

Our currently limited understanding of the mechanism of biological dinitrogen fixation[1] – i.e. the reduction of N2 to two equivalents of ammonia by addition of protons and electrons – motivates a sustained effort towards the preparation of functional models of the nitrogenase MoFe-cofactor. One plausible mechanism for N2 reduction is a so-called distal or Chatt-type cycle[2], in which three hydrogen-atom equivalents are successively added to the distal N-atom of a metal-bound dinitrogen molecule (M–N≡N), resulting in the elimination of one equivalent of NH3 to yield an intermediate nitrido complex (M≡N). The latter is in turn converted to ammonia by reaction with three additional protons and electrons. Indeed, such a cycle has been suggested as operative for a mononuclear, molybdenum-based catalyst.[3] The increasing evidence for substrate coordination at iron in the MoFe cofactor[1b] warrants the continued investigation of iron-based models in a related context.[4]
A Chatt-type cycle at an iron center would require a single ligand scaffold to stabilize low-valent complexes with a π-acidic dinitrogen molecule as well as high valent complexes with a π-basic nitride ligand. Our group has investigated this chemistry in two different ligand-imposed geometries that model the local trigonal symmetry of the coordination environment of iron in the MoFe cofactor. The pseudotetrahedral geometry has been successfully used to stabilize terminal imide[5, 6] and nitride[7, 8] ligands because the π-antibonding d-orbitals of e parentage are usually empty, but most of the N2 complexes in this geometry are dinuclear, bridged species rather than terminal ones due to the difficulty of implementing sufficient steric protection.[9, 10] On the other hand, the trigonal-bipyramidal geometry enforced by tetradentate tris(phosphino)silyl ligands has been shown to stabilize terminal N2 complexes of iron in three different oxidation states,[11] but is less well suited for accomodating Fe–N multiple bonds because of the presence of low-lying, π-antibonding orbitals.[12]
It has been shown that both the trigonal-bipyramidal and tetrahedral geometries can be accessed with a single NL3-type scaffold (L = phosphine[13], N-heterocyclic carbene[8c]) in which the nitrogen donor is labile. We reasoned that a similar behaviour could be accessed with a Lewis-acidic borane in the apical position and therefore turned our attention to the tris(phosphino)borane (TPB) ligand 1 (Scheme 1), which has been introduced recently by the group of Bourissou.[14] Ligand 1 has been shown to form a range of C3-symmetrical metallaboratranes with an (M–B)10 configuration,[15] in which the B–M distance varies from 2.168 Å in [(TPB)Ni] to 2.540 Å in [(TPB)AgCl], pointing to some flexibility in adapting the metal–boron interaction to the Lewis-basicity of the metal.[14b,c] Herein we explore the iron chemistry of ligand 1, showing that the (TPB)Fe fragment accommodates both terminal N2 complexes in low oxidation states and an imidoiron complex at the iron(II) state.
Scheme 1.
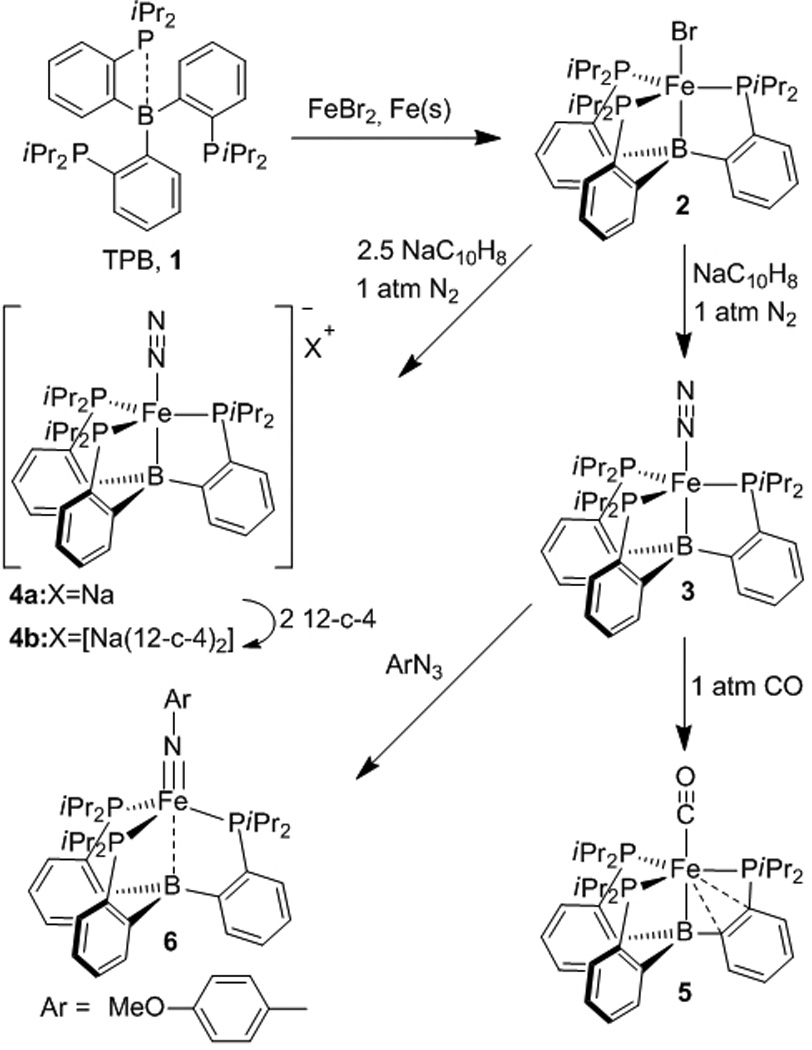
Synthesis and reactivity of ferraboratranes derived from tris(phosphino)borane (TPB) ligand 1.
A convenient entry into the iron chemistry of the TPB ligand was found to be the iron bromide complex 2 (Scheme 1), which was obtained as a greenish-brown powder from the comproportionation of iron(II) bromide and iron powder in the presence of TPB (THF, 90 °C, 66 h). Broad 1H NMR resonances ranging from −23.1 to 92.5 ppm and the solution magnetic moment of 4.1 µB (Evans method) indicate an S = 3/2 ground state. The X-ray diffraction (XRD) crystal structure[16] of 2 (Figure 1) exhibits a slightly distorted trigonal-bipyramidal geometry, with rather long Fe–Br (2.4138(7) Å) and Fe–P (2.3832(13) – 2.4351(11) Å) distances as expected for a high spin complex. The (M–B)7 electron configuration of 2 is, to our knowledge, unprecedented in metallaboratrane chemistry.
Figure 1.

Solid state structures of 2, 4, 5b, and 6. Thermal ellipsoids set at 50%. For clarity, hydrogen atoms as well as the [Na(12-crown-4)2]+ counterion and an unbound diethylether molecule in structure 5b are omitted. Only the main component of a disordered isopropyl group in 2 and only one of the two independent molecules of 6 are plotted.
Reduction of 2 with a slight excess of sodium naphthalide (THF, −60 °C to RT) afforded a brown compound that exhibits a strong IR absorption at 2011 cm−1 and a solution magnetic moment of 2.8 µB. These data are consistent with its formulation as the terminal (Fe–B)8 dinitrogen complex 3 with an S = 1 ground state. Exposure of a yellowish brown solution of 3 to vacuum results in a reversible color change to dark reddish brown accompanied by major changes in the 1H-NMR spectrum, indicating a labile N2 ligand. Crystals of 3 invariably suffered from severe twinning that prevented an unequivoqual structure determination, but its monomeric nature was confirmed by a DOSY experiment showing that 2 and 3 have virtually equal diffusion coefficients.
The nature of complex 3 was further confirmed by reacting it with one atmosphere of CO to afford a brown compound that exhibits a single IR absorption at 1857 cm−1 and is consequently formulated as [(TPB)Fe(CO)] (4). Despite the isoelectronic nature of N2 and CO, compound 4 does not have the expected spin-triplet ground state, but is diamagnetic. The 1H NMR spectrum of 4 resembles that of a C3 symmetrical species, but a broad 31P resonance at δ 87 ppm suggests fluxionality. Upon cooling to −90 °C, this signal splits into three mutually-coupled resonances at δ 94, 87.5, and 16.7 ppm, indicating an asymmetric geometry. This was confirmed by an XRD crystal structure[16] (Figure 1), which reveals an additional interaction of the iron center with two carbon atoms of an aromatic ring of the TPB ligand (Fe–C1 2.337(2) Å; Fe–C2 2.321(2) Å), resulting in an overall η4 coordination of one arm. Partial dearomatization of the iron-bound ring is evident from C–C bond length alternation (See SI). There are a few examples of related η3-coordinated phenylborane moieties,[17] but incorporation of this motif in a metallaboratrane cage structure had been, to the best of our knowledge, unknown.
A quasi-reversible wave at –2.19 V vs Fc/Fc+ in the cyclic voltammogram (CV) of 3 suggested the accessibility of the anionic N2 complex 5, which could indeed be isolated as its sodium salt 5a from the reaction of 2 with 2.5 equivalents of sodium naphthalide (THF, −60 °C to RT). The paramagnetic compound 5a has an S = 1/2 ground state, as shown by its solution magnetic moment of 1.6 µB and a quasi-axial EPR signal (X-Band, toluene/THF glass, 20K) with g1 = 2.23, g2 = 2.09, g3 = 2.05. The IR spectrum of 5a in THF solution exhibits two intense bands at 1918 and 1877 cm−1, which are attributed to the N–N stretch of the free anion Fe–N≡N− and tight ion pair Fe–N≡N−∙∙∙Na+, respectively. In accord with this assignment, only the second band is observed in the less-coordinating solvent diethylether (1862 cm−1) and in the solid state (1879 cm−1). This was additionally confirmed by treating 5a with two equivalents of 12-crown-4 to afford the complex salt 5b, which exhibits only the free-anion band at 1918 cm−1 in THF and at 1905 cm−1 in solid state. XRD crystal structures[16] were obtained for both 5a (SI) and 5b (Figure 1). Both exhibit a distorted trigonal-bipyramidal geometry with P–Fe–P angles of 107.3, 110.3, and 134.6° in 5a and 105.4, 112.3, and 135.0° in 5b, consistent with a Jahn-Teller distortion arising from having three electrons in degenerate orbitals of dxy, and dx2−y2 character. The main difference between 5a and 5b is that the sodium counterion in 5a is terminally bound to the N2 unit while the complex cation [Na(12-crown-4)2]+ is isolated, which appears to have little effect on the N–N distance (1.149(2) in 5a vs 1.144(3) in 5b).
To test the ability of the (TPB)Fe platform to support a two electron redox process, compound 3 was treated with p-methoxyphenyl azide (benzene, RT) to yield the stable diamagnetic imido complex 6 (Scheme 1), which possesses the unusual (Fe–B)6 configuration. Compound 6 was thoroughly characterized by multinuclear NMR, UV-Vis spectroscopy, and XRD.[16] Two independent molecules were found in the asymmetric unit of 6, with an average Fe–N distance (1.668 Å) only marginally longer than that of 1.651 Å found in the pseudotetrahedral iron(II) imide [(BP3)Fe≡N(1-Ad)][nBu4N] ((BP3) = [PhB(CH2PPh2)3]−).[5c] The large Fe–N–C angle (170.2°) additionally supports the description of the Fe–N linkage as a triple bond. The very long Fe–B distance (2.608 Å) causes us to describe the coordination geometry as derived from pseudotetrahedral, as confirmed by a sum of P–Fe–P angles (333.0°) close to the value of 328.4° for a regular tetrahedron.
More insight into the electronic structure of 6 is gained by considering the frontier Kohn-Sham orbitals (Figure 2) obtained from density functional theory (DFT) calculations[18] on the closely related compound [(TPB)Fe≡NPh]. The highest occupied molecular orbital (HOMO) is of dz2 parentage but still possesses some Fe–B σ-bonding character arising from a stabilizing interaction with the empty boron-centered orbital. Two orbitals of dxy, and dx2−y2 parentage, respectively, lie within a 0.4 eV interval of the HOMO. Accordingly, the two orbitals of π-antibonding character derived from dxz and dyz are unoccupied, in accord with the presence of a Fe≡NAr triple bond.
Figure 2.

Frontier Kohn-Sham orbitals of [(TPB)Fe≡NPh] calculated at the B3LYP/6-31G(d) level. Energies in eV.
While metallaboratranes have now been isolated for all transition metals from groups 8 to 11,[20] examples of metal-based reactivity in such compounds are scarce. These include oxidative cleavage of Fe–B[21] and Ni–B[22] bonds as well as reversible B–H bond formation.[23] To our knowledge, the only previously known redox reactions that preserve the metal–boron bond are deprotonation of the (Pt–B)8 cation {[B(mt)3]Pt(H)(PPh3)}+ (mt = methimazolyl) and oxidative additions to the resulting (Pt–B)10 complex {[B(mt)3]Pt(PPh3)}.[24] The series of compounds described herein is unique in that they span four different electronic configurations ((Fe–B)6 in 6 to (Fe–B)9 in 5a,b) that can be interconverted via formal one or two-electrons processes.
This property largely stems from the ability of the Fe–B interaction to respond to changes in the electronic properties of the metal, as is evident from the structural properties summarized in Table 1 and the schematic orbital correlation diagram depicted in Figure 3. In the highly reduced compound 5a, the short Fe–B distance (2.311(2) Å) and strong pyramidalization of the boron atom (Σ(C–B–C) = 329.8°) indicate a strong metal–boron interaction that pulls the iron center into the P3 plane (Σ(P–Fe–P) = 352.2°). As a result, the σ(Fe–B) bonding orbital is expected to be lower in energy than the d orbitals and the electronic structure of 5a,b parallels that of a d7 metal in a trigonal-bipyramidal geometry. This is in accord with the covalent bond classification (CBC) system[19], in which all σ-bonding electrons are substracted from the d-electron count. In contrast, the Fe–B distance of 2.608 Å in the imidoiron(II) complex 6 points to a weak interaction resulting in a pseudotetrahedral geometry (Σ(P–Fe–P) = 333.0°) in which the two sets of d orbitals of e parentage are inverted as compared to 5a,b. The similarity between this orbital diagram and that previously obtained for the related pseudotetrahedral iron(II) imide [(BP3)Fe≡N(1-Ad)][nBu4N][5c] suggests that 6 closely resembles a low-spin d6 compound, even though the dz2 orbital has some σ-bonding character. The geometry of 2 – and presumably 3 – is intermediate between the trigonal-bipyramidal and pseudotetrahedral extremes, and the consequent energetic proximity of the two sets of degenerate orbitals is consistent with the S = 3/2 ground state of 2. Finally, the neutral carbonyl complex 4 adds another twist with the η4-BCCP coordination that results in a Fe–B distance (2.227(2) Å) even shorter than that in 5a,b. It is worth noting that all the observed Fe–B bond length are significantly longer than that of 2.108 Å in the octahedral complex {[B(mttBu)3]Fe(CO)2}.[21] This can be attributed to the lower angular flexibility of the tertiary phosphine donors of TPB (<C(Ar)–P–Fe> = 107.5° in 5a) as compared to the terminal thione (<C–S–Fe> = 99.2° in {[B(mttBu)3]Fe(CO)2)} of the [B(mttBu)3] ligand.
Table 1.
Geometrical Features of Compounds 2, 4, 5a,b, and 6 Relevant to the Fe–B Interaction.
| Compound | Fe–B [Å] | Σ(C–B–C) | Σ(P–Fe–P) |
|---|---|---|---|
| 2 | 2.459 | 341.9° | 341.9° |
| 4 | 2.227 | 352.0° | 357.4° |
| 5a | 2.311 | 329.8° | 352.2° |
| 5b | 2.293 | 331.0° | 352.7° |
| 6 | 2.608[a] | 338.3°[a] | 333.0°[a] |
Averaged over two independent molecules.
Figure 3.

Schematic orbital correlation diagram for compounds 2, 3, 5a,b, and 6.
It is of interest to compare the chemistry of the neutral TPB ligand with that of the closely related anionic (SiPiPr3) ((SiPiPr3) = [2-(iPr2P)C6H4]3Si−).[11] Both ligands are able to stabilize low-valent iron–N2 compounds in a trigonal-bipyramidal geometry, and the degree of activation of the N2 is similar for compounds having the same electrical charge: νNN = 2011 and 1905 cm−1 for 3 and 5b versus 2008 and 1920 cm−1 for [(SiPiPr3)Fe(N2)] and [(SiPiPr3)Fe(N2)][Na(12-crown-4)2], respectively. This observation may at first appear surprising because the (SiPiPr3) complexes have one more valence electron than their TPB counterparts, but it can be easily understood by considering that the additional electron lies in an orbital of dxy or dx2−y2 parentage that has no overlap with the σ and π orbitals of N2. A major difference between TPB and (SiPiPr3) resides in the greater flexibility of the Fe–B bond length as compared to Fe–Si,[11] which explains why the triply-bonded imido complex 6 is stable while imidoiron complexes of (SiPiPr3) are short-lived intermediates in the formation of azo compounds from aryl azides.[12, 25]
In summary, the TPB ligand has been shown to stabilize both low-valent iron–dinitrogen complexes and a mid-valent imido species with a Fe≡NAr triple bond, thanks to its ability to shuttle between trigonal-bipyramidal and pseudotetrahedral geometries by elongation of the apical iron–boron bond. The adaptability of the coordination environment in ferraboratranes derived from triphosphinoborane ligands makes them promising candidates as functional models for biological N2 fixation, which will be the subject of further investigation in our laboratories.
Footnotes
This work was supported by the NIH (GM 070757). M.-E. M. acknowledges a Fellowship for Prospective Researchers from the Swiss National Science Foundation. We thank Charlene Tsay and Larry Henling for crystallographic assistance, as well as David VanderVelde for assistance with DOSY experiments and Angelo Di Bilio for EPR measurements.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the authors.
References
- 1.a) Hoffman BM, Dean DR, Seefeldt LC. Acc. Chem. Res. 2009;42:609–619. doi: 10.1021/ar8002128. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DR. Acc. Chem. Res. 2005;38:208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]; c) Einsle O, Tezcan FA, Andrade SL, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 2.Chatt J, Dilworth JR, Richards RL. Chem. Rev. 1978;78:589–625. [Google Scholar]
- 3.a) Yandulov DV, Schrock RR. Science. 2003;301:76–78. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]; b) Yandulov DV, Schrock RR. Inorg. Chem. 2005;44:1103–1117. doi: 10.1021/ic040095w. [DOI] [PubMed] [Google Scholar]
- 4.a) Hazari N. Chem. Soc. Rev. 2010;39:4044–4056. doi: 10.1039/b919680n. [DOI] [PubMed] [Google Scholar]; b) Crossland JL, Tyler DR. Coord. Chem. Rev. 2010;254:1883–1894. [Google Scholar]; c) Holland PL. Dalton Trans. 2010;39:5415–5425. doi: 10.1039/c001397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Brown SD, Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:322–323. doi: 10.1021/ja028448i. [DOI] [PubMed] [Google Scholar]; b) Brown SD, Peters JC. J. Am. Chem. Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]; c) Brown SD, Peters JC. J. Am. Chem. Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]; d) Thomas CM, Mankad NP, Peters JC. J. Am. Chem. Soc. 2006;128:4956–4957. doi: 10.1021/ja0604358. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mehn MP, Peters JC. J. Inorg. Biochem. 2006;100:634–643. doi: 10.1016/j.jinorgbio.2006.01.023. [DOI] [PubMed] [Google Scholar]; f) Mehn MP, Brown SD, Jenkins DM, Peters JC, Que Jr L. Inorg. Chem. 2006;45:7417–7427. doi: 10.1021/ic060670r. [DOI] [PubMed] [Google Scholar]; g) Lu CC, Saouma CT, Day MW, Peters JC. J. Am. Chem. Soc. 2007;129:4–5. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Verma AK, Nazif TN, Achim C, Lee SC. J. Am. Chem. Soc. 2000;122:11013–11014. [Google Scholar]; b) Nieto I, Ding F, Bontchev RP, Wang H, Smith JM. J. Am. Chem. Soc. 2008;130:2716–2717. doi: 10.1021/ja0776834. [DOI] [PubMed] [Google Scholar]
- 7.a) Betley TA, Peters JC. J. Am. Chem. Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]; b) Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc. Natl. Acad. Sci. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Scepaniak JJ, Fulton MD, Bontchev RP, Duesler EN, Kirk ML, Smith JM. J. Am. Chem. Soc. 2008;130:10515–10517. doi: 10.1021/ja8027372. [DOI] [PubMed] [Google Scholar]; b) Scepaniak JJ, Young JA, Bontchev RP, Smith JM. Angew. Chem. 2009;121:3204–3206;. doi: 10.1002/anie.200900381. Angew. Chem. Int. Ed.2009, 48, 3158-3160; [DOI] [PubMed] [Google Scholar]; c) Vogel C, Heinemann FW, Sutter J, Anthon C, Meyer K. Angew. Chem. 2008;120:2721–2724. doi: 10.1002/anie.200800600. Angew. Chem. Int. Ed. 2008, 47, 2681-2684. [DOI] [PubMed] [Google Scholar]
- 9.Betley TA, Peters JC. J. Am. Chem. Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]
- 10.a) Chomitz WA, Arnold J. Chem. Commun. 2007:4797–4799. doi: 10.1039/b709763h. [DOI] [PubMed] [Google Scholar]; b) Chomitz WA, Arnold J. Dalton Trans. 2009:1714–1720. doi: 10.1039/b821954k. [DOI] [PubMed] [Google Scholar]
- 11.a) Mankad NP, Whited MT, Peters JC. Angew. Chem. 2007;119:5870–5873. doi: 10.1002/anie.200701188. Angew. Chem. Int. Ed. 2007, 46, 5768-5771; [DOI] [PubMed] [Google Scholar]; b) Whited MT, Mankad NP, Lee Y, Oblad PF, Peters JC. Inorg. Chem. 2009;48:2507–2517. doi: 10.1021/ic801855y. [DOI] [PubMed] [Google Scholar]; c) Lee Y, Mankad NP, Peters JC. Nat. Chem. 2010;2:558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mankad NP, Müller P, Peters JC. J. Am. Chem. Soc. 2010;132:4083–4085. doi: 10.1021/ja910224c. [DOI] [PubMed] [Google Scholar]
- 13.a) di Vaira M, Ghilardi CA, Sacconi L. Inorg. Chem. 1976;15:1555–1561. [Google Scholar]; b) MacBeth CE, Harkins SB, Peters JC. Can. J. Chem. 2005;83:332–340. [Google Scholar]
- 14.a) Bontemps S, Bouhadir G, Dyer PW, Miqueu K, Bourissou D. Inorg. Chem. 2007;46:5149–5151. doi: 10.1021/ic7006556. [DOI] [PubMed] [Google Scholar]; b) Bontemps S, Bouhadir G, Gu W, Mercy M, Chen C-H, Foxman BM, Maron L, Ozerov OV, Bourissou D. Angew. Chem. 2008;120:1503–1506. doi: 10.1002/anie.200705024. Angew. Chem. Int. Ed. 2008, 47, 1481-1484. [DOI] [PubMed] [Google Scholar]; c) Sircoglou M, Bontemps S, Bouhadir G, Saffon N, Miqueu K, Gu W, Mercy M, Chen C-H, Foxman BM, Maron L, Ozerov OV, Bourissou D. J. Am. Chem. Soc. 2008;130:16729–16738. doi: 10.1021/ja8070072. [DOI] [PubMed] [Google Scholar]
- 15.To keep track of the electron count without arbitrarily assigning the σ(Fe–B) bonding electrons to either Fe or B, we make use of the (M–B)n notation recently introduced by Hill Hill AF. Organometallics. 2006;25:4741–4743.
- 16.CCDC 797913 (2), 797914 (4), 797915 (5a), 797916 (5b), and 797917 (6) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 17.a) Oakley SR, Parker Kyle D., Emslie DJH, Vargas-Baca I, Robertson CM, Harrington LE, Britten JF. Organometallics. 2006;25:5835–5838. [Google Scholar]; b) Sircoglou M, Bontemps S, Mercy M, Miqueu K, Ladeira S, Saffon N, Maron L, Bouhadir G, Bourissou D. Inorg. Chem. 2010;49:3983–3990. doi: 10.1021/ic901896z. [DOI] [PubMed] [Google Scholar]
- 18.Geometry optimized at the the B3LYP/6-31G(d) level using Gaussian 03, Revision D.01; M. J. Frisch et al., Gaussian, Inc., Wallingford CT, 2004. Full reference in Supp. Info.
- 19.a) Green MLH. J. Organomet. Chem. 1995;500:127–148. [Google Scholar]; b) Parkin G. Organometallics. 2006;25:4744–4747. [Google Scholar]; c) Landry VK, Melnick JG, Buccella D, Pang K, Ulichny JC, Parkin G. Inorg. Chem. 2006;45:2588–2597. doi: 10.1021/ic051975t. [DOI] [PubMed] [Google Scholar]
- 20. Hill AF, Owen GR, White AJP, Williams DJ. Angew. Chem. 1999;111:2920–2923. Angew. Chem. Int. Ed.1999, 38, 2759-2761; Braunschweig H, Dewhurst RD, Schneider A. Chem. Rev. 2010;110:3924–3957. doi: 10.1021/cr900333n. and references therein Fontaine F-G, Boudreau J, Thibault M-H. Eur. J. Inorg. Chem. 2008:5439–5454. and references therein.
- 21.Figueroa JS, Melnick JG, Parkin G. Inorg. Chem. 2006;45:7056–7058. doi: 10.1021/ic061353n. [DOI] [PubMed] [Google Scholar]
- 22.Pang K, Tanski JM, Parkin G. Chem. Commun. 2008:1008–1010. doi: 10.1039/b714466k. [DOI] [PubMed] [Google Scholar]
- 23.a) Crossley IR, Hill AF. Dalton Trans. 2008:201–203. doi: 10.1039/b712058c. [DOI] [PubMed] [Google Scholar]; b) Crossley IR, Hill AF, Willis AC. Organometallics. 2010;29:326–336. [Google Scholar]
- 24.a) Crossley IR, Hill AF. Organometallics. 2004;23:5656–5658. [Google Scholar]; b) Crossley IR, Hill AF, Willis AC. Organometallics. 2008;27:312–315. [Google Scholar]
- 25.The heavier analogues [(SiPiPr3)MIII(NAr)] (M = Ru, Os) are stable, but they have a significant N-centered radical character which indicates a low M–N bond order. Takaoka A, Gerber LCH, Peters JC. Angew. Chem. 2010;122:4182–4185. Angew. Chem. Int. Ed. 2010, 49, 4088-4091.