substitution within the N-finger of GATA-1 [5]. It was thought that this mutation reduces the hydrophilic interaction by substitution of a positively charged amino acid, which caused more profound destabilization of the GATA1 and FOG-1 interaction, than other amino acid substitutions at the same location. Thus it was hypothesized that a patient carrying this mutation would have a more severe clinical presentation. This mutation was initially identified in a 17 year old boy, the third child born from the family [5]. Subsequently, there have been two reported cases of the same mutation, one who on bone marrow analysis at age 12 had 5% ringed sideroblasts, but without ringed sideroblasts in the other [17,18]. Some patients also carried a diagnosis of idiopathic thrombocytopenia purpura prior and some had splenectomy.
The finding of co-existing previously unknown hemizygous R479Q mutation in the ALAS2 gene is interesting. Mutations in the ALAS2 gene are linked to X-linked hereditary sideroblastic anemia [19]. The patient with known mutations mainly presented with microcytic anemia, iron overload and ringed sideroblasts. However, the R479Q mutation identified in our patient is a novel variant at a conserved region. Although in-silico analysis indicates that this mutation is unlikely to be damaging to the ALAS2 protein (per GeneDx report), a functional study might be helpful in further understanding this mutation. Continued clinical follow up might be helpful as some patients might present in late childhood or even in late adulthood [20]. Clinically, our patient has normocytic anemia initially, and later macrocytic anemia without evidence of iron overload.
The relationship of GATA1 mutation and ALAS2 mutation in our patient is unknown. Studies have shown that GATA1 binds to ALAS2 at a GATA consensus sequence in intron 8 of the gene [21], presumably to regulate heme synthesis. The mutation in this patient is located in the coding region in ALAS2 gene, not the GATA1 binding sequence. While the patient’s mutation in ALAS2 alone may not affect protein function, it remains to be determined what kind of interaction there may be between the dysfunctional GATA1 protein and the ALAS2 protein. It is possible that the hemizygous GATA1 mutation plays an insignificant role in the ALAS2 regulated heme synthesis, as long as the ALAS2 mutation does not affect the protein function.
Another interesting aspect of this case is that the initial bone marrow biopsy at 2 months of age did not reveal typical morphologic features that are suggestive of a typical GATA1 mutation. The reasons for this are not clear. Transfusions and the underlying medical conditions in the perinatal period might mask the peripheral blood and bone marrow findings. In addition, the role of GATA1 and ALAS2 in fetal and postnatal hematopoiesis is not well understood. This case presents the challenge in diagnosing certain genetic disorders in hematopoiesis in a newborn.
Disclosure of conflict of interest
The authors have no conflicts of interest to disclose.
References
- 1.Tsang AP, Fujiwara Y, Hom DB, Orkin SH. Failure of megakaryopoiesis and arrested erythropoiesis in mice lacking the GATA-1 transcriptional cofactor FOG. Genes Dev. 1998;12:1176–1188. doi: 10.1101/gad.12.8.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wall L, deBoer E, Grosveld F. The human beta-globin gene 3’ enhancer contains multiple binding sites for an erythroid-specific protein. Genes Dev. 1988;2:1089–1100. doi: 10.1101/gad.2.9.1089. [DOI] [PubMed] [Google Scholar]
- 3.Crispino JD. GATA1 in normal and malignant hematopoiesis. Semin Cell Dev Biol. 2005;16:137–147. doi: 10.1016/j.semcdb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nature Genet. 2002;32:148–152. doi: 10.1038/ng955. [DOI] [PubMed] [Google Scholar]
- 5.Del Vecchio GC, Giordani L, De Santis A, De Mattia D. Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol. 2005;114:113–116. doi: 10.1159/000086586. [DOI] [PubMed] [Google Scholar]
- 6.Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, Weiss MJ. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24:266–270. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehaffey MG, Newton AL, Gandhi MJ, Crossley M, Drachman JG. X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood. 2001;98:2681–2688. doi: 10.1182/blood.v98.9.2681. [DOI] [PubMed] [Google Scholar]
- 8.Balduini CL, Pecci A, Loffredo G, Izzo P, Noris P, Grosso M, Bergamaschi G, Rosti V, Magrini U, Ceresa IF, Conti V, Poggi V, Savoia A. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb Haemost. 2004;91:129–140. doi: 10.1160/TH03-05-0290. [DOI] [PubMed] [Google Scholar]
- 9.Phillips JD, Steensma DP, Pulsipher MA, Spangrude GJ, Kushner JP. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood. 2007;109:2618–2621. doi: 10.1182/blood-2006-06-022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.May BK, Dogra SC, Sadlon TJ, Bhasker CR, Cox TC, Bottomley SS. Molecular regulation of heme biosynthesis in higher vertebrates. Prog Nucleic Acid Res Mol Biol. 1995;51:1–51. doi: 10.1016/s0079-6603(08)60875-2. [DOI] [PubMed] [Google Scholar]
- 11.Bottomley SS, May BK, Cox TC, Cotter PD, Bishop DF. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J Bioenerg Biomembr. 1995;27:161–168. doi: 10.1007/BF02110031. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci U S A. 1996;93:12355–12358. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood. 1999;93:2867–2875. [PubMed] [Google Scholar]
- 14.Shivdasani RA, Fujiwara Y, McDevitt MA, Orkin SH. A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 1997;16:3965–3973. doi: 10.1093/emboj/16.13.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millikan PD, Balamohan SM, Raskind WH, Kacena MA. Inherited thrombocytopenia due to GATA-1 mutations. Semin Thromb Hemost. 2011;37:682–689. doi: 10.1055/s-0031-1291378. [DOI] [PubMed] [Google Scholar]
- 16.Stachura DL, Chou ST, Weiss MJ. Early block to erythromegakaryocytic development conferred by loss of transcription factor GATA-1. Blood. 2006;107:87–97. doi: 10.1182/blood-2005-07-2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dührsen U, Kratz CP, Flotho C, Lauenstein T, Bommer M, König E, Brittinger G, Heimpel H. Long-term outcome of hemizygous and heterozygous carriers of a germline GATA1 (G208R) mutation. Ann Hematol. 2011;90:301–306. doi: 10.1007/s00277-010-1088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kratz CP, Niemeyer CM, Karow A, Volz-Fleckenstein M, Schmitt-Graff A, Strahm B. Congenital transfusion-dependent anemia and thrombocytopenia with myelodysplasia due to a recurrent GATA1 (G208R) germline mutation. Leukemia. 2008;22:432–434. doi: 10.1038/sj.leu.2404904. [DOI] [PubMed] [Google Scholar]
- 19.Bottomley SS, May BK, Cox TC, Cotter PD, Bishop DF. Molecular defects of erythroid 5-aminolevulinate synthase in X-linked sideroblastic anemia. J Bioenerg Biomembr. 1995;27:161–168. doi: 10.1007/BF02110031. [DOI] [PubMed] [Google Scholar]
- 20.Cotter PD, Baumann M, Bishop DF. Enzymatic defect in “X-linked” sideroblastic anemia: molecular evidence for erythroid delta-aminolevulinate synthase deficiency. Proc Natl Acad Sci U S A. 1992;89:4028–4032. doi: 10.1073/pnas.89.9.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surinya KH, Cox TC, May BK. Identification and characterization of a conserved erythroid-specific enhancer located in intron 8 of the human 5-aminolevulinate synthase 2 gene. J Biol Chem. 1998;273:16798–16809. doi: 10.1074/jbc.273.27.16798. [DOI] [PubMed] [Google Scholar]