

Abstract
Purpose
Genetic analysis of bladder cancer (BC) has revealed a number of frequently altered genes, including frequent alterations of the telomerase (TERT) gene promoter; though few altered genes have been functionally evaluated. Our objective is to characterize alterations observed by exome sequencing and sequencing of the TERT promoter, and to examine the functional relevance of KDM6A, a frequently mutated histone demethylase, in BC.
Experimental Design
We analyzed BC samples from 54 U.S. patients by exome and targeted sequencing and confirmed somatic variants using normal tissue from the same patient. We examined the biological function of KDM6A using in vivo and in vitro assays.
Results
We observed frequent somatic alterations in BAP1 in 15% of tumors, including deleterious alterations to the deubiquitinase active site and the nuclear localization signal. BAP1 mutations contribute to a high frequency of tumors with BRCA pathway alterations and were significantly associated with papillary histologic features in tumors. BAP1 and KDM6A mutations significantly co-occurred in tumors. Somatic variants altering the TERT promoter were found in 69% of tumors but were not correlated with alterations in other BC genes. We examined the function of KDM6A, altered in 24% of tumors, and show depletion in human BC cells enhanced in vitro proliferation, in vivo tumor growth, and cell migration.
Conclusions
This study is the first to identify frequent BAP1 and BRCA pathway alterations in BC, show TERT promoter alterations are independent of other BC gene alterations, and show KDM6A loss is a driver of the BC phenotype.
Keywords: bladder neoplasms, mutation, biomarker, survival analysis, next generation sequencing, tumor suppressor, chromatin remodeler, TERT, KDM6A, UTX, BAP1, BRCA
Introduction
Bladder cancer (BC) is the 5th most common cancer worldwide (1) with 386,300 new cases and 150,200 deaths in 2008 (1). BC is classified into two types that are thought to be driven by mutations in different sets of genes (2). The low-grade and non-muscle invasive (NMI) form is successfully treated with transurethral surgery and intravesical immunotherapy (2) and accounts for ∼80% of all cases. Alterations associated with this form include mutation or over expression of HRAS, fibroblast growth factor receptor 3 (FGFR3), and the histone lysine (K)-specific demethylase 6A (KDM6A/UTX)(2-4). The remaining 20% of cases are muscle invasive (MI) and, despite aggressive treatment with cystectomy, have a 5 year survival rate of <50% (2). Over expression or mutation of human epidermal growth factor receptor-2 (HER2), epithelial growth factor receptor (EGFR), TP53, and RB1 are associated with MI BC (2-4).
Next generation sequencing (NGS) of the exomes of nine BC tumors revealed frequent somatic alterations in 54 genes, including 16 new BC genes mutated in ≥5% of cases (3). Eight were genes encoding chromatin modifying/remodeling enzymes, ARID1A, CHD6, CREBBP, EP300, MLL, MLL3, NCOR1, and KDM6A. Recently, analysis of 99 BC tumors by whole genome, whole exome, and transcriptome NGS (5) revealed additional altered genes, including STAG2 and ESPL1, encoding proteins involved in the spindle checkpoint of the cell cycle, and a recurrent fusion involving FGFR3 and TACC3, another spindle checkpoint gene (6).
All tumors analyzed to date by NGS were from Chinese patients. Thus, findings may reflect the regional ethnicity of the patients, differences in exposure to damaging agents, or lifestyle factors. Here we determine by exome sequencing of 14 tumors if novel alterations could be found in a cohort of non-Asian patients diagnosed with BC in the United States (US). Given the recent report of TERT gene promoter mutations in BC (7), we examined the TERT promoter using targeted sequencing in the 14 tumors and in an additional 40 tumors. Healthy, non-cancerous tissue DNA was available to determine variants that were tumor specific. Finally, given the frequent alteration of KDM6A found by this study and in Chinese patient BC tumors, we examined whether KDM6A expression has functional in vitro, in vivo, and clinical prognostic impact.
Results
Exome Sequencing of 14 Bladder Tumors from U.S. Patients
We subjected 14 primary urothelial bladder tumors to exome sequencing to characterize genomic variants in U.S. BC patients (Supplementary Table S1). This yielded an average of >7.6 gigabases per sample and an average mean NGS read depth of 92× (Supplementary Table S2). Grouping NGS-predicted variants by the type of nucleotide change revealed significant enrichment in C>T and G>A changes, similar to previous reports in bladder and other cancers (Fig. 1A, top panel) (3, 8). We identified putative somatic variants by removing variants observed at a frequency >1% in the 1000 Genomes Project and variants in known segmental duplications (9). This yielded 9312 missense, 348 nonsense, 216 splice junction (SJ), and 653 coding insertion/deletion (indel) candidate variants.
Figure 1. Altered BC genes.
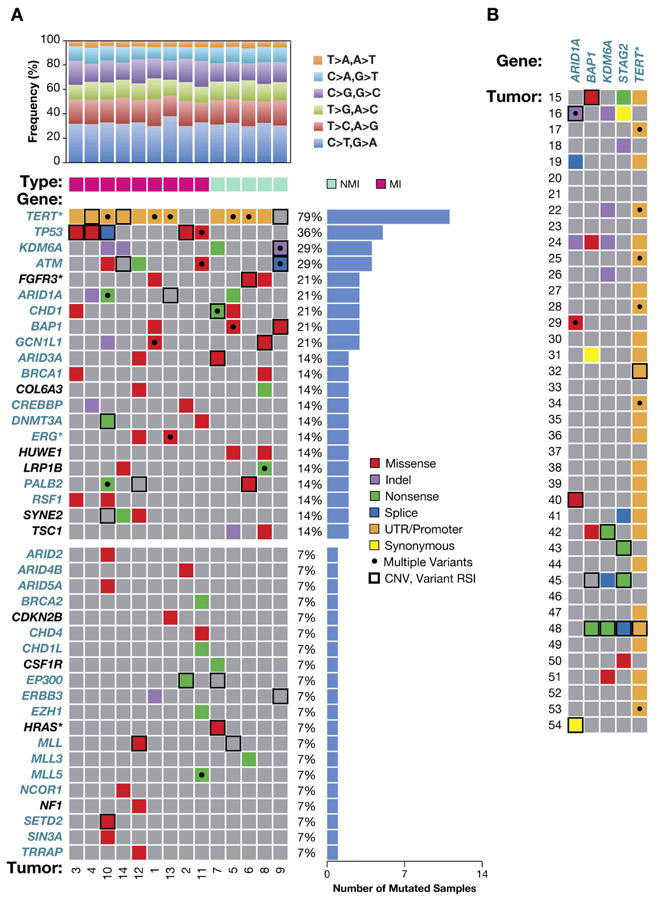
A, 41 BC genes with somatic nonsynonymous and SJ alterations. Top histogram, NGS predicted variants by nucleotide change for non-muscle invasive (NMI, stage Ta, T1) and muscle invasive (MI, stage T2-T4) tumors. Central panel, genes (left) and CRM-DA functions (teal color, see text); asterisk, a proven or likely oncogene; black dot, tumor has ≥1 somatic mutation; histogram, right side, percent of tumors with somatic gene mutation; bold or hatched box, CNV; bottom, tumor IDs (1-14). B, Somatic alterations in five CRM-DA BC genes (top) in 40 validation tumors (left side), annotated as in panel A.
We analyzed matched tumor and normal tissue DNA (Supplementary Table S1) using PCR and Sanger sequencing to confirm NGS-predicted variants and identify somatic from germline alterations. Nonsynonymous and SJ variants selected for validation were in genes with >1 NGS-predicted alteration, known cancer and related genes, genes with clearly deleterious variants (frameshift and nonsense), and several randomly selected genes. We examined 316 variants and confirmed 228 (72%), 112 were somatic (Supplementary Table S3) and 116 were germline, including 43 novel germline variants (Supplementary Table S4).
Somatic Alterations in Four Novel Bladder Cancer Genes
Validated somatic variants were observed in 67 genes including 10 indels, 22 nonsense, 78 missense, and 2 variants in SJs. Twenty genes were mutated in >1 tumor, including genes known to be mutated in BC such as FGFR3 (10), TP53 (11), and TSC1 (12) (Fig. 1A, central panel; Supplementary Table S3). Eight genes were altered in ≥3 tumors and are referred to as frequently mutated genes (FMG), including previously identified BC genes, KDM6A and ARID1A, altered in four and three tumors, respectively (3). Twenty additional genes with a somatic alteration are also included in Fig. 1A (bottom) based on previous identification as a cancer or cancer-related gene. This selection is further supported by the types of somatic alterations. For example, MLL3 is altered in tumor 6 by a somatic c.C8695T (p.Q2899X) and was altered in 25% of TCGA BC tumors, and EP300 is altered in tumor 2 by a somatic c.G3052T (p.E1018X) and was altered in 17% of TCGA BC tumors (13, 14).
To our knowledge, four altered genes are novel BC FMGs when compared to other studies (5): BRCA1 associated protein-1 (BAP1) located on chromosome (chr) 3p21, chromodomain helicase DNA binding protein 1 (CHD1, chr. 5q21), chromodomain helicase DNA binding protein 1-like (CHD1L, chr. 1q21), and GCN1 general control of amino-acid synthesis 1-like 1 (GCN1L1, chr. 12q24). These genes are of particular interest as they encode proteins that have distinct roles in chromatin remodeling (CRM)(Fig. 1A, gene names in teal). These FMGs increase the number of BC genes encoding proteins with CRM and DNA-associated (DA) (CRM-DA) functions. An average of 4.7 somatic sequence changes to CRM-DA genes occurred in each tumor, range 2-13, indicating a substantial contribution from altered CRM-DA genes to BC.
Somatic CHD1, CHD1L, and GCN1L1 Alterations in Bladder Cancer
Four alterations in CHD1 were confirmed as somatic (Fig. 1A). Recurrent somatic C>T variants in tumors 3 and 5 altered a highly conserved proline 1684 to serine (p.P1684S) and a somatic G>T in tumor 7 introduced a nonsense codon (p.E272X), truncating most of the 1710 amino acid (aa) protein (NM_001270). CHD1 encodes a CHD family-related protein that alters chromatin structure and influences transcription via SNF2-related helicase/ATPase and chromo (chromatin organization modifier) domains (15). Somatic alterations were observed in genes related to CHD1, including a truncation (p.Q728X) of CHD1-like (CHD1L) and a p.Q1833E in CHD4. Altered expression of CHD1L has been implicated in BC (16) and germline alterations are associated with congenital anomalies of the kidney and urinary tract (17) suggesting CHD1L is a valid BC gene. A total of 4/14 tumors (29%) showed somatic alteration of CHD genes. CHD2, CHD4, CHD5, and CHD6 were altered in 4%, 3%, 4%, and 7%, respectively, of Chinese patient tumors in a total of 15/99 tumors (15%)(5).
Four somatic alterations of GCN1L1 were observed and these may lead to loss of function, as indicated by a single base deletion in tumor 10 causing a frameshift and truncation after codon 1777 (2671 aa full length, NM_006836)(Fig. 1A). Two somatic, synonymous changes were observed previously in 99 Chinese patient tumors (5). The current data indicates, to our knowledge, the first evidence of somatic nonsynonymous GCN1L1 alterations in BC. GCN1L1 may link AA metabolism to chromatin remodeling and gene expression through roles in regulating GCN2 kinase and as a transcriptional co-regulator in Mediator complexes (18). Future analyses of CHD1, CHD1L, CHD-related genes, and GCN1L1 in additional BC tumors are planned to determine accurate somatic mutation frequencies in BC.
BAP1 and BRCA Pathway Defects in Bladder Cancer
Exome sequencing revealed four somatic missense variants in BAP1 in 3 tumors (Fig. 1, Supplementary Table S3). To confirm BAP1 as a BC gene, PCR and Sanger sequencing was performed on an additional 40 bladder tumors with matched normal tissue from U.S. patients (Supplementary Table S1). Five additional somatic alterations were observed and, in total, BAP1 was altered in 8/54 tumors (15%).
The likely functional effects of BAP1 mutations are predominantly loss of function, such as introduction of a stop codon (p.E257X) and two somatic changes altering histidine 169 to an arginine (p.H169R) or a glutamine (p.H169Q)(Fig. 2A). Histidine 169 has a critical function as the predicted proton donor residue in the ubiquitin hydrolase active site (Uniprot DB). A comparative three dimensional model of BAP1 was generated at ModBase and the structure confirms that p.H169 occupies a critical position directly across from the active site nucleophile, cysteine 91 (Fig. 2B, adapted from ModBase)(19). Both substitutions alter the active site: histidine to an arginine introduces a significantly longer side chain and histidine to a glutamine introduces an uncharged for a positively charged residue. BAP1 was also altered in two tumors by recurrent, nonconservative substitutions of a glycine for arginine 718 (p.R718G). Arginine 718 is one of four charged arginine residues in a highly conserved nuclear localization signal (NLS) consisting of AAs 717-722 (Fig. 2C) (19, 20). Previously published alteration of arginines to alanines in the NLS, including p.R718, completely blocked cytoplasmic to nuclear shuttling of the protein and abolished BAP1 deubiquitinase activity. These observations are consistent with deleterious somatic alterations targeting a tumor suppressor in BC, similar to previous observations of BAP1 in other cancers (21).
Figure 2. Chromatin remodeling genes frequently altered in BC.
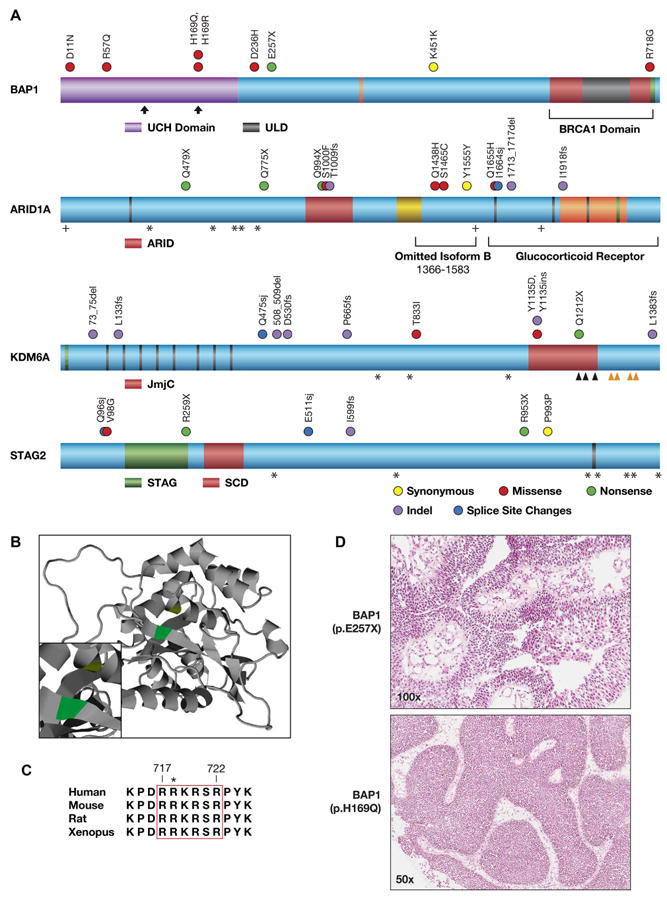
A, Locations of somatic alterations on annotated proteins. BAP1: ubiquitin C terminal hydrolase domain (UCH, purple); HCF-1 binding motif (orange line); BRCA1 binding region (BRCA1 Domain, red); UCH37-like domain (ULD, gray); NLS, aa 717-722 (green); active site residues (vertical arrows). Adapted from (21, 51). ARID1A: LXXLL motif (black line); AT-interaction domain (ARID, red); HIC1 binding domain (yellow); Pfam homology domain (orange); B/C box (green); acetylation site (+); region omitted in isoform B, aa 1366-1583 (left bracket); GR binding domain (Glucocorticoid Receptor, right bracket). KDM6A: alanine rich region (green); 8 TPR repeats (black lines); Jumonji C domain (JmjC, red); iron (ˆ) and zinc (ˆ) binding sites. STAG2: STAG domain (STAG, green); SCD domain (SCD, red); serine rich region (black line). In all proteins, recurrent phosphorylation sites are shown (*). B, A ribbon diagram of BAP1 residues 5-291 (UniProtKB Q92560) is based on the crystal structure of homologous UCHL3-UbVME (19). Active site residues, H169 (proton donor, green) and cysteine 91 (nucleophile, yellow) are shown (inset at higher magnification). C, Alignment of NLS residues in BAP1 orthologues. Residue numbers are shown for human BAP1 (top), p.R718 mutation (asterisk). Adapted from (20). D, Papillary features in H&E stained tumor sections from bladder tumors with somatic BAP1 mutations. Top: tumor 48, a low grade papillary urothelial carcinoma; bottom: tumor 9, a low grade urothelial carcinoma, inverted papillary type.
BAP1 protein binds breast cancer 1 (BRCA1)(22) and loss of function BAP1 mutations may target the BRCA DNA repair pathway (23). This is supported by 16 somatic alterations in BAP1 and four BRCA DNA repair pathway genes in 9/14 tumors (64%)(Fig. 1A; Supplementary Table S3). A tenth tumor was homozygous for a truncating BRCA2 germline allele, totaling a surprising 10/14 (71%) BC tumors with BRCA pathway alterations (Supplementary Table S4). Eight tumors were altered by clearly deleterious mutations, including four truncations, and approximately one pathway gene per tumor was inactivated (6 tumors). Ataxia telangiectasia mutated (ATM) was altered by six somatic sequence changes in four tumors, including alteration of a conserved SJ (c.5497-1G>C) and a p.Q1636X in tumors 9 and 12, respectively. BRCA1 was altered by somatic missense p.P364A and p.S1286T in tumors 3 and 8, respectively. Breast cancer 2 (BRCA2) was altered by a homozygous germline p.K3326X and a somatic p.Q3066X in tumors 7 and 11, respectively. Finally, partner and localizer of BRCA2 (PALB2) was altered by somatic p.S535C and p.Q613X in tumors 6 and 10, respectively. These alterations define a majority of BC tumors with a common DNA repair deficiency that might be exploited by targeted therapies such as PARP inhibitors (23).
BAP1 mutation in kidney tumors is associated with rhabdoid morphology (21) and we examined this phenotype in BC tumors. Blinded to the BAP1 status, one author (S.M.L) classified a set of 16 hematoxylin and eosin–stained (H&E) stained tumor sections, eight with a BAP1 mutation and eight wild type (WT). This revealed papillary features in six tumors (Fig. 2D, Supplementary Table S5), of which five had a somatic BAP1 alteration. Three tumors with BAP1 mutations did not exhibit papillary features in the tumor section analyzed, though additional tumor sections were not available to examine other regions of these tumors. Our data suggests that papillary features in some bladder tumors are associated with somatic alteration of BAP1.
We compared BAP1 somatic mutations in 54 U.S. patient tumors to 99 Chinese patient tumors (5) and found BAP1 was altered at a significantly greater frequency in the U.S. cohort (15% vs. 1%, p=0.003). This remained marginally significant after accounting for differences in tumor grade and stage (p=0.037). To determine if other CRM-DA genes were altered at different frequencies between these cohorts, we sequenced the three most frequently mutated CRM-DA BC genes, ARID1A, KDM6A, and STAG2 (3, 5, 24) in the same set of 40 validation tumors (Fig. 2A, Supplementary Table S1). ARID1A, KDM6A, and STAG2 were altered by somatic sequence changes in 17%, 24%, and 17%, respectively, of 54 U.S. patient tumors (Supplementary Table S3). Cumulatively, the four genes were altered in 25/54 (46%) of U.S. tumors. Guo et al. (5) observed ARID1A, KDM6A, and STAG2 alterations in 15%, 30%, and 11%, respectively, of 99 Chinese patient tumors. The somatic mutation frequencies of ARID1A, KDM6A, and STAG2 were similar and only BAP1 was significantly different. Thus, BAP1 appears to be preferentially altered in U.S. BC patients, perhaps due to patient ethnicity, exposure, or lifestyle factors.
Copy Number Alterations in Bladder Cancer Genes
Exome and Sanger sequencing revealed allelic imbalance at sites of somatic and heterozygous germline sequence variants in tumor versus normal tissue DNA, which could be classified as copy number variation (CNV)(Fig. 1, bold boxes). Somatic and heterozygous germline variants with high relative signal intensity (RSI) scores ≥ 0.7 (mutant allele signal / WT + mutant allele signal) are reliable indicators of CNVs in cancer (25, 26). The 41 BC genes in Figure 1A panel were altered by 27 CNVs with an average of 2 per tumor, range 0-5. These genes were altered by 100 somatic sequence changes with an average of 7.1/tumor, range 4-16. In total, an average of 9.1 somatic alterations/tumor, range 4-20, were observed which were predominantly sequence changes (78%) compared to CNVs (21%). Two or more alterations potentially affecting both alleles of a BC gene were detected in 30 genes with an average of 2.1 multiply-altered genes/tumor, range 0-4. Similar results were observed for five genes analyzed in panel B (Fig. 1B). Somatic alterations (n=77) were comprised of 64 sequence changes (83%) and 13 CNVs (17%), and 17 tumors (43%) displayed at least one gene with >1 alteration. This data indicates homozygous loss of function of tumor suppressor genes or homozygous activation of oncogenes affects 2 genes/tumor and indicates potential high priority therapeutic targets.
Rare, Deleterious Germline Alleles in Bladder Cancer
Exome and targeted sequencing of 54 tumors identified 11 deleterious germline variants resulting in nonsense, frameshift truncation, and SJ alterations. Seven alleles caused frameshift truncations, three introduced nonsense codons, and one altered a highly conserved SJ nucleotide (bold, Supplementary Table S4). Two previously identified cancer genes were altered, BRCA2 (p.K3326X, rs11571833) and RB1 (p.Q846fs, novel). One novel allele in tumor 9 truncated the fibroblast growth factor (FGF) binding protein 1 (FGFBP1)(c.6delG, p.K2fs), potentially indicating a new (likely rare) disease-associated allele in the FGF signaling pathway. Lastly, we describe tumors with germline TERT promoter variants, including identical variants confirmed as either germline or somatic in distinct tumors. This data suggests a greater than expected contribution to these supposed sporadic cases of BC from rare germline variants.
Somatic and Germline Alteration of the TERT Promoter
Somatic TERT promoter alterations were recently reported in melanoma, glioma, and in small sample sets from other cancers including bladder (7, 27, 28). We examined the proximal promoter of TERT in 54 bladder tumors and identified 93 single nucleotide substitutions in 48/54 tumors, comprised of 29 unique variants (Fig. 1, 3; Supplementary Table S6). Of these, 42 nucleotide substitutions, consisting of 20 unique variants, were germline in 30/54 tumors (56%) and 19 were novel (asterisk, Fig. 3B). The most common variant, c.-245T>C (rs2853669), was observed in 32/54 tumors (59%). This variant was confirmed as germline in tumors from 23 patients and as a novel somatic (tumor-specific) change in 9 tumors. Novel variants at two other sites (c.-113C>T, and c.-212C>G and c.-212C>T) were also confirmed as both germline and somatic in distinct tumors.
Figure 3. TERT promoter variants in BC.
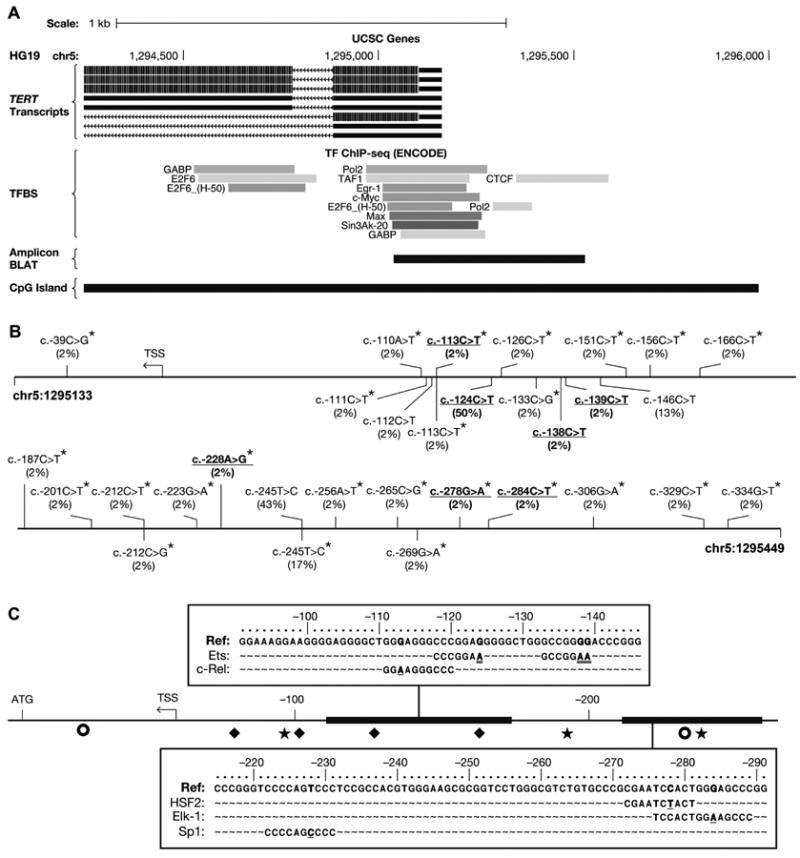
A, The proximal TERT locus in the UCSC Genome Browser with HG19 genomic coordinates (Hg19), TERT isoforms (TERT transcripts), TFBS from ENCODE ChIP-seq, the amplicon location (Amplicon BLAT), and a CpG-rich region (CpG Island, green). B, The TERT promoter amplicon (5′, top left, to 3′, bottom right) with germline variants (above) and somatic variants (below) the genomic reference sequence (solid line). Variant annotation is based on the TERT coding strand (not shown). Frequencies of variants (n=54, in parentheses), novel variants (asterisk), and variants in new TFBS (blue, details in Fig. 3C) are indicated. C, TFBS in the TERT promoter (bold horizontal line, center) are shown relative to the transcription start site (TSS) and the protein-coding start codon (ATG): ◆, Sp1; ○, E-box; and ✶, Ets TFBS. New TFBS created by novel (blue) and previously identified variants (red) are shown in enlarged regions.
We observed 51 somatic alterations in 37/54 tumors (69%) consisting of 11 unique variants, including four novel variants, c.-111C>T, -113C>T, -133C>G, and -212C>G (asterisk, Fig. 3B, Supplementary Table S6). Eleven tumors (20%) had >1 somatic alteration and one sample (tumor 17) had four variants in a cluster, c.-111, c.-112, c.-113, and c.-124. Three somatic variants previously reported in other cancers (7, 27, 28) were observed in multiple bladder tumors, c.-124C>T in 27 tumors (50%), c.-245T>C in nine tumors (17%) tumors, and c.-146C>T in seven tumors (13%), suggesting these nucleotides are recurrent targets in BC. Cytosine to thymine alterations comprised 39/51 (76%) of the observed somatic TERT promoter alterations.
Similar to a previous report (28), a subset of somatic and germline variants were predicted to create new transcription factor (TF) binding sites (TFBS) (Fig. 3C). Variant c.-113C>T creates a potential c-Rel binding site; variants c.-124C>T, c.-138C>T, and c.-139C>T potential Ets binding sites; variant c.-228A>G, a potential Sp1 binding site; variant c.278G>A, a potential HSF2 binding site, and variant c.-284C>T, a potential Elk-1 binding site. Additionally, variants altered sequences shown to bind TFs by chromatin immunoprecipitation followed by NGS (ENCODE data tracks in the UCSC Genome Browser)(Fig. 3A), including Pol2, TAF1, EGR-1, Myc, Max, Sin3A, and CTCF. Variants c.-110A>T, c.-111C>T, c.-112C>T, c.-113C>T, c.-126C>T, and c.-133C>G altered multiple Sp1 binding sites causing an expected loss of TF binding (29). Lastly, three promoter variants altered CpG dinucleotides or neighboring residues (+/-1 bp), including a somatic c.-269G>A, germline c.-306G>A, and germline c.-329C>T. These may affect regulation of promoter methylation.
Association of Mutations with Patient and Tumor Variables
Next we examined five genes for which we had somatic mutation status in 54 tumors, ARID1A, BAP1, KDM6A, STAG2, and TERT, for correlations in somatic mutations and for associations between mutation status and clinical factors. We found only BAP1 and KDM6A mutations were correlated (p=0.017, Supplementary Table S7). Somatic alteration of the TERT promoter occurred in tumors both with and without mutations in each of the other genes. This data indicates likely independent contributions of the mutations in these five genes to the tumor cell phenotype. No significant associations between the mutation status of the five genes and clinical factors were observed (not shown).
Network Analysis Reveals the Importance of KDM6A in Bladder Cancer
To better understand the potential interactions between altered BC genes, we performed network analysis on genes with confirmed somatic mutations in the current study and a recent study of 99 Chinese bladder tumor exomes (5). This identified several robust interacting networks and biological functional groups (Supplementary Table S8). The highest scoring network (p<10-68) was comprised of 34 mutated genes known to bind Ubiquitin C (UBC)(Supplementary Figure S1). The second highest scoring network (p<10-46) contained proteins encoded by the third through the fifth most mutated genes in the Chinese cohort (ARID1A, CREBBP, and EP300)(5) (Supplementary Figure S2). Genes encoding enzymes associated with the deubiquitinylating pathway, including BAP1 which is mutated in this study (BAP1, DUB, USP21, USP26, USP31, USP34, USP36, USP38, and USP48) and DNA methylation (ARID4B, CHD4, CHD3, MTA, and SIN3A), were well represented in this network. The third highest scoring network (p<10-42) contained TP53 along with four genes involved in DNA methylation (DNMT1, DNMT3A, MLL3, and MLL5) and multiple genes associated with chromosomal structural elements and post-translational modifiers of histones H3 and H4 (Fig. 4A).
Figure 4. Biological and co-mutational connections of KDM6A to a network of genes with somatic mutations in BC.
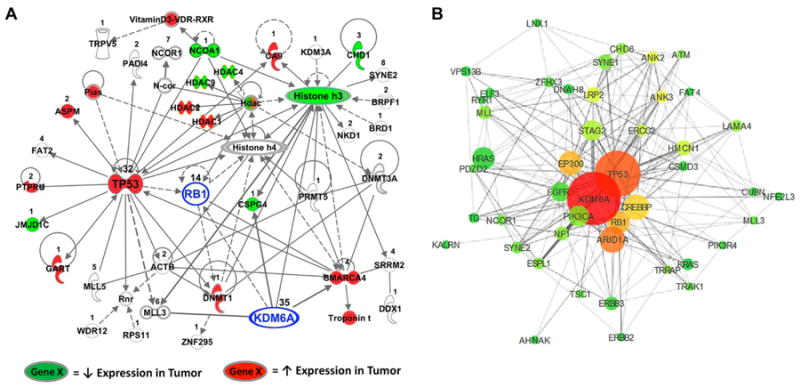
A, The third most significant network (Fisher's Exact Test, p<10-42) constructed by IPA from genes with confirmed somatic mutations in BC, including KDM6A and RB1 (highlighted in blue). The number of independently identified mutations is listed above each respective gene. Evidence of direct (solid lines) or indirect (dashed lines) relationships between gene products is shown. Colored genes indicate decreased (green) or increased (red) gene expression in tumors compared to normal bladder samples in at least two out of three patient cohorts (FDR < 5%). B, The network was created using the co-mutation rate and the frequency of mutation to plot the mutations in 3D space. The size of each gene (node) is representative of the times it was mutated in different tumors. The connection lines (edges) in the network indicate whether the genes were found mutated together in the same tumor, with the length and thickness of each line shorter and wider with an increasing co-mutation rate. The nodes and edges of our model have been filtered to show only those genes found mutated in five or more tumors and edges between genes that were mutated together more than once. Node color corresponds to a gene's relative centrality score, with genes closer to the center of the network red and genes farther away turning to yellow then green at the periphery.
KDM6A is frequently mutated in both U.S. and Chinese tumors and alterations are significantly correlated with somatic BAP1 alterations. KDM6A has limited protein-protein interaction data, however, it could be included in the third highest scoring network since this network is the only one of the three that contains proteins known to interact with KDM6A. Inclusion of KDM6A in a network different from BAP1 makes sense since mutations in these genes are correlated and therefore they likely contribute different selective advantages to BC cells. We also included the RB1 tumor suppressor in this network, based on previously described relationships with TP53 and KDM6A (30). We found that KDM6A interacts with several other proteins encoded by genes mutated in Chinese tumors. KDM6A directly binds to MLL3, CSPG4, and SMARCA4 (BRG1) (31, 32) and regulates RB1 expression (30). MLL3, RB1, and SMARCA4 are known tumor suppressors (33-35) while CSPG4 is thought to be oncogenic (32, 36). Using three publicly available microarray datasets (Supplementary Table S9), we identified genes that were consistently up- or down-regulated in tumors in at least two datasets (FDR < 5%) and highlighted them on the mutation network (Fig. 4A). In addition to being a known tumor suppressor interacting with KDM6A, SMARCA4 expression was up-regulated in tumors in all three cohorts. KDM6A indirectly interacts with several proteins consistently deregulated in BC, such as histone H3, which is required for EGF induced transformation (37). Network analysis revealed that KDM6A interacts with the products of genes that harbor BC driver mutations and with genes consistently differentially expressed in BC, suggesting its central importance as a driver of BC. We also used Cytoscape (www.cytoscape.org) to plot these genes in three-dimensional space using the co-mutational rate and the frequency of mutation as parameters in our model. This puts KDM6A as the most central and well-connected gene in this mutational network (Fig. 4B).
Functional Relevance of KDM6A in Bladder Cancer
KDM6A is frequently mutated in BC and is important in signaling networks as we show above. Therefore, we developed a gene mutation signature for KDM6A (38) where a high KDM6A signature score corresponded to a likely somatic mutation. We examined three patient cohorts (Supplementary Table S9) and found higher scores corresponded to NMI BC stratified patients with a more favorable prognosis. KDM6A signature scores are higher in bladder tumor samples compared to normal urothelial cells (Fig. 5), indicating the signature likely detected altered KDM6A-related pathways in BC. While these studies suggest that KDM6A is a driver of BC and has prognostic value, the direct functional role of KDM6A in BC has not yet been experimentally established.
Figure 5. Relationship between a KDM6A signature score and BC.
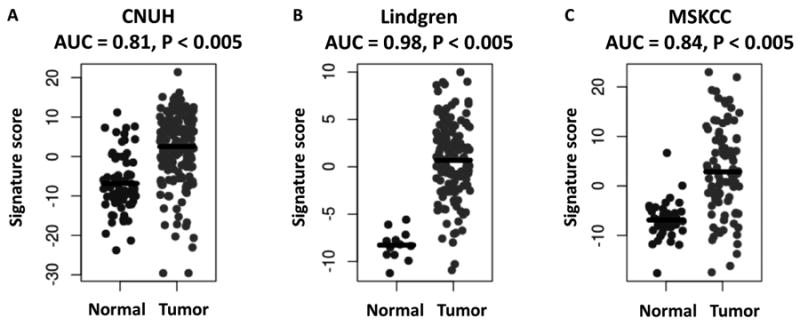
The KDM6A signature score is plotted for normal and tumor samples in A, CNUH (n = 197), B, Lindgren (n=156), and C, MSKCC (n=129) cohorts. AUC, area under receiving operator characteristics curve, with AUC>0.50 corresponding to a signature score higher in tumor than normal samples. P-values were calculated by the Wilcoxon rank-sum test.
Hence we determined the role of KDM6A in shaping the cancer cell phenotype using two well established human BC models, T24T (39) and MGHU3 (40). These tumor-derived cell lines were selected based on KDM6A mutation status. MGHU3 was WT, and T24T had a homozygous G>T nonsense mutation introducing a premature stop codon (p.E895X, NM_021140, 1401 aa full length). The stop codon truncated 506 C-terminus AAs (36% of the full length protein) including the catalytic jumonji C domain (see Methods). shRNA and cDNA constructs were used to deplete and over-express WT KDM6A in MGHU3 and T24T, respectively. The effect of these treatments on KDM6A expression was verified by qPCR (Fig. 6A). KDM6A depletion in MGHU3 enhanced anchorage independent growth (p=0.04) and cell migration (p<0.001) but not monolayer growth (Fig. 6B-D). In T24T cells, KDM6A over-expression diminished anchorage independent growth (p=0.02) and cell migration (p=0.055) but not monolayer growth (Fig. 6B-D). In vivo, depletion of KDM6A in MGHU3 led to significantly enhanced tumor growth (p=0.002) confirming the functional relevance of intact KDM6A as a tumor growth inhibitor (Fig. 6D). The deleterious mutations observed in tumors in this and other studies (3, 5) and the functional data presented here indicate KDM6A is a frequently mutated gene encoding a tumor suppressor in BC.
Figure 6. KDM6A loss drives the BC phenotype.
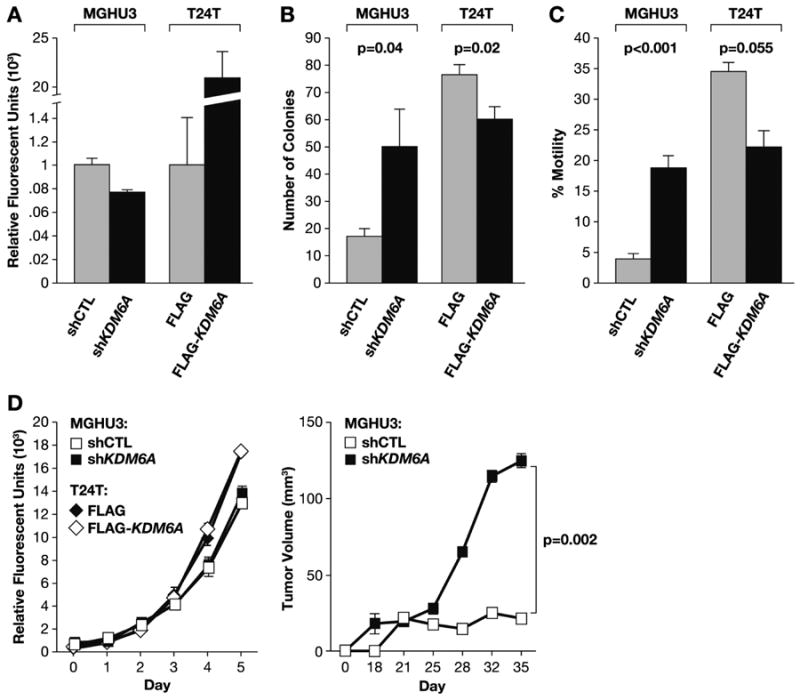
KDM6A WT MGHU3 cells were treated with short hairpin RNA (shRNA) targeting KDM6A (shKDM6A) or a scrambled shRNA (shCTL). These were compared to KDM6A-mutant T24T cells transiently re-expressing FLAG-tagged KDM6A (FLAG-KDM6A) or empty FLAG vector (FLAG). A, Relative KDM6A mRNA expression by quantitative PCR. B, Anchorage independent growth assay (n=6 wells/line). C, Transwell cell migration (n=4 wells/line). D, Monolayer growth of cells (left) assessed by CYquant fluorescence assay (n=4 wells/line) and subcutaneous tumor growth (right, n=20 mice/line). Results are shown as the mean +/- SEM. P-values were calculated using a Student's t-test on the final day of the assay. Assay details are described or referenced in the Materials and Methods.
Discussion
This study contributes to the catalog of genes altered in human BC by evaluating 54 tumors from U.S. patients. We identified 41 genes altered by somatic variants that may be relevant to disease, including four genes, CHD1, CHD1L, GCN1L1, and BAP1, not previously reported as mutated in BC. Interestingly, all four impact chromatin remodeling and/or regulation.
CHD-family gene alterations may play an important role in BC due to their influence on chromatin structure and transcription (15). Over expression of CHD1L has been implicated in BC (16) and germline alterations are associated with congenital anomalies of the kidney and urinary tract (17), indicating a likely role in bladder development. CHD1 somatic alterations have been reported in prostate (41) and other cancers (14, 42) and a review of the COSMIC DB shows CHD1L is altered in endometrial (4%), stomach (2%), and lung cancers (2%). Alteration of GCN1L1 may target its role in AA metabolism and excretion (43). The COSMIC DB shows GCN1L1 is mutated in 4-7% of endometrial, cervical, urinary tract (bladder), colon, lung, and stomach cancers. We plan to examine additional BC tumors to confirm accurate mutation frequencies for CHD1, CHD1L, and GCN1L1.
BAP1 acts to remodel chromatin via an ubiquitin hydrolase catalytic function that removes ubiquitin from histone H2A. Somatic and germline BAP1 alterations have been observed in melanoma, mesothelioma, and kidney cancers (44-46). Somatic alterations of BAP1 define a subtype of clear cell kidney cancer characterized by papillary features (21). Interestingly, review of bladder tumor samples revealed that papillary features were more common in bladder tumors with mutant BAP1, indicating similar phenotypic characteristics between BAP1-mutant bladder and kidney tumors. BAP1 mutations are correlated with KDM6A alterations, suggesting these alterations may provide complementary advantages to tumor cells, similar to co-occurrence of PBRM1 and SETD2 mutations in RCC (47). Interestingly, BAP1 mutations were complementary to alterations in genes encoding proteins of the BRCA DNA repair pathway (ATM, BRCA1, BRCA2, and PALB2). Components of this pathway are altered in Fanconi anemia, an inherited, autosomal recessive disease characterized by leukemia and an increased susceptibility to multiple types of cancer. The pathway regulates the cellular responses to DNA damage and, via ATM, associated cell cycle checkpoints. To our knowledge, frequent alteration of the BRCA pathway in BC has not been previously reported. Mutations in BRCA pathway genes may indicate tumors that are deficient in DNA repair and therefore vulnerable to DNA lesions created by chemotherapeutic drugs (23), especially if combined with poly ADP ribose polymerase (PARP) (48) inhibitors.
A high frequency of somatic TERT promoter alterations was recently reported in several cancers (7, 27, 28). While we found germline and somatic alterations in BC that had not been previously identified, somatic TERT promoter alterations were not associated with alterations in other BC genes or with tumor stage or grade. The variants observed may affect the TERT promoter in several ways. TERT expression is known to be regulated by methylation and hydroxymethylation of promoter CpGs (49, 50). Several variants altered CpGs or neighboring nucleotides suggesting potential disruption of epigenetic regulation of TERT. In addition, ENCODE data shows germline and somatic variants altered DNA sequences known to bind TFs. TF binding may also be altered by variants predicted to create new TF binding sites, such as c.-113C>T which creates a consensus sequence for c-Rel. Hence, germline and somatic variants may work in concert to alter TERT gene expression.
It is not clear why certain cancer genes vary in mutation frequency across patient cohorts with the same disease. When compared to bladder tumors from Chinese patients, BAP1 is mutated at a higher frequency in tumors from U.S. patients, while the mutation frequencies of KDM6A, ARID1A, and STAG2 were similar between these cohorts. Thus, BAP1 and perhaps BRCA pathway genes may represent genes targeted for alteration due to specific ethnic, lifestyle, or geographic differences. Given its frequent and similar mutation frequency in both patient cohorts, we sought to determine the functional and prognostic role of KDM6A in human BC. In vitro and in vivo experiments examining KDM6A depletion and over-expression in bladder tumor cells support a role for KDM6A as a suppressor of tumor growth and cell migration.
Taken together, our work provides further insights on the genomic landscape of BC while providing clues to possible prognostic (KDM6A status signature score) and companion biomarkers (BAP1 and BRCA pathway gene mutations) suggesting novel therapeutic strategies (PARP inhibitors combined with DNA damaging agents) that may be effective against BC.
Methods
Human Subjects, Tumor and Normal Tissue Samples, and Cell Lines
The study was approved by the University of Colorado Denver (UCD), Institutional Review Board (Colorado Multiple Institutional Review Board, CB F490, protocols 10-1365 and 09-913). Clinical details on patients and tumors used for exome and targeted sequencing are summarized in Supplementary Table S1. Tissue samples were snap frozen in liquid nitrogen and stored at -80°C until DNA was isolated using proteinase K digestion followed by phenol/chloroform extraction. H&E sections of tumors were prepared by standard methods and evaluated by a board certified genitourinary pathologist (S.M.L.). Only tumor samples with ≥85% purity were processed for DNA. DNA was isolated from normal adjacent bladder tissue that was manually reviewed by a pathologist (S.M.L.) to confirm samples were free of urothelial carcinoma in situ and low-grade urothelial dysplasia. Functional experiments utilized T24T and MGHU3, two human BC cell lines that have been used extensively were selected. PCR and Sanger sequencing of T24T DNA revealed a KDM6A homozygous c.G2683T (p.E895X, NM_021140) introducing a stop codon. This truncates 506 C-terminus AAs (36% of 1401 AA total), including the catalytic jumonji C domain (aa 1099 – 1241). Since T24T was derived from a female patient, the WT copy of KDM6A was also lost. The KDM6A protein coding sequences and SJs were WT in MGHU3. T24T cells were cultured in Dulbecco modified Eagle medium (DMEM)/F12 with 2.5 mmol/L L-glutamine solution adjusted with 2.4 g/L sodium bicarbonate and 5% fetal bovine serum. MGHU3 were cultured in minimal essential medium (MEM) with 2 mmol/L L-glutamine, Earle's balanced salt solution (2.2 g/L sodium bicarbonate), and 10% fetal bovine serum. Cell lines have been tested and authenticated by SNP analysis.
Exome Capture, Sequencing, and Analysis
Bladder tumor genomic DNA (3 μg, quantitated by fluorometer and agarose gel) was fragmented and the exome captured using probes for ∼180,000 protein-coding exons and microRNA loci based on curated genes in the Consensus Coding Sequence (CCDS) database (SureSelect Human All Exon Kit, Agilent, Santa Clara, CA). Libraries were sequenced on a HiSeq 2000 platform (Illumina, San Diego, CA) and base calls on paired-end, 100 bp reads were generated using the Genome Analyzer Pipeline, v. 1.3, and standard parameters. Additional details are available in the Supplementary Methods.
Variant Validation
Selected variants identified in tumor exomes were examined in DNA from the tumor and normal adjacent bladder tissue. Primers were designed using Primer 3 and ExonPrimer (UCSC Genome Browser) as described in the Supplementary Methods. PCR utilized FastStart reagents (Roche; South San Francisco, CA) and GeneAmp 9700 thermal cyclers (ABI; Foster City, CA) at a 58°C annealing temperature for 30 cycles preceded by a 10 cycle, 5°C touchdown. PCR products were evaluated by 2% agarose gel electrophoresis and sequenced using Big Dye v.3.1 reagents (ABI) and a 3730 Genetic Analyzer (ABI). Mutation Surveyor (Softgenetics, State College, PA) and Sequencher, v.4.8 (GeneCodes; Ann Arbor, MI) were used for sequence analysis.
Copy Number Assessment
Copy number was assessed using the RSI of somatic and heterozygous germline variants in Sanger chromatograms (25, 26) which is an estimate of mutant/mutant+WT nucleotide peak heights in sequencing chromatograms. RSI are semi-quantitative measures of relative allelic fractions in heterogeneous cancer samples comparable to allele frequencies obtained after subcloning. High RSI scores indicate allelic imbalance due to somatic loss of WT alleles or amplification of oncogenic mutant alleles in cancer samples.
Annotation of CRM-DA Proteins and BAP1
CRM-DA proteins were annotated using UniProtKB/Swiss-Prot, Genecards, PhosphoSitePlus, Modbase, and The Protein Model Portal databases for selected isoforms: 729 aa BAP1 adapted from (51); ARID1A, 2285 aa; KDM6A, 1401 aa; and STAG2, 1231 aa. Phosphorylation sites are recurrent observations by mass spectrometry (PhosphoSitePlus). The BAP1 ribbon diagram includes residues 5-291 (UniProtKB Q92560) based on the 2.5 angstrom crystal structure of homologous UCHL3-UbVME (ModBase template code 3ihrA) (19) and was viewed using The Protein Model Portal. Aligned BAP1 NLS residues include orthologous proteins: human, AAH01596; mouse, NP_081364; rat, NP_001100762; and Xenopus, NP_001008206 (adapted from (20).
TERT Promoter Analysis
The TERT promoter was analyzed similar to a recent publication (7). Additional primers were designed for nested PCR using 1 μl of a 1:10 dilution of the first round PCR and sequencing. Forward nested, GATTCGACCTCTCTCCGCTG; Reverse nested, CCTCGCGGTAGTGGCTGCGC. TFBS were analyzed using Tfsearchv1.3 (http://www.cbrc.jp/research/db/TFSEARCH.html) and 15 bases on either side of each variant. Multiple variants in the same sample were tested individually and in combination.
Gene Expression Datasets and Network Analysis
Gene expression datasets are described in Supplementary Information. The genes with somatic mutations confirmed by Sanger sequencing in a recent work (5) and the current manuscript were imported into Ingenuity Pathway Analysis (IPA)(Ingenuity Systems, Redwood City, CA) for bioinformatic analysis. IPA was used to group these mutated genes into gene-limited networks (35 genes maximum) based on evidence of direct or indirect relationships between genes according to the IPA Knowledge Base. The IPA network algorithm seeks to maximize the interconnectivity within a group of selected genes and scores networks based on a right tailed Fisher's Exact Test that calculates the probability that the given relationships can be explained by a random model. The networks do not include all possible relationships for each member, due to size constraints placed on the network, and specific genes may appear in multiple networks. We identified differentially expressed genes in BC (relative to normal bladder samples) using publicly available microarray datasets. P-values were calculated by the non-parametric Wilcoxon rank-sum test and each p-value adjusted to obtain the false discovery rate. The expression patterns of genes consistently up- or down-regulated in two or more cohorts (false discovery rate < 5%) were then overlaid on the networks constructed by IPA from the mutation data.
The co-mutational network was based on a gene matrix table where total mutations are found at a gene's intersection with itself and gene co-mutations in the remaining columns/rows. From this table we parse into Cytoscape (www.cytoscape.org) compatible, tab-delimited tables using Python. The output from the script is imported into Cytoscape 3.0.2 generating the BC interaction network. For the network in Fig. 4B, we removed genes with fewer than 5 total mutations and less than two co-mutation interactions bringing the count from 423 genes with 9317 interactions down to 46 genes with only 596 interactions. Edge-weights based on co-mutation rates between genes were used to organize the network, pulling genes with higher rates of co-mutation closer together. Node size corresponds to a gene's total mutations and color to a node's relative centrality score (genes in the center, red; periphery, green). The raw count data, script, script output, addition sample metadata, and saved Cytoscape session are available (https://github.com/brwnj/interaction_network).
A KDM6A Mutation Signature and BC Diagnosis
We previously identified a 26-gene signature reporting on KDM6A mutation status that consists of genes differentially expressed between KDM6A WT and mutant tumors (38). The ability of this signature to discriminate between normal urothelial and BC cells was assessed by calculating a signature score. Microarray probes were matched to gene symbols according to Affymetrix or Illumina annotation. When multiple probes mapped to a single signature gene, the probe with the highest mean expression was selected. The expression values of all genes were normalized to have mean 0 and variance 1 across all samples in a cohort. The signature score is the sum of the (normalized) expression of all genes up-regulated in mutants minus the sum of the expression of all genes down-regulated in mutants. Therefore, a high signature score indicates a sample is more similar to a tumor with a mutated gene. The relationship between signature score and cancer diagnosis (tumor versus normal) was evaluated by the area under the receiver operating characteristic curve (AUC), with AUC > 0.50 indicating the signature score is higher in tumor samples (compared to normal urothelial cells). The Wilcoxon rank-sum test was used to calculate a p-value comparing AUC to 0.50 (i.e., what would be predicted by a random model).
KDM6A Constructs, Transfections, RT-PCR, and Functional Assays
Short hairpin RNA (shRNA) targeting human KDM6A (shKDM6A) and a scrambled shRNA control (shCTL) in non-targeting plasmid pLKO.1-puro (Sigma-Aldrich) was used to examine KDM6A in MGHU3 cells. Mammalian expression vectors allowing re-expression of KDM6A (FLAG-KDM6A) or empty control vector (FLAG) were constructed using a modified Gateway Multisite Recombination system (Life Technologies, Carlsbad, CA) and transfected into T24T cells. KDM6A depletion and re-expression was validated by quantitative Reverse Transcriptase-PCR. Anchorage dependent and independent growth and cell migration was assessed as previously described (39). For in vivo assessments, 5-week-old male NCrnu/nu mice were injected with 2 × 106 MGHU3 cells stably expressing shKDM6A or shCTL and were assessed as described in the Supplementary Methods.
Statistical Analysis
Pearson's correlation coefficient was used to assess pairs of altered genes in 54 U.S. tumors (ARID1A, BAP1, STAG2, KDM6A, and TERT). We used a Fisher's exact test or logistic regression for univariate analysis and logistic regression for multivariate analysis to investigate association of clinical factors with gene mutation status. Nonsynonymous, coding indels, UTR, and intronic variants within 10 bp of the intron/exon border were included in the mutation correlation and the clinical association analyses. Statistical analysis was performed using R software (http://cran.r-project.org).
Supplementary Material
Statement of Translational Relevance.
Sequencing of bladder cancers reveals hitherto unappreciated mutations in BAP1 and BRCA pathway genes while finding that somatic TERT promoter alterations were independent of somatic alterations in other genes. This data suggests novel strategies with PARP inhibitors combined with DNA damaging agents may be effective in specific patients.
Acknowledgments
We would like to thank the Theodorescu and Dean lab members and Dr. Berton Zbar (National Cancer Institute) for comments; and Dr. Silvia Jiménez Morales (Instituto Nacional de Medicina Genómica, Mexico), Dr. Dom Esposito, David Wells, Alana Ebert-Zavos, Andrew Warner, and Allen Kane (National Cancer Institute), and Dr. Jean-Noel Billaud (Ingenuity) for technical assistance. This work is supported in part by National Institutes of Health (NIH) grants CA075115 and CA104106 (D.T.) and by the Intramural Research Program of the NIH, the National Cancer Institute, Center for Cancer Research (M.L.N., M.D.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- BC
bladder cancer
- NMI
non-muscle invasive BC
- MI
muscle invasive BC
- KM
Kaplan-Meier
- HR
hazard ratio
- NGS
next-generation sequencing
- chr
chromosome
- CRM
chromatin remodeling
- DA
DNA associated
- AUC
area under the receiver operating characteristic curve
- AA
amino acid
- H&E
hematoxylin eosin–stained
- WT
wild type
- indel
insertion or deletion
- TF
Transcription factor
- TFBS
TF binding sites
- FDR
false discovery rate
- UTR
untranslated region
- SJ
splice junction
- TSS
transcription start site
Footnotes
Conflicts of Interest: The authors have no conflicts to disclose.
Author's Contributions: Designed research: Michael L. Nickerson, Dan Theodorescu
Performed research: Michael L. Nickerson, Garrett M. Dancik, Kate M. Im, Sevilay Turan, Charles Owens, Christina Ruiz-Rodriguez, Lee E. Moore, Scott M. Lucia,
Contributed new reagents or analytic tools: Guangwu Guo, Shirley X. Tsang, Yingrui Li, Quan Zhou, Zhiming Cai.
Analyzed data: Michael L. Nickerson, Garrett M. Dancik, Kate M. Im, Michael G. Edwards, Charles Owens, James C Costello, Scott M. Lucia
Wrote the paper: Michael L. Nickerson, Garrett M. Dancik, Michael Dean, Dan Theodorescu
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nat Rev Cancer. 2005;5:713–25. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 3.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurst CD, Platt FM, Taylor CF, Knowles MA. Novel tumor subgroups of urothelial carcinoma of the bladder defined by integrated genomic analysis. Clin Cancer Res. 2012;18:5865–77. doi: 10.1158/1078-0432.CCR-12-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013:1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, et al. Frequent truncating mutations of STAG2 in bladder cancer. Nat Genet. 2013;45:1428–30. doi: 10.1038/ng.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]; 45:1459–1463. [Google Scholar]
- 7.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci U S A. 2011;108:17087–92. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- 11.Cordon-Cardo C, Dalbagni G, Saez GT, Oliva MR, Zhang ZF, Rosai J, et al. p53 mutations in human bladder cancer: genotypic versus phenotypic patterns. Int J Cancer. 1994;56:347–53. doi: 10.1002/ijc.2910560309. [DOI] [PubMed] [Google Scholar]
- 12.Knowles MA, Habuchi T, Kennedy W, Cuthbert-Heavens D. Mutation spectrum of the 9q34 tuberous sclerosis gene TSC1 in transitional cell carcinoma of the bladder. Cancer research. 2003;63:7652–6. [PubMed] [Google Scholar]
- 13.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. Characterization of the CHD family of proteins. Proc Natl Acad Sci U S A. 1997;94:11472–7. doi: 10.1073/pnas.94.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian F, Xu F, Zhang ZY, Ge JP, Wei ZF, Xu XF, et al. Expression of CHD1L in bladder cancer and its influence on prognosis and survival. Tumour Biol. 2013;34:3687–3690. doi: 10.1007/s13277-013-0951-4. [DOI] [PubMed] [Google Scholar]
- 17.Brockschmidt A, Chung B, Weber S, Fischer DC, Kolatsi-Joannou M, Christ L, et al. CHD1L: a new candidate gene for congenital anomalies of the kidneys and urinary tract (CAKUT) Nephrol Dial Transplant. 2012;27:2355–64. doi: 10.1093/ndt/gfr649. [DOI] [PubMed] [Google Scholar]
- 18.Ebmeier CC, Taatjes DJ. Activator-Mediator binding regulates Mediator-cofactor interactions. Proc Natl Acad Sci U S A. 2010;107:11283–8. doi: 10.1073/pnas.0914215107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgie SE, Bingman CA, Soni AB, Phillips GN., Jr Structural characterization of human Uch37. Proteins. 2012;80:649–654. doi: 10.1002/prot.23147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Research. 2008;68:6953–62. doi: 10.1158/0008-5472.CAN-08-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–9. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen DE, Proctor M, Marquis ST, Gardner HP, Ha SI, Chodosh LA, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16:1097–112. doi: 10.1038/sj.onc.1201861. [DOI] [PubMed] [Google Scholar]
- 23.O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31:961–7. doi: 10.1093/carcin/bgq069. [DOI] [PubMed] [Google Scholar]
- 24.Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333:1039–43. doi: 10.1126/science.1203619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickerson ML, Im KM, Misner KJ, Tan W, Lou H, Gold B, et al. Somatic Alterations Contributing to Metastasis of a Castration Resistant Prostate Cancer. Hum Mutat. 2013;34:1231–1241. doi: 10.1002/humu.22346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–61. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 28.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–9. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiesen HJ, Bach C. Target Detection Assay (TDA): a versatile procedure to determine DNA binding sites as demonstrated on SP1 protein. Nucleic Acids Res. 1990;18:3203–9. doi: 10.1093/nar/18.11.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terashima M, Ishimura A, Yoshida M, Suzuki Y, Sugano S, Suzuki T. The tumor suppressor Rb and its related Rbl2 genes are regulated by Utx histone demethylase. Biochem Biophys Res Commun. 2010;399:238–44. doi: 10.1016/j.bbrc.2010.07.061. [DOI] [PubMed] [Google Scholar]
- 31.Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–50. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- 32.Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clem BF, Chesney J. Molecular Pathways: Regulation of Metabolism by RB. Clin Cancer Res. 2012;18:6096–100. doi: 10.1158/1078-0432.CCR-11-3164. [DOI] [PubMed] [Google Scholar]
- 34.Marschalek R. Mixed lineage leukemia: roles in human malignancies and potential therapy. Febs J. 2010;277:1822–31. doi: 10.1111/j.1742-4658.2010.07608.x. [DOI] [PubMed] [Google Scholar]
- 35.Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat. 2008;29:617–22. doi: 10.1002/humu.20730. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Wang Y, Yu L, Sakakura K, Visus C, Schwab JH, et al. CSPG4 in Cancer: Multiple Roles. Curr Mol Med. 2010;10:419–29. doi: 10.2174/156652410791316977. [DOI] [PubMed] [Google Scholar]
- 37.Choi HS, Choi BY, Cho YY, Mizuno H, Kang BS, Bode AM, et al. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer Research. 2005;65:5818–27. doi: 10.1158/0008-5472.CAN-05-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dancik GM, Owens CR, Iczkowski KA, Theodorescu D. A cell of origin gene signature indicates human bladder cancer has distinct cellular progenitors. Stem Cells. 2014;32:974–82. doi: 10.1002/stem.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gildea JJ, Golden WL, Harding MA, Theodorescu D. Genetic and phenotypic changes associated with the acquisition of tumorigenicity in human bladder cancer. Genes Chromosomes Cancer. 2000;27:252–63. doi: 10.1002/(sici)1098-2264(200003)27:3<252::aid-gcc5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 40.Williams RD. Human urologic cancer cell lines. Invest Urol. 1980;17:359–63. [PubMed] [Google Scholar]
- 41.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 43.McGregor RF, Crawford R, Johnson DE, Brown B, Sharon MS, Johnston D. Urinary amino acid excretion: comparison of normal individuals and patients with bladder cancer. Urology. 1977;9:538–42. doi: 10.1016/0090-4295(77)90247-3. [DOI] [PubMed] [Google Scholar]
- 44.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465:243–7. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–5. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–21. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pena-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and Antagonism among Cancer Genes: The Renal Cancer Paradigm. Cancer research. 2013;73:4173–9. doi: 10.1158/0008-5472.CAN-13-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 49.Guilleret I, Benhattar J. Demethylation of the human telomerase catalytic subunit (hTERT) gene promoter reduced hTERT expression and telomerase activity and shortened telomeres. Exp Cell Res. 2003;289:326–34. doi: 10.1016/s0014-4827(03)00281-7. [DOI] [PubMed] [Google Scholar]
- 50.Pettigrew KA, Armstrong RN, Colyer HA, Zhang SD, Rea IM, Jones RE, et al. Differential TERT promoter methylation and response to 5-aza-2′-deoxycytidine in acute myeloid leukemia cell lines: TERT expression, telomerase activity, telomere length, and cell death. Genes Chromosomes Cancer. 2012;51:768–80. doi: 10.1002/gcc.21962. [DOI] [PubMed] [Google Scholar]
- 51.Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. 2013;11:1061–71. doi: 10.1158/1541-7786.MCR-13-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.