

Abstract
Purpose
A diagnostic feature of focal cortical dysplasia (FCD) type II on magnetic resonance imaging (MRI) is increased subcortical white matter (WM) signal on T2 sequences corresponding to hypomyelination, the cause of which is unknown. We aimed to quantify WM pathology in FCD type II and any deficiency in the numbers and differentiation of oligodendroglial (OL) cell types within the dysplasia.
Methods
In 19 cases we defined four regions of interests (ROIs): ROI1 = abnormal WM beneath dysplasia, ROI2 =dysplastic cortex, ROI3 = normal WM, and ROI4 = normal cortex. We quantified axonal and myelin density using immunohistochemistry for neurofilament, myelin basic protein and quantified mature OL with NogoA, cyclic nucleotide 3-phosphodiesterase (CNPase) and OL precursor cell (OPC) densities with platelet derived growth factor receptor (PDGFR)α, β and NG-2 in each region.
Key Findings
We observed a significant reduction in myelin and axons in the WM beneath dysplasia relative to normal WM and there was a correlation between relative reduction of myelin and neurofilament in each case. OL and OPC were present in the WM beneath dysplasia and although present in lower numbers with most markers, were not significantly different from normal WM. Neurofilament and myelin labeling highlighted disorganized orientation of fibers in dysplastic cortex but there were no significant quantitative differences compared to normal cortex. Clinical correlations showed an association between the severity of reduction of myelin and axons in the WM of FCD and duration of epilepsy.
Significance
These findings indicate a reduction of myelinated axons in the WM of FCD type II rather than dysmyelination as the primary pathologic process underlying WM abnormalities, possibly influenced by duration of seizures. The range of OPC to OL present in FCD type II does not implicate a primary failure of cell recruitment and differentiation of these cell types in this pathology.
Keywords: Focal cortical dysplasia type II, White matter, Myelination, Oligodendroglia
In the first descriptions of the neuropathology now known as focal cortical dysplasia type II (FCD II), Corsellis and Bruton noted that the adjacent white matter (WM) was poorly myelinated (Taylor et al., 1971). Despite numerous subsequent histopathologic studies based on epilepsy surgical series, this component of the pathology, in particular with regard to the origin of the reduced myelin, has remained relatively unexplored. (Blumcke et al., 2011).
Diagnostic magnetic resonance imaging (MRI) features of FCD II take into account WM abnormalities, visualized as blurring of the gray-white interface or increased subcortical signal on T2 and fluid-attenuated inversion recovery (FLAIR) images (Urbach et al., 2002; Blumcke et al., 2011). FCD II on MRI can be limited to the bottom of a sulcus (Barkovich et al., 1997), with local increased WM signal intensity (Hofman et al., 2011), or form an extensive “transmantle dysplasia” where abnormal signal extends to the margin of the ventricle (Barkovich et al., 1997). Furthermore, in some pathology-proven cases of FCD II, MRI changes are subtle or overlooked (Oster et al., 2012; Regis et al., 2011). These observations suggest that the extent of WM pathology within the spectrum of FCD II lesions is highly variable. Diffusion tensor imaging (DTI) studies in FCD have aimed to specifically address the extent of WM pathology (Eriksson et al., 2001; Widjaja et al., 2007; Diehl et al., 2010), which in addition to diagnostic value may be of functional relevance to the exploration of abnormal cortical connections (Riley et al., 2010).
FCD II is widely regarded as a developmental abnormality with several lines of evidence pointing to a disturbance in the migration and differentiation of radial glial stems cells and their progeny to the cortical plate (Andres et al., 2005; Cepeda et al., 2006; Lamparello et al., 2007; Sisodiya et al., 2009; Hadjivassiliou et al., 2010). The contribution of myelinating oligodendroglia (OL), and their progenitor and precursor cell populations oligodendroglial progenitor cells (OPCs), has not been specifically investigated in FCD II lesions and, in particular, if aberrant maturation could be implicated in the pathoetiology of abnormal myelination.
Our aim was to examine the patterns of myelination in a series of FCD II lesions operated on in childhood and adulthood for the treatment of drug-resistant epilepsy as well as cases confirmed at postmortem. We aimed to quantify the extent of the WM abnormalities and the composition of OL and OPC populations in these regions.
Methods
Case selection
Seventeen cases with a neuropathologic diagnosis of FCD II (Blumcke et al., 2011) were selected from the epilepsy surgical databases in the departments of neuropathology, Institutes of Neurology and Child Heath, operated over a period from 1996 to 2009 in addition to two cases diagnosed at postmortem examination. The study has been approved by the Joint Research Ethics Committee of the National Hospital for Neurology and Neurosurgery and the Institute of Neurology. Thirteen cases were operated in adulthood and four in childhood. In one of the cases the histologic diagnosis was FCD type IIA and in the remaining 18 cases, type IIB with balloon cells (Blumcke et al., 2011). We included the one type IIA case because although no balloon cells were identified on serial sections, white matter abnormalities were present similar to typical type IIB cases. Cases were selected that had undergone more extensive resections, where in addition to the region of dysplasia, more normally laminated cortex was available in the same specimen for comparison. All patients had histories of drug-resistant epilepsy, and standard presurgical investigations were carried out, including MRI, prior to surgical resection. The preoperative diagnosis on MRI in the adult surgical cases had been FCD, although an uncertain diagnosis or possible dysembryoplastic neuroepithelial tumor was proposed in three cases. However, in adult surgical cases, a retrospective review of preoperative MRI confirmed WM signal abnormalities on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences in the region of the dysplasia, typical of FCD II in all but one patient. Clinical details of all 19 cases, including seizure history and outcome data following resection, are presented in Table 1.
Table 1.
Clinical data of the 19 cases of FCD type II
| Case | FCD type | Age at seizure onset (years) | Seizure type | Localization | Age at Surgery or at deatha (years) | Outcome at follow-up at 2 years |
|---|---|---|---|---|---|---|
| 1 | FCD IIB | 0.9 | SPS, CPS | Left frontal | 24 | 1a |
| 2 | FCD IIB | 1.4 | CPS; SGS | Left frontal | 52 | 1 |
| 3 | FCD IIB | 1 | SPS; CPS; SGS (rare) | Right frontal | 22 | 1 |
| 4 | FCD IIB | 4 | SPS; CPS; SGS | Right frontal | 46 | 1 |
| 5 | FCD IIB | 5 | SPS; CPS; SGS | Left frontal | 26 | 5 |
| 6 | FCD IIA | 7 | SPS; CPS; SGS | Right parietal | 18 | 4 |
| 7 | FCD IIB | 7 | CPS; SGS (in past) | Temporal | 30 | 4 |
| 8 | FCD IIB | 3 | SPS; CPS; SGS | Right temporal | 52 | 4 |
| 9 | FCD IIB | 12 | CPS; SGS | Right temporal | 26 | 2 |
| 10 | FCD IIB | 11 | CPS, SGS | Left parietal | 25 | 3a |
| 11 | FCD IIB | 12 | SGS, right head versive seizures, left leg tonic seizures, | Left frontal | 29 | Unknown |
| 12 | FCD IIB | 12 | SGS, atonic seizures | Parietal | 32 | 2 |
| 13 | FCD IIB | 7 | SGS, CPS | Frontal | 34 | 1 |
| 14 | FCD IIB | 5 | SGS | Frontal | 81a | Not applicable |
| 15 | FCD IIB | 3 | CPS, SGS, SPS | Right frontal | 59a | Not applicable |
| 16 | FCD IIB | 4 | FS, SGS | Right parietal | 13 | 1 Not seizure-free (11 years) |
| 17 | FCD IIB | 0.5 | CPS, SE | Right frontal | 6 | Not seizure-free (10 years) |
| 18 | FCD IIB | 0.08 | Clonic jerks | Left frontal | 3 | 1 |
| 19 | FCD IIB | 0 | SGS, drop attacks, atonic seizures. | Left frontal | 18 | Unknown |
SPS, simple partial seizure; SGS, secondary generalized seizures; CPS, complex partial seizures; FS, febrile seizures.
The postoperative outcome is classified according to International League Against Epilepsy (ILEA) 2001 outcome criteria for adult surgical cases; class 1 being seizure-free to class 5 having 50% reduction in seizures. The data are recorded at 2 years follow-up. For two cases a data were available for only 1 year follow-up, and in seven patients longer follow-ups of up of 14 years (where there was a change in ILAE outcome this is noted in the table). In two patients there was no postoperative clinical follow-up and on the two postmortem patients (cases 14 and 15) this was not applicable as they had not had surgery.
Immunohistochemistry
Sections were cut at a thickness of 7 μm from one representative block; in two cases, two blocks were selected that contained the region of dysplasia in one and more normal cortex in another. Immunohistochemistry was carried out using standard methods with a panel of primary antibodies (Table 2), visualized with diaminobenzidine chromagen (Dako Envision Dako, Cambridge, United Kingdom).
Table 2.
Details of immunohistochemistry panel and protocols
| Antibody: Target cell/structure | Antibody clone | Source | Dilution | Pretreatments |
|---|---|---|---|---|
| Phosphorylated neurofilaments: Axons | SMI31 | Sternberger Monoclonals, Baltimore, MD, U.S.A. | 1:5,000 | None |
| Nonphosphorylated neurofilaments: Axons and neurons | SMI32 | Sternberger Monoclonals | 1:500 | EDTA heat-mediated epitope retrieval, 100°C for 20 min |
| Myelin basic protein: Mature myelinated axons | SMI94 | Sternberger Monoclonals | 1:2,000 | Enzyme pretreatment at room temperature for 10 min |
| MAP2, microtubule associated protein: Dendritic labeling | Map2 | Sigma-Aldrich, Dorset, United Kingdom | 1:1,000 | Citrate heat-mediated epitope retrieval, 100°C for 30 min |
| Reticulon family protein: Mature oligodendroglia | NogoA | Chemicon, Temecula, CA, U.S.A. | 1:500 | Citrate Heat-mediated epitope retrieval, 100°C for 20 min |
| Cyclic nucleotide 3-phosphodiesterase: Mature oligodendroglia, myelin, and oligodendroglial precursor cells | CNPase | Chemicon | 1:400 | Vector citrate-based antigen retrieval buffer, microwave for 15 min |
| Chondroitin sulphate proteoglycan: oligodendroglial precursor cells | NG2 anti-rabbit polyclonal | Chemicon | 1: 100 | Vector citrate-based antigen retrieval buffer, microwave for 18 min |
| Platelet derived growth factor receptor alpha: oligodendroglial precursor cells | PDGF R α anti-rabbit polyclonal | Gift, B Stallcup | 1:600 | Vector citrate-based antigen retrieval buffer, microwave for 18 min |
| Platelet derived growth factor receptor beta: oligodendroglial precursor cells | PDGF R β | Abcam plc, Cambridge, United Kingdom | 1:800 | Vector citrate-based antigen retrieval buffer, microwave for 12 min |
In addition, double-labeling immunofluorescence was carried out on selected cases for combinations of: platelet derived growth factor receptor (PDGFR)α/β, PDGFRα/glial fibrillary acidic protein (GFAP), PDGFRα/CD45, and PDGFRα/HLADR. In brief, sections were incubated overnight with primary antibody. Dako Envision horseradish peroxide solution was applied for 30 min before fluorescein-labeled antibody. Sections were thoroughly washed before second antibody applied. Alexa Fluor 564 conjugated anti-rabbit (1:100; Molecular Probes, Invitrogen, Carlsbad, CA, U.S.A.) and/or Alexa Fluor 633 conjugated antimouse secondary antibodies (1:50; Molecular Probes, Invitrogen) in Dako diluent were incubated on sections for 3 h at room temperature. Sections were coverslipped in DAPI-mounting media (Vector Laboratories, Burlingame, CA, U.S.A.). Immunofluorescent-labeled sections were viewed under a confocal laser scanning microscope (Zeiss LSM610 Meta, Cambridge, United Kingdom) equipped with blue diode (405 nm), argon (458, 477, 488, and 514 nm), and helium/neon (546 and 633 nm) lasers.
Qualitative analysis
Qualitative analysis of myeloarchitecture was undertaken on Luxol Fast Blue (LFB) myelin-stained sections in addition to the immunohistochemistry stains. The region of dysplasia and underlying WM were compared to the more normal adjacent cortex.
Quantitative analysis
Four regions of interest (ROIs) were defined in each case as follows: ROI1, subcortical WM in region of dysplasia; ROI2, dysplastic cortex (full thickness) overlying ROI1; ROI3, normal WM in adjacent cortex; ROI4, normal cortex (full thickness) overlying ROI3 (Fig. 1).The ROIs were defined on LFB-stained sections in each case and corresponding regions outlined on immunostained sections.
Figure 1.
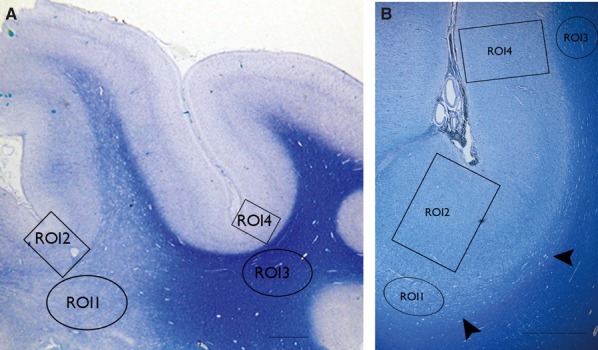
Low power views of myelin stained sections (LFB) form two cases of FCD type IIB illustrating the regions of interest (ROIs) used for the analysis. (A) The white matter pallor extends from the depth of sulcus deep to the white matter, whereas in (B) only the immediate subcortical zone, that of the U-fibers shows pallor that forms a band running along the bottom of the cortex (arrowheads) and the overlying cortex shows excess myelination. The ROI indicated are ROI 1 subcortical white matter (WM) in region of dysplasia, ROI2 dysplastic cortex (full thickness) overlying ROI1, ROI 3 normal WM in adjacent cortex, ROI4 normal cortex (full thickness) overlying ROI 3. (The ROI shown here provide an approximation of the size of the freehand drawn ROI on the immunostained sections.) The scale bars in A = 800 and B = 1,500 μm.
For image acquisition, each section was viewed under a Zeiss microscope (Carl Zeiss, Axioscope). Each of the four ROIs were outlined by a freehand-drawn shape using an image analysis system (Image Pro Plus, Media Cybernetics, Marlow, United Kingdom and Histometrix, Kinetic Imaging, Liverpool, United Kingdom) at objective ×2.5 magnification. Images were systematically acquired from each drawn ROI at high magnification (×20 or ×40 objective) using 100% field sampling. The areas of the ROI1–4 varied between and within cases from 4.4 to 9.5 mm2.
We used threshold-based analysis to quantify the density of immunostaining for myelin (myelin basic protein/SMI94 and cyclic nucleotide 3-phosphodiesterase [CNPase]), axons (phosphorylated neurofilament/SMI31), and dendrites (microtubule associated protein MAP2) for each ROI (using Image Pro Plus). A threshold mask was set with red, green, blue (RGB) parameters to maximize recognition of fiber staining but elimination of nonaxonal structures. In particular, staining of neuronal cell bodies with SMI31 was excluded from the analysis. The same threshold mask was applied to all images of each ROI of the same immunostained section of each case. The data from each ROI was summarized as a percentage of overall staining (labeling index).
Systematic cell counting was carried out in immunostained sections for OL (NogoA and CNPase) and OPC (NG-2, PDGFRα and PDGFRβ). Images were acquired as above for each ROI, and only immunopositive cells (not processes or fibers) were systematically counted through manual tagging. The total number of immunopositive cells for each ROI was expressed in relation to the total area of ROI.
Statistical analysis
Statistical analysis was carried out using analysis program SPSS version 18 for Windows (IBM, Armonk, NY, U.S.A.). Mann-Whitney U-test and Wilcoxon signed-rank test were used to compare data between ROIs and Pearson∼s test for clinical pathologic correlations.
Results
Qualitative findings
LFB and MBP (SMI94) sections
A reduction of WM myelinated fibers in the region of dysplasia compared to normal WM was observed to varying degree (Figs. 1A,B and 2A,B). In four cases, this involved the immediate subcortical zone, in the region of the “U” fibers, whereas in other cases, myelin loss extended more deeply (Fig. 1A,B). In the normal cortex, radial bundles of myelinated fibers were clearly defined with SMI94 in the deeper cortical layers (Fig. 2D), whereas in the region of dysplasia, the cortical myeloarchitecture was disorganized, often with prominent horizontal fiber networks obscuring this normal radial pattern (Fig. 2C).
Figure 2.
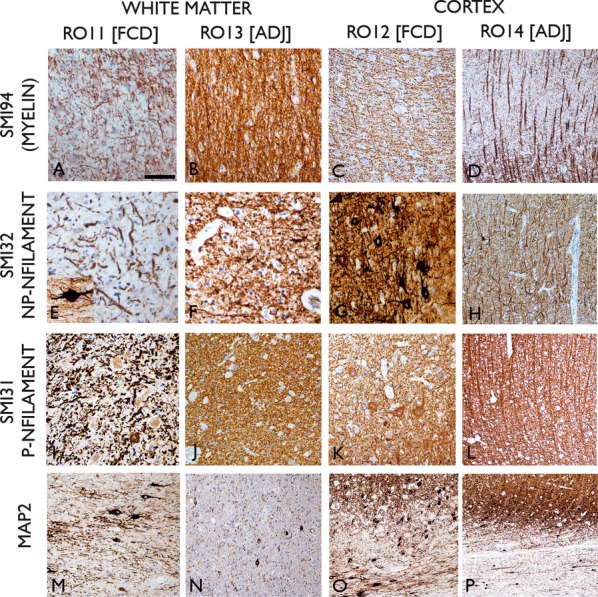
Immunohistochemistry for myelin basic protein (SMI94; A–D), nonphosphorylated neurofilament (NP-NFilament SMI32; E–H), phosphorylated neurofilament (P-Nfilament SMI31; I–L) and Map2 (microtubule associated protein) in ROI1 (FCD WM), ROI3 (normal WM), ROI2 (FCD cortex), and ROI4 (normal cortex). Reduction of number of processes was noted in ROI1 with SMI31,32, & 94 antibodies with thick, tortuous fibres present, particularly in SMI32. Inset in (E) shows a dysmorphic neuron in the immediate subcortical region with thick bipolar processes running horizontally to the cortex. In ROI3 (B, F, J) normal density and size of axons were seen with all antibodies. In the dysplastic cortex, prominent horizontal fibers were seen with SMI94 (C), obscuring the normal radial orientation observed in normal cortex (D). Similarly in neurofilament stains, disorganized axonal and dendritic processes were seen in the dysplasia (G, K) relative to the radial organized patterns of normal cortex (H, L). In Map2 stained sections in the WM of the region of dysplasia (M), dysmorphic neurons and dendrites were present compared to infrequent, small white matter neurons and fine dendrites in adjacent normal WM (N). In the region of dysplasia (O) Map2 staining highlights the ill-defined border between the gray and white matter interface with numerous unstained balloon cells and prominent horizontal neurons in the subcortical zone. In the adjacent cortex, sharper demarcation of cortex and white matter is observed (P). ROI, Region of interest; FCD, Focal cortical dysplasia; WM, white matter; ADJ, adjacent normal cortex. Bar = 60 microns in A to N and 140 microns in O & P.
Neurofilament stained sections (SMI32 and SMI31)
Reduced labeling of axons and processes in the white matter in the region of dysplasia was observed (Fig. 2E,I) compared to adjacent white matter (Fig. 2F,J). In addition, WM axons in the region of dysplasia often appeared thicker and more tortuous (Fig. 2E,I). Dysmorphic horizontal neurons in the immediate subcortical WM, exhibited coarse, bipolar processes often running parallel to the cortex (Fig. 2E inset). In the dysplastic cortex, axon stains revealed a disorganized network of processes (Fig. 2G,K) compared to the radial bundles of axons in the normal cortex (Fig. 2H,L).
MAP2 sections
Dysmorphic neurons with coarse dendrites or surrounding processes were observed in the WM in the region of dysplasia compared to scattered small, single neurons with fine processes in the normal white matter (Fig. 2M,N). In the dysplastic cortex, MAP2 highlighted the ill-defined boundary between the gray and white matter with prominent, horizontally orientated neurons in the immediate subcortical region (Fig. 2O) in contrast to a sharper gray-white boundary in the adjacent normal cortex (Fig. 2P).
NG-2, PDGFRα, and β sections
Positive cytoplasmic labeling of cells with similar morphology were identified in all ROIs (Fig. 3), with small, round nuclei and fine, short multipolar processes with branch points, particularly visible with NG2 (Fig. 3H) and PDGFRβ (Fig. 3A,I). Additional labeling of vascular structures was present on PDGFRβ sections. Double labeling confirmed colocalization between PDGFRα and β (Fig. 3I), but no colocalization between PDGFRα and GFAP, HLADR, or CD45. The morphology of these multipolar cells was therefore considered compatible with oligodendroglial precursor or progenitor cell types (OPCs) (Jakovcevski et al., 2009). There was no distinct labeling of balloon cells in the white matter with these markers.
Figure 3.
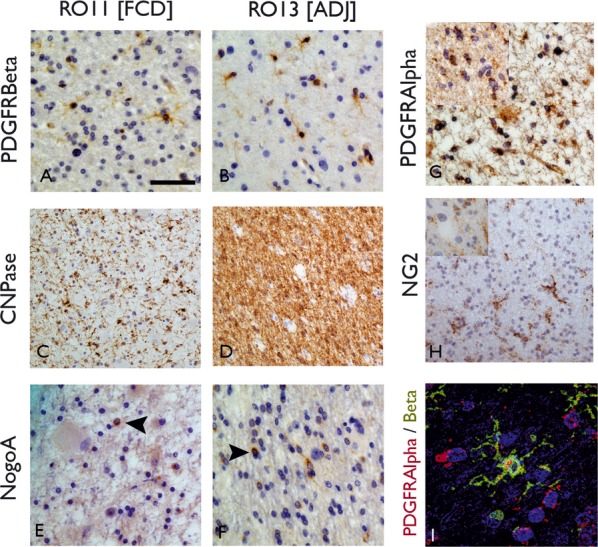
Immunohistochemistry for oligodendroglial (OL) and precursor cell types (OPC). Comparison of ROI1 (white matter in the region of dysplasia [A, C, E]) with ROI3 (adjacent white matter [B, D, F]) positive labeling of cells with PDGFRβ (A, B) CNPase (C, D) and NogoA (E, F) are seen in both ROI. With PDGFRβ, small round cells were labeled with fine branching processes, compatible with the described morphology of OPC, and were visible in both ROI; with CNPase, labeling of small OL in addition to fibers was noted with a marked reduction in the labeling of fibers in ROI1 (C) compared to ROI3 (D). NogoA labeled infrequent small OL cells in all ROI with a small, peripheral rim of cytoplasmic labeling. (G) PDGFRα also showed positive round cells in ROI1 and (inset) ROI3. (H) NG2 labeled cells with similar morphology, with fine branching process in ROI3 and in (inset) ROI1 near to an unlabeled balloon cell. (I) Confocal microscopy confirmed overlap of labeling of PDGFRα and β in cells with multipolar morphology. Bar = 15 microns (A, B, E, F, G, H, I [including insets]) and 35 microns (C, D).
CNPase sections
Small, OL cells showed cytoplasmic labeling in all regions, in addition to labeling of myelinated fibers in the normal white matter (Fig. 3C,D) with prominent demonstration of the cortical radial fiber bundles and horizontal myelinated fibers and oligodendroglial in layer I in the normal cortex. There was a qualitative impression of a reduction of CNPase labeling in the white matter underlying the dysplasia and disorganized fiber arrangement in the cortex.
NogoA sections
Similar small round cells, albeit fewer in number than with CNPase, were visible in all ROIs, with labeling restricted to a thin rim of cytoplasmic staining around the nucleus (Fig. 3E,F).
Quantitative analysis
There was a significant reduction in the mean MBP labelling with SMI94, CNPase, and neurofilament (SMI31) in ROI1 compared to ROI3 in FCD cases (p < 0.0001; p < 0.01 and p < 0.05, respectively) (Table 3). No significant differences in mean cortical MBP labeling or neurofilament (between ROIs 2 and 4) were noted (p = 0.41 and p = 0.21) despite the abnormal distribution of fibers observed in the dysplastic zone. Myelin staining values with SMI94 were lower in ROI1 than three in 16 of the 19 cases and for neurofilament (SMI31) in 14 of the 19 cases. In the 19 cases, there was a significant correlation between the MBP (SMI94) and neurofilament (SMI31) labeling index in ROI1 (p < 0.01) and SMI94 and CNPase (p < 0.05). Increased mean dendritic staining with Map2 was observed in ROI1 compared to ROI3 (Table 3), but the differences were not significant.
Table 3.
Results of quantitative evaluation of FCD cases with mean values shown for each region of interest (ROI) in the FCD cases
| FCD region | Adjacent cortex | ||||
|---|---|---|---|---|---|
| Target structure/immunomarker | ROI WM Mean [SD] | ROI 2 Cortex Mean [SD] | ROI 3 WM Mean [SD] | ROI 4 Cortex Mean [SD] | Significance (between ROI and ROI 3) |
| Myelin labeling (myelin basic protein) | |||||
| SMI94% labeling | 33.4 [17.5] N = 19 | 20.4 [16.6] N = 19 | 55.5 [15.0] N = 19 | 22.6 [13.7] N = 19 | p < 0.0001 |
| Axonal labeling (neurofilament) | |||||
| SMI31% labeling | 19.7 [10.3] N = 19 | 27.4 [15.4] N = 19 | 32.3 [13.1] N = 19 | 29.6 [18.4] N = 19 | p < 0.01 |
| Dendritic labeling (microtubule-associated protein) | |||||
| MAP2% labeling | 12.2 [9.4] N = 15 | – | 10.6 [8.8] N = 15 | – | |
| Mature oligodendroglia | |||||
| CNPase% labeling | 24.9 [15.9] N = 12 | – | 42.5 [16.9] N = 12 | – | p < 0.05 |
| CNPase Cell density × 10−5/μm2 | 22.30 [25.5] N = 9 | 10.03 [8.47] N = 9 | 26.97 [28.8] N = 10 | 8.96 [7.9] N = 10 | |
| NOGOA Cell density × 10−5/μm | 0.85 [1.6] N = 13 | 0.17 [0.3] N = 13 | 5.2 [12.4] N = 12 | 0.08 [0.15] N = 12 | |
| Immature oligodendroglia | |||||
| PDGFR-α Cell density × 10−5/μm2 | 8.971 [7.75] N = 9 | 6.31 [5.38] N = 9 | 12.751 [9.69] N = 10 | 8.305 [5.67] N = 10 | |
| PDGFR-β Cell density × 10−5/μm2 | 3.05 [4.5] N = 14 | 4.4 [5.8] N = 10 | 2.7 [0.46] N = 13 | 3.2 [5.3] N = 9 | |
| NG-2 Cell density × 10−5/μm2 | 0.058 [0.076] N = 6 | 0.062 [0.088] N = 2a | 0.5662 [0.67] N = 7 | 0.097 [0.16] N = 3 | |
The two main measurements were the overall percentage of ROI labeling (i.e., the “field fraction” or “labeling index”) or the number of positive labeled cells per ROI (cell density expressed per square micron [μm]). WM = white matter, N = number of cases evaluated for each test. In a proportion of cases, particularly with
NG2 labeling in cortical region, enumeration of positive cells with image analysis was not possible due to differentiation of background labeling from positive cell labeling. Statistical analysis between ROI 1 and 3 is shown using nonparametric tests and only p-values of <0.05 are shown
Analysis of OL cell numbers with mature (CNPase, NogoA) and immature cell markers (PDGFRα, PDGFRβ, and NG2) revealed lower mean cell densities in ROI1 than ROI3 for all markers apart from PDGFRβ, which was increased in ROI1 (Table 3), although the differences were not statistically significant. There was a correlation between the myelin staining in ROI1 with SMI94 and the number of mature oligodendroglia with NogoA (p < 0.05).
The findings above remained significant for the 18 FCD type IIB cases alone, excluding the single FCD type IIA case.
Clinical correlations
There was a significant negative correlation between duration of seizures (age of onset of epilepsy to age at surgery) and neurofilament (SMI31) (p < 0.001) and MPB (p < 0.05) labeling index in the white matter in the region of dysplasia (ROI1). There were also significant positive correlations between the relative changes in the white matter labeling [(RO3-RO1)/ROI3] for SMI31 with duration of epilepsy (p < 0.05) and for CNPase with age of onset of epilepsy (p < 0.05) (Fig. 4A).
Figure 4.
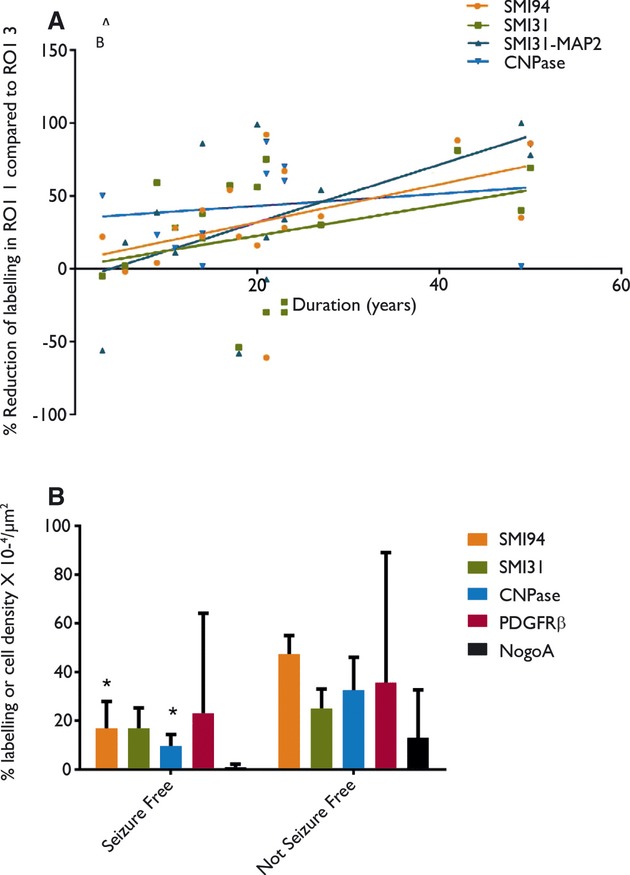
(A) The relative reduction of labeling fraction between region of interest (ROI) 1 and 3 [(ROI3-1)/ROI3] is plotted for myelin (SMI94 & CNPase), axons (SMI31) and axons with labeling of dendrites subtracted in each ROI (SMI31-MAP2) against duration of epilepsy (time between onset of seizures and surgery) for the 17 surgical cases in the study. The relative reduction in values provides more comparable data between cases, taking into account any variations of staining due to tissue processing and fixation. There was a significant positive correlation noted for SMI31 and CNPase. (B) A graph of the mean values for the field fraction of labeling with myelin basic protein (SMI94), neurofilament (SMI31), CNPase myelin, and cell density of oligodendroglial precursors (PDGF) and mature oligodendroglia (NogoA) in the white matter associated with FCD II in patients who were seizure-free at last follow-up compared to patients who continue to have seizures. Significantly lower myelin staining (with SMI94 and CNPase) was observed in the seizure-free patients in this small study group.
Outcome data available in the 17 operated cases, taken at the time of last clinical follow-up, showed that six patients remained seizure-free and nine patients continued to have seizures, with no data for two patients (Table 1). Correlation with the pathology measures showed significantly lower mean myelin labeling index with SMI94 (p < 0.0001) and CNPase (p < 0.005) in ROI1 in patients who remained seizure-free compared to the non–seizure-free group (Fig. 4B). Mean SMI31 labelling was lower in the seizure-free group in ROI1 but not significantly so, whereas OL cell density (with PDGFRβ and NOGOA) were marginally increased.
Discussion
We have confirmed, through quantitative analysis, a significant reduction in myelin in the WM in FCD II. This was accompanied by a parallel reduction in neurofilament staining that correlated with the myelin reduction in individual cases. The less marked reduction in neurofilament than myelin observed, could be an effect of increased neurofilament-positive dystrophic dendrites in the WM in FCD, as noted in previous studies (Cepeda et al., 2003).We demonstrated this in the present study with increased MAP2 labeling in the region of dysplasia, which specifically label dendrites, but not axons, and this could explain the less significant reduction in neurofilament compared to myelin. Taking this into consideration, we interpret these findings that a reduction of myelinated axons (compared to normal white matter) rather than reduced myelination of axons likely represents the predominant pathologic cause of the “hypomyelinated” white matter associated with FCD.
One explanation for this observation is that axon projections from the overlying dysplastic cortex take abnormal routes. We noted abnormal organization of myelinated cortical axons and dendrites in FCD, often with an excess of horizontal or transverse processes. This may be secondary to the abnormal orientation of neurons in FCD, as previously shown with intracellular biocytin tracing methods (Cepeda et al., 2003). The normal polarized state of a neuron is actively maintained by transcription factors and closely linked to the mechanisms regulating axonal pathways as well as the distribution of its dendrites (de la Torre-Ubieta & Bonni, 2011), and respecification of a dendrite as an axon may also occur in pathologic conditions (Gomis-Ruth et al., 2008). One possibility, therefore, is that dysregulation of these processes occurs in cortical dysplasia, either as a primary or secondary mechanism, with the formation of the observed abnormal intracortical axodendritic networks and consequent reduction in WM axons.
A more likely hypotheses, however, is that the reduction in WM axons reflects a reduction in neuronal number in the overlying dysplastic cortex. We have previously demonstrated lower mean cortical neuronal densities in FCD II compared to adjacent normal cortex (Thom et al., 2005), more recently confirmed by another study (Muhlebner et al., 2012). Our previous study also showed a trend for a decline in cortical neuronal density in FCD II, with age of patient and duration of seizures (Thom et al., 2005). In addition, in this current study we have observed a decline of white matter axons in relation to seizure duration in support of this hypothesis, which suggests that there is progressive degeneration in FCD II with ongoing neuronal and axonal (and myelin) loss.
We also examined OPC and OL populations in FCD. Loss of OL function has been implicated in animal models of tuberous sclerosis with hypomyelination (Ess, 2010). There is a body of evidence that the neuronal and glial cytopathology in FCD may reflect abnormal cellular maturation and differentiation, with persisting expression of stem cell markers demonstrated on balloon cells (Ying et al., 2005; Najm et al., 2007). Balloon cells have properties of pathologic progenitor cells (Yasin et al., 2010), and studies applying developmental lineage markers suggest that balloon cells and dysmorphic neurons likely derive from radial glia or radial migrating ventricular zone progenitors (Lamparello et al., 2007; Hadjivassiliou et al., 2010). Related theories propose FCD is a result of events in the late stages of corticogenesis with localized failure of elimination of immature subplate and radial glial elements (Cepeda et al., 2006). OL and their progenitor cells have, however, been little investigated, although a recent study of FCD IIB demonstrated a reduction in Olig2-positive cells in the white matter in two-thirds of cases and a correlation between myelin reduction and oligodendroglial numbers (Muhlebner et al., 2012).
OPC migration and maturation into OL occurs in three waves and from different origins including the ganglionic eminence as well as the radial glial cells of the sub-ventricular zone (Jakovcevski et al., 2009). Their differentiation and maturation is shown by sequential expression of lineage markers from PDGFα/NG2 in early OPC to NogoA and MBP in mature OL (Jakovcevski et al., 2009; Bradl & Lassmann, 2010; Muhlebner et al., 2012). Of possible relevance to the hypomyelination in FCD, during mid-gestation, OPCs locate to the transient subplate zone beneath the cortex, an interlude considered to be relevant to their maturation and myelination of local axonal projections (Jakovcevski et al., 2009). Unlike other precursor cell types, all stages of OPC persist in the cortex and WM through adult life to replenish OL numbers (Jakovcevski et al., 2009). Previous studies confirm that NG2-positive cells represent the largest proliferating cell pool in epilepsy surgical tissues (Geha et al., 2010). In the current study we were able to identify the range of OPC and OL cell types in FCD II with our immunohistochemistry panel. Although for most markers there were reduced numbers in the region of dysplasia, with a greater reduction in the WM than dysplastic cortex, the differences were not numerically significant to control regions. In our study, PDGFRβ immunohistochemistry revealed cells with similar cyto-morphology to NG2 and PDGFRα labelling, the latter being more recognized OPC lineage markers. PDGFRβ has previously been identified as a marker of pericytes in human brain angiogenesis (Virgintino et al., 2007). We also noted vascular staining with PDGFRβ, but this marker has not previously been reported to label OPC-like cells. Of note, the morphology of the OL cell types with all markers, in contrast to a previous study (Muhlebner et al., 2012) appeared normal and we did not identify any significant labelling of balloon cells with any OPC markers. Therefore, although we identified some reduction in OL/OPC number in addition to the myelin in FCD II white matter, the OL numbers were present in an appropriate ratio to the level of myelination, in keeping with findings in the previous study of FCD II by Muhlebner et al. (2012). There is also limited evidence from our data to support a significant failure of OL maturation or cytomorphology to implicate this cell lineage as the primary or developmental cause of the local myelin and axon deficiency.
Human myelination of the WM proceeds from the region of the central sulcus by 15 months towards the fontal and temporal poles by the 23rd postnatal month (Kinney et al., 1988). Completion of myelination continues over decades, projection pathways typically myelinating before association pathways (Ullen, 2009). We noted the reduction of myelinated axons was limited to the immediate subcortical territory of the U-fibres of Meynert in some FCD cases. The U-fibres, travel in a tangential rather than radial orientation, forming local cortical-cortical connections as recently mapped by DTI tractography (Oishi et al., 2008). In other FCD cases, pallor of deep WM likely represents reduction of longer range afferent and efferent cortical projections. In Taylor∼s original paper on FCD they also describe cases where the myelin pathology extended deeply from the cortex and other cases, where only the immediate or subjacent WM, was affected (Taylor et al., 1971). In subsequent FCD series there has been little descriptive neuropathological information regarding the topography of myelin depletion, although is presence frequently recorded (Urbach et al., 2002; Mackay et al., 2003; Blumcke et al., 2011). We noted a relationship between age of onset of epilepsy and severity of reduction of myelin with CNPase in FCD. It is possible that early seizures interfere with these stages of myelin maturation which requires investigation in a larger series, ideally incorporating neuroimaging. DTI studies in cortical malformations have approached the extent and nature of WM tract changes (Eriksson et al., 2001; Diehl et al., 2010) with alterations in diffusivity suggested to correlated with loss of myelin integrity, axonal density or directional order of WM (Widjaja et al., 2007). However, there is a lack of detailed pathological-imaging correlation. In the current study, MRI abnormalities, as blurring of the grey-white matter junction and abnormal WM signal intensity on T2-weighted or FLAIR images was noted. It was not possible to carry out a quantitative neuroimaging correlation in the current series as the patients had been operated and imaged over a 13 year period using different MR modalities and retrospective coregistration of tissue sample with MRI was not feasible. Furthermore, myelin abnormalities are also present histologically in other FCD subtypes (Blumcke et al., 2011), with abnormal superficial cortical myelination noted in FCD IIIa (Thom et al., 2009) and WM hypomyelination in FCD IIIb (Thom et al., 2011), the later which may be misinterpreted as FCD II in conventional MRI (Campos et al., 2009), including cases in the present study. Further investigation of differences (or similarities) in myelin abnormalities between FCD subtypes, with pathology-imaging coregistration, are warranted to improve pre-operative recognition and discrimination of these lesions.
In regard to patient outcome in this small series, we showed significantly lower measures of white matter myelination in the patients with seizure-free outcome at last follow-up. It has been reported that completeness of resection of the dysplastic cortex but not the underlying WM is necessary for seizure freedom (Wagner et al., 2011) implying that the extent of WM pathology is not relevant to outcome. It is possible, in the present series, that the presence of white matter pathology allowed better discrimination of the extent of the lesion on MRI and a more complete cortical resection, compared to cases without this feature. However, this is a study of a small number of cases and the prognostic value of white matter pathology would require verification in a larger series.
In conclusion, in this study quantifying the pathological basis of dysmyelination abnormalities in FCD we confirm primarily a loss of myelinated WM axons but with disorganized patterns of cortical myelination and overall preservation and representation of OL cells and their precursors. Our study has highlighted several future lines of investigation to pursue as to the cause and effects of these integral and diagnostic pathological changes in the context of FCD.
Acknowledgments
We are very grateful to Professor W. Stallcup for the gift of his characterized antibodies for oligodendroglial progenitor cells. This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health∼s NIHR Biomedical Research Centres∼ funding scheme and was supported by a grant from the MRC (MR/J01270X/1). TSJ is supported by a HEFCE Clinical Senior Lecturer Award and Great Ormond Street Hospital Children∼s Charity.
Disclosure
The authors have no conflicts of interest to declare. We confirm that we have read the Journal∼s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- Andres M, Andre VM, Nguyen S, Salamon N, Cepeda C, Levine MS, Leite JP, Neder L, Vinters HV, Mathern GW. Human cortical dysplasia and epilepsy: an ontogenetic hypothesis based on volumetric MRI and NeuN neuronal density and size measurements. Cereb Cortex. 2005;15:194–210. doi: 10.1093/cercor/bhh122. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Kuzniecky RI, Bollen AW, Grant PE. Focal transmantle dysplasia: a specific malformation of cortical development. Neurology. 1997;49:1148–1152. doi: 10.1212/wnl.49.4.1148. [DOI] [PubMed] [Google Scholar]
- Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ, Battaglia G, Becker A, Cepeda C, Cendes F, Colombo N, Crino P, Cross JH, Delalande O, Dubeau F, Duncan J, Guerrini R, Kahane P, Mathern G, Najm I, Ozkara C, Raybaud C, Represa A, Roper SN, Salamon N, Schulze-Bonhage A, Tassi L, Vezzani A, Spreafico R. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–174. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AR, Clusmann H, von Lehe M, Niehusmann P, Becker AJ, Schramm J, Urbach H. Simple and complex dysembryoplastic neuroepithelial tumors (DNT) variants: clinical profile, MRI, and histopathology. Neuroradiology. 2009;51:433–443. doi: 10.1007/s00234-009-0511-1. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Flores-Hernandez J, Hernandez-Echeagaray E, Klapstein GJ, Boylan MK, Calvert CR, Jocoy EL, Nguyen OK, Andre VM, Vinters HV, Ariano MA, Levine MS, Mathern GW. Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J Neurosci Res. 2003;72:472–486. doi: 10.1002/jnr.10604. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Andre VM, Levine MS, Salamon N, Miyata H, Vinters HV, Mathern GW. Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav. 2006;9:219–235. doi: 10.1016/j.yebeh.2006.05.012. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Bonni A. Transcriptional regulation of neuronal polarity and morphogenesis in the mammalian brain. Neuron. 2011;72:22–40. doi: 10.1016/j.neuron.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl B, Tkach J, Piao Z, Ruggieri P, LaPresto E, Liu P, Fisher E, Bingaman W, Najm I. Diffusion tensor imaging in patients with focal epilepsy due to cortical dysplasia in the temporo-occipital region: electro-clinico-pathological correlations. Epilepsy Res. 2010;90:178–187. doi: 10.1016/j.eplepsyres.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Eriksson SH, Rugg-Gunn FJ, Symms MR, Barker GJ, Duncan JS. Diffusion tensor imaging in patients with epilepsy and malformations of cortical development. Brain. 2001;124:617–626. doi: 10.1093/brain/124.3.617. [DOI] [PubMed] [Google Scholar]
- Ess KC. Tuberous sclerosis complex: a brave new world? Curr Opin Neurol. 2010;23:189–193. doi: 10.1097/WCO.0b013e32832c4ff5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geha S, Pallud J, Junier MP, Devaux B, Leonard N, Chassoux F, Chneiweiss H, Daumas-Duport C, Varlet P. NG2+/Olig2+ cells are the major cycle-related cell population of the adult human normal brain. Brain Pathol. 2010;20:399–411. doi: 10.1111/j.1750-3639.2009.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Ruth S, Wierenga CJ, Bradke F. Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol. 2008;18:992–1000. doi: 10.1016/j.cub.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Hadjivassiliou G, Martinian L, Squier W, Blumcke I, Aronica E, Sisodiya SM, Thom M. The application of cortical layer markers in the evaluation of cortical dysplasias in epilepsy. Acta Neuropathol. 2010;120:517–528. doi: 10.1007/s00401-010-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman PA, Fitt GJ, Harvey AS, Kuzniecky RI, Jackson G. Bottom-of-sulcus dysplasia: imaging features. AJR Am J Roentgenol. 2011;196:881–885. doi: 10.2214/AJR.10.4423. [DOI] [PubMed] [Google Scholar]
- Jakovcevski I, Filipovic R, Mo Z, Rakic S, Zecevic N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front Neuroanat. 2009;3:5. doi: 10.3389/neuro.05.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney HC, Brody BA, Kloman AS, Gilles FH. Sequence of central nervous system myelination in human infancy. II. Patterns of myelination in autopsied infants. J Neuropathol Exp Neurol. 1988;47:217–234. doi: 10.1097/00005072-198805000-00003. [DOI] [PubMed] [Google Scholar]
- Lamparello P, Baybis M, Pollard J, Hol EM, Eisenstat DD, Aronica E, Crino PB. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain. 2007;130:2267–2276. doi: 10.1093/brain/awm175. [DOI] [PubMed] [Google Scholar]
- Mackay MT, Becker LE, Chuang SH, Otsubo H, Chuang NA, Rutka J, Ben-Zeev B, Snead OC, III, Weiss SK. Malformations of cortical development with balloon cells: clinical and radiologic correlates. Neurology. 2003;60:580–587. doi: 10.1212/01.wnl.0000044053.09023.91. [DOI] [PubMed] [Google Scholar]
- Muhlebner A, Coras R, Kobow K, Feucht M, Czech T, Stefan H, Weigel D, Buchfelder M, Holthausen H, Pieper T, Kudernatsch M, Blumcke I. Neuropathologic measurements in focal cortical dysplasias: validation of the ILAE 2011 classification system and diagnostic implications for MRI. Acta Neuropathol. 2012;123:259–272. doi: 10.1007/s00401-011-0920-1. [DOI] [PubMed] [Google Scholar]
- Najm IM, Tilelli CQ, Oghlakian R. Pathophysiological mechanisms of focal cortical dysplasia: a critical review of human tissue studies and animal models. Epilepsia. 2007;48(Suppl. 2):21–32. doi: 10.1111/j.1528-1167.2007.01064.x. [DOI] [PubMed] [Google Scholar]
- Oishi K, Zilles K, Amunts K, Faria A, Jiang H, Li X, Akhter K, Hua K, Woods R, Toga AW, Pike GB, Rosa-Neto P, Evans A, Zhang J, Huang H, Miller MI, van Zijl PC, Mazziotta J, Mori S. Human brain white matter atlas: identification and assignment of common anatomical structures in superficial white matter. Neuroimage. 2008;43:447–457. doi: 10.1016/j.neuroimage.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster JM, Igbokwe E, Cosgrove GR, Cole AJ. Identifying subtle cortical gyral abnormalities as a predictor of focal cortical dysplasia and a cure for epilepsy. Arch Neurol. 2012;69:257–261. doi: 10.1001/archneurol.2011.1002. [DOI] [PubMed] [Google Scholar]
- Regis J, Tamura M, Park MC, McGonigal A, Riviere D, Coulon O, Bartolomei F, Girard N, Figarella-Branger D, Chauvel P, Mangin JF. Subclinical abnormal gyration pattern, a potential anatomic marker of epileptogenic zone in patients with magnetic resonance imaging-negative frontal lobe epilepsy. Neurosurgery. 2011;69:80–93. doi: 10.1227/NEU.0b013e318212bb1a. discussion 93–84. [DOI] [PubMed] [Google Scholar]
- Riley JD, Franklin DL, Choi V, Kim RC, Binder DK, Cramer SC, Lin JJ. Altered white matter integrity in temporal lobe epilepsy: association with cognitive and clinical profiles. Epilepsia. 2010;51:536–545. doi: 10.1111/j.1528-1167.2009.02508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya SM, Fauser S, Cross JH, Thom M. Focal cortical dysplasia type II: biological features and clinical perspectives. Lancet Neurol. 2009;8:830–843. doi: 10.1016/S1474-4422(09)70201-7. [DOI] [PubMed] [Google Scholar]
- Taylor DC, Falconer MA, Bruton CJ, Corsellis JA. Focal dysplasia of the cerebral cortex in epilepsy. J Neurol Neurosurg Psychiatry. 1971;34:369–387. doi: 10.1136/jnnp.34.4.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Martinian L, Sen A, Cross JH, Harding BN, Sisodiya SM. Cortical neuronal densities and lamination in focal cortical dysplasia. Acta Neuropathol. 2005;110:383–392. doi: 10.1007/s00401-005-1062-0. [DOI] [PubMed] [Google Scholar]
- Thom M, Eriksson S, Martinian L, Caboclo LO, McEvoy AW, Duncan JS, Sisodiya SM. Temporal lobe sclerosis associated with hippocampal sclerosis in temporal lobe epilepsy: neuropathological features. J Neuropathol Exp Neurol. 2009;68:928–938. doi: 10.1097/NEN.0b013e3181b05d67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B, Dean A, McEvoy A, Sisodiya SM, Brandner S. One hundred and one dysembryoplastic neuroepithelial tumors: an adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol. 2011;70:859–878. doi: 10.1097/NEN.0b013e3182302475. [DOI] [PubMed] [Google Scholar]
- Ullen F. Is activity regulation of late myelination a plastic mechanism in the human nervous system? Neuron Glia Biol. 2009;5:29–34. doi: 10.1017/S1740925X09990330. [DOI] [PubMed] [Google Scholar]
- Urbach H, Scheffler B, Heinrichsmeier T, von Oertzen J, Kral T, Wellmer J, Schramm J, Wiestler OD, Blumcke I. Focal cortical dysplasia of Taylor∼s balloon cell type: a clinicopathological entity with characteristic neuroimaging and histopathological features, and favorable postsurgical outcome. Epilepsia. 2002;43:33–40. doi: 10.1046/j.1528-1157.2002.38201.x. [DOI] [PubMed] [Google Scholar]
- Virgintino D, Girolamo F, Errede M, Capobianco C, Robertson D, Stallcup WB, Perris R, Roncali L. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- Wagner J, Urbach H, Niehusmann P, von Lehe M, Elger CE, Wellmer J. Focal cortical dysplasia type IIb: completeness of cortical, not subcortical, resection is necessary for seizure freedom. Epilepsia. 2011;52:1418–1424. doi: 10.1111/j.1528-1167.2011.03158.x. [DOI] [PubMed] [Google Scholar]
- Widjaja E, Blaser S, Miller E, Kassner A, Shannon P, Chuang SH, Snead OC, III, Raybaud CR. Evaluation of subcortical white matter and deep white matter tracts in malformations of cortical development. Epilepsia. 2007;48:1460–1469. doi: 10.1111/j.1528-1167.2007.01105.x. [DOI] [PubMed] [Google Scholar]
- Yasin SA, Latak K, Becherini F, Ganapathi A, Miller K, Campos O, Picker SR, Bier N, Smith M, Thom M, Anderson G, Helen Cross J, Harkness W, Harding B, Jacques TS. Balloon cells in human cortical dysplasia and tuberous sclerosis: isolation of a pathological progenitor-like cell. Acta Neuropathol. 2010;120:85–96. doi: 10.1007/s00401-010-0677-y. [DOI] [PubMed] [Google Scholar]
- Ying Z, Gonzalez-Martinez J, Tilelli C, Bingaman W, Najm I. Expression of neural stem cell surface marker CD133 in balloon cells of human focal cortical dysplasia. Epilepsia. 2005;46:1716–1723. doi: 10.1111/j.1528-1167.2005.00276.x. [DOI] [PubMed] [Google Scholar]