
Figure 1.
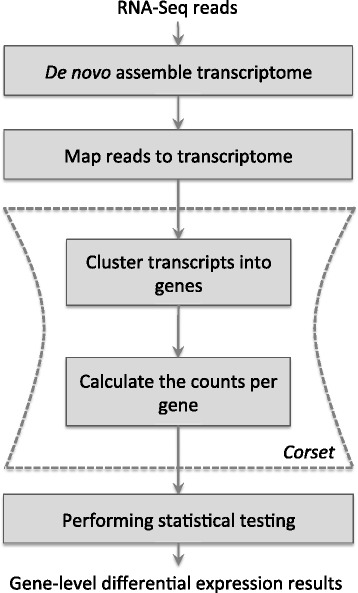
The pipeline for performing a count-based gene-level differential expression analysis on non-model organisms. Cleaned RNA-seq reads are first de novo assembled into contig sequences. Reads are mapped back to the transcriptome and the association between contigs and genes must be established (clustering of contigs). Then the abundance of each gene is estimated. Finally, statistical testing is performed on the count data to determine which genes are differentially expressed. Corset performs the clustering and counting (dashed box) in a single step.