

Abstract
Adhesive interactions between neutrophils and endothelium involve chemokine-induced neutrophil spreading and subsequent crawling on the endothelium to sites of transmigration. We investigated the importance of cell topography in this process using immunofluorescence, scanning electron microscopy, and live-cell imaging using total internal reflectance microscopy to observe redistribution of key membrane proteins, both laterally and relative to surface topography, during neutrophil spreading onto glass coated with interleukin 8. During formation of the lamellipod, L-selectin is distributed on microvilli tips along the top of the lamellipodium, whereas the interleukin 8 receptors CXCR1 and CXCR2 and the integrin LFA-1 (αLβ2) were present at the interface between the lamellipodium and the substrate. Total internal reflection fluorescence imaging indicated that LFA-1 and both chemokine receptors redistributed into closer contact with the substrate as the cells spread onto the surface and remodeled their topography. A geometric model of the surface remodeling with nonuniform distribution of molecules and a realistic distribution of microvilli heights was matched to the data, and the fits indicated a 1000-fold increase in the concentration of chemokine receptors and integrins available for bond formation at the interface. These observations imply that topographical remodeling is a key mechanism for regulating cell adhesion and surface-induced activation of cells.
Introduction
Mechanisms by which cells regulate their adhesive interactions are central to a broad range of biological activities, not the least of which is the recruitment of leukocytes to tissues during inflammation and the immune response. As early as the 19th century, the leukocyte recruitment cascade resulting in the infiltration of cells into inflamed tissue was described as consisting of three sequential events: rolling, adhesion, and transmigration (1–3). With the discovery of selectins, integrins, chemokines, and their ligands, these steps were specified as selectin-mediated rolling, chemokine-induced activation, and integrin-dependent adhesion and transmigration (4–6). More recently, an intermediate step, cell spreading and adhesion strengthening, was identified as an important part of the process (7). Similar mechanisms are at work during stem cell homing and cancer metastasis. Although a great deal of research has focused on the regulation of molecular affinity of principal adhesion molecules (particularly integrins) and their surface expression, significantly less attention has been paid to understanding the role that physical factors can play in limiting adhesive interactions. Surface topography can have significant effects on bond formation, as shown by Williams and colleagues (8), who demonstrated a 50-fold difference in bond formation rate for the same ligand expressed on smooth or wrinkled cell surfaces.
It is well established that leukocytes have ruffled surfaces (9,10) and that the effects of surface ruffling on adhesion can be compounded when molecules are not uniformly distributed relative to that surface topography. It has been demonstrated by scanning electron microscopy (SEM) and total internal reflection fluorescence (TIRF) microscopy studies that L-selectin is located at the tips of microvilli on a resting neutrophil, whereas integrins are excluded from the microvilli and are predominantly localized in the valleys between microvillus ridges (11–14). The location relative to the surface topography of other important molecules, such as chemokine receptors, has not yet been characterized. Knowing the spatial distribution of selectins, integrins, and chemokine receptors on neutrophil surfaces is important for a detailed understanding of the mechanisms by which leukocyte interaction with endothelium might be modulated, particularly those related to the topography, surface deformation, and distribution of molecules on the interacting surfaces.
Recent literature recognizes cell spreading and crawling as important intermediate steps in leukocyte recruitment that occurs after cell arrest and before transmigration, as the cell finds its way to sites of egress through the endothelium (7,15,16). In this report, we focus on the process of cell spreading, an essential step between arrest and crawling during which dramatic changes in the molecular interactions between cell surfaces can occur. Using SEM, fluorescence microscopy, and TIRF microscopy, we observe and quantify the dynamic lateral and topographical redistribution of key adhesion molecules and chemokine receptors as neutrophils spread onto a surface presenting interleukin 8 (IL8, CXCL8), a principal chemokine for neutrophils. We also introduce a model of dynamic changes in cell surface topography that is consistent with our experimental observations and demonstrates that a simple collapse of the microvillus structure can produce a dramatic increase (three orders of magnitude) in the engagement of the β2 integrins and the chemokine (IL8) receptors CXCR1 and CXCR2.
Materials and Methods
The overall strategy of the experiments is illustrated schematically in Fig. 1. Fluorescently labeled neutrophils were brought into contact with IL8-coated glass coverslips or glass beads and the distribution of fluorescence was monitored as the cell spread onto the surface.
Figure 1.
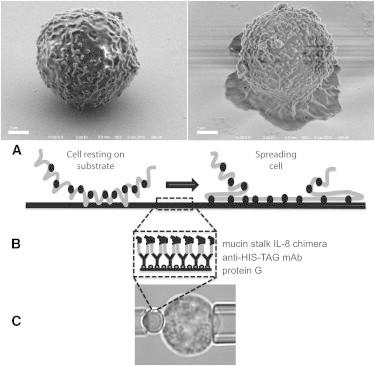
Experimental approaches used in the study. (A) Scanning electron micrographs showing the cell morphology before and during spreading. Scale bar, 1.0 μm. Also shown is a schematic of the rough cell surface becoming smooth during spreading and bringing molecules into closer contact with the substrate. (B) Schematic of the chemical coating on the surface. Circles at the surface represent protein G, y-shaped structures are antibodies to His-tag, and the chimeric protein is shown in two shades, light gray for the fractalkine stalk and black for the IL-8 portion. Note that the same chemistry is used on both glass slides and beads. (C) Video micrograph showing the use of micropipettes to bring cells into contact with IL8 immobilized on the bead surface.
Antibodies and chemicals
Five mouse anti-human monoclonal antibodies were used: DREG-56 (eBioscience, San Diego, CA), which binds to CD62L (L-selectin); clone 38 (Ancell, Bayport, MN), which binds to CD11a (LFA-1); clone 42705 (R&D Systems, Minneapolis, MN), which binds to CXCR1 (IL8 RA); clone 48311 (R&D Systems), which binds to CXCR2 (IL8 RB); and IB4 (Ancell), which binds to CD18 (β2 integrin subunit). All antibodies were conjugated with Alexa Fluor 488 or Qdot 625 using antibody conjugation kits from Molecular Probes (Invitrogen, Grand Island, NY). All the antibodies were diluted at 0.5 mg/mL for labeling. As a nonspecific control, the cell surface was labeled with Alexa Fluor 488 carboxylic acid, tetrafluorophenyl ester (Invitrogen).
For surface preparation, protein G was purchased from Calbiochem (La Jolla, CA), anti-His⋅Tag monoclonal antibody from Novagen (Madison, WI), dimethyl pimelimidate dihydrochloride, triethanolamine, and TRIS from Sigma (St. Louis, MO), and recombinant human IL8/mucinlike stalk chimera and ICAM-1 chimera from R&D Systems.
Surface preparation
For chemokine immobilization, human IL8 was obtained as a chimera with the mucinlike stalk of human fractalkine. At the opposite end of the mucinlike stalk, a His⋅Tag sequence was encoded. To attach these molecules to glass coverslips or beads, protein G (20 μg/mL) was adsorbed onto the surface of acid-cleaned coverslips by 1-h incubation at room temperature. Anti-His⋅Tag antibody (60 μg/mL) was then added and the preparation again was incubated for 1 h at room temperature. After three washes with 0.2 M triethanolamine (pH 8.2), 20 mM dimethyl pimelimidate dihydrochloride in triethanolamine was added to covalently link the Fc portion of the antibody to the protein G. After a 1-h incubation at room temperature, the reaction was stopped by adding 50 mM Tris (pH 7.5). After three washes with 0.1% bovine serum albumin in phosphate-buffered saline, IL8 chimera was added (10 μg/mL) and the coverslips or beads were stored at 4°C until use. A schematic of the resulting surface chemistry is shown in Fig. 1 B.
Cell preparation
Neutrophils were obtained from healthy donors by diluting 1 μL of peripheral blood from a finger prick in 80 μL of balanced saline solution (BSS) consisting of 5 mM KCl, 146 mM NaCl, and 5.5 mM glucose with 10 mM HEPES (Sigma) and 4% fetal bovine serum (HyClone, Logan, UT) made with low-endotoxin water (Invitrogen) and supplemented with 1 mM Mg2+ and 1 mM Ca2+, pH 7.4, 290 mOsm. When labeling was required, 10 μL of the appropriate antibody was added to the cells. After 15 min incubation at room temperature, cells were washed three times with BSS and placed on the microscope stage.
As a nonspecific control, neutrophils were labeled with Alexa Fluor 488 by diluting 1 μL of peripheral blood from a finger prick in 1 mL BSS containing Alexa Fluor 488 carboxyl acid, tetrafluorophenyl ester (component A from the Antibody Labeling Kit (Invitrogen)). After 10 min incubation at room temperature, cells were washed twice with 4% fetal bovine serum in BSS and used for the experiment.
Experimental procedures
For immunofluorescence imaging of cell spreading onto coverslips, the cells were placed in a chamber consisting of a U-shaped spacer enclosed with two coverslips. The top coverslip was untreated, whereas the bottom coverslip had two separate regions, one coated with immobilized IL8 and one uncoated. Using a micropipette, labeled neutrophils were transferred from the uncoated region to the region coated with IL8 and dropped onto the coverslip (Fig. 1 A). Experiments were performed on a Nikon Eclipse TE 2000-E microscope, equipped with epifluorescence and TIRF illumination. The microscope objective was focused at the coverslip surface to observe cell behavior at the cell/substrate interface, and the perfect focus feature of our Nikon microscope was engaged to stabilize the focal plane. A series of brightfield, epifluorescence, and TIRF images were recorded every 8 s and saved to the hard drive for offline analysis. Only cells that responded to the IL8 surface within 5 min were analyzed. (Fewer than 1 in 20 cells tested did not respond to the substrate within 5 min, and even these almost invariably did respond after a longer delay.) When testing for the role of β2 integrins, the experiments were performed in the presence of CD18 blocking antibody (clone IB4) at 20 μg/mL final concentration.
For the SEM experiments, neutrophils were brought into contact with the surface and allowed to spread as described above. At different stages of spreading, 2.5% glutaraldehyde (Electron Microscopy Science, Fort Washington, PA) was used to fix the cells. After three washes in distilled water, cells were dehydrated using increasing concentrations of ethanol and dried using hexamethyldisilazane (Electron Microscopy Science).
Analysis of TIRF images
In previous reports, we used the ratio of the TIRF signal to the epifluorescence signal to estimate the fraction of molecules in the interface that are in close proximity to the substrate (14). In this case, the shape of the cell is undergoing dramatic changes, leading to potential artifacts in the brightness of the epifluorescent image because of changes in out-of-focus fluorescence coming from the cell above the interface. To minimize these possible effects, we normalize the TIRF signal intensity by the epifluorescence intensity over a 2.0-μm-radius region at the center of the contact zone (Epicenter), where the volume directly above the membrane is occupied by the cell interior throughout the spreading process: Therefore, the normalized TIRF signal was calculated based on four regions of interest measured for each time point: the epifluorescent image at the cell center (Epicenter), a region of interest in the epifluorescent image far from the contact area containing the background signal (Epibkgd), a region of interest containing the TIRF image of the cell (TIRFsignal), and a region of interest in the TIRF image far from the contact area containing the background signal (TIRFbkgd).
| (1) |
At time zero, this ratio gives an estimate of the relative proximity of the molecules to the substrate in the resting cell (as described in Hocdé et al. (14)). After time zero, it enables us to observe the redistribution of different molecules at the interface on a common scale.
Fluorescence redistribution over the cell contour
The lateral redistribution of fluorescence on the cell body was assessed in two ways. In one, fluorescent images of cells fixed and labeled for the SEM experiments were acquired as serial Z-stacks and assembled into three-dimensional (3D) reconstructions using NIS-Elements software (Nikon Instruments, Melville, NY). In the second, the lateral redistribution of fluorophores was observed during spreading onto IL8-coated beads. After labeling with fluorescent antibody, cells were held in a micropipette and a second pipette was used to bring the bead into contact with the cell. A series of epifluorescence images were taken as the cell first spread onto and eventually engulfed the bead (Fig. 1 C). This latter approach involves spreading onto a curved surface but has the advantage of higher resolution along the axis of symmetry.
Model calculations
Microvillus shape
Model calculations were performed to determine how nonuniform distributions of molecules on a ruffled cell surface might explain the increase in TIRF signal as the cells spread, forming a smooth interface in the contact zone. (Additional details about the modeling procedures are provided in the Supporting Material.) We populated our model surface with an array of two-dimensional Gaussian-like microvilli, where one dimension was given a larger variance to create an elongated ridgelike shape (σy ≈ 10 σx). The local height of the Gaussian relative to the cell surface, zg, was determined as
| (2) |
where hi is the peak height of a given microvillus (see the Supporting Material). Different formulations for the shape of the microvilli were tested. Using a traditional Gaussian formulation (with x and y to the second power) resulted in shapes that were too pointed, and using x and y to the sixth power resulted in more flattened, plateaulike shapes. Neither of these shapes resembled the appearance of microvilli in electron micrographs.
The microvilli on the cell surface are of different heights, and therefore an array of peak microvillus heights was chosen that replicated the heights observed experimentally. The original data of Bruehl were based on sections taken through fixed neutrophils and viewed in transmission electron microscopy. The authors observed a log-normal distribution of microvillus heights. We constructed a distribution of different microvillus heights and weighted their appearance on the cell surface such that, when sectioned mathematically, they gave a distribution that matched that observed by Bruehl. The peak value of the distribution was adjusted to obtain a match to our own TIRF measurements performed on cells with a uniform membrane label (see the Supporting Material).
Time course of spreading
To model spreading, we assumed that any microvillus in contact with the surface would undergo a decrease in height on an exponential time course (see the Supporting Material). Thus, the longest microvilli began to collapse first and shorter microvilli began their height decrease as they came into contact with the surface. (We also experimented with a linear decrease in height with time, but the exponential time course provided better agreement with the data.) During spreading, the width of the region over which the integrated signal was calculated was increased to maintain approximately constant surface area. The relative distribution of fluorescent label from the base to the tip of each microvillus was assumed to remain the same, compressing in the z direction as the height decreased.
We used measurements of the changes in TIRF intensity during spreading of uniformly labeled cells to determine the characteristic height of the microvilli and the exponential constant used to characterize the time course of the change in height. This involved fitting the model predictions to the data using two free parameters (see the Supporting Material).
Nonuniform distribution of fluorescence
The distribution of molecules was expressed as the probability of finding a fluorescent molecule at a position x relative to the ridgelike peak of a microvillus. This probability was assumed to be uniform for the control Alexa-488-labeled cells and to follow an inverted Gaussian-like function for CXCR1, CXCR2, and LFA-1:
| (3) |
where σf represents the width of the distribution of fluorophore, an adjustable variable in the fit to the data. (We originally allowed the fluorescence intensity to vary in both x- and y-directions, but found that the fits were insensitive to the coefficient for the y-direction.) In this case also, we experimented with different mathematical formulations for the distributions. Different powers of x (2 or 4) in the exponential term failed to provide a good match to the data. We also performed calculations using a β distribution and obtained results similar to that obtained with Eq. 3 (see the Supporting Material).
Evanescent wave intensity and calculation of TIRF signal
At each time point, the probabilistic distribution of fluorophores was convolved with an exponentially decaying evanescent wave with an intensity E that fell off with distance from the surface (z) according to
| (4) |
The parameters I1, I2, γ1, and γ2 were determined for our microscope system by calibration (see the Supporting Material). Knowing E(z), the predicted fluorescence intensity was obtained by integrating over the appropriate microvillus projected area:
| (5) |
Additional details are given in the Supporting Material. Note that the total fluorescence signal was determined as a weighted sum of the above integral evaluated for each different microvillus height in the distribution constructed to match the data of Bruehl (11). The resulting prediction was matched to the data for the uniform label using two parameters (the characteristic microvillus height and the characteristic rate of height decrease), and for nonuniform distributions of fluorescence, these parameters were held constant and the prediction was matched to the data using one free parameter characterizing the nonuniformity of the distribution.
Results
We evaluate two principal physical contributors to increased ligand binding that result from cell spreading. The first is a simple increase in contact area and the second is the smoothing of the surface topography.
Changes in contact area
When neutrophils were dropped onto an IL8-coated glass surface, the cells rested gently on the surface for a period of time, then actively spread onto the surface forming a more or less circular lamellipodium. Once the cell approached a maximum spreading diameter, it began to crawl across the surface. During the spreading process, the diameter of the lamellipodium increased almost linearly, with a logarithmic deceleration as the diameter became large. The precise time at which spreading began was difficult to observe directly, because the site where spreading began was often obscured by the body of the cell above the contact region. Thus, to determine the start of spreading, we extrapolated backward from the measured time course of the increase in cell diameter observed in brightfield images, where the boundary of the lamellipodium was clearly visible (Fig. 2 A). An empirical function of the form
| (6) |
was used for the extrapolation, where B1 and B2 are fixed constants and are chosen to match the data in the observable range (D = 8–13.5 μm), and A and t0 are fitted parameters (Fig. 2 B). The spreading rate was determined by evaluating the slope of this function at D = 10.0 μm. Mean values typically ranged from 0.15 to 0.17 μm/s, except when CXCR1 was labeled, in which case the spreading velocity was slower (0.13 μm/s; Table 1). Analysis of variance revealed that the decrease in the spreading rate when CXCR1 was blocked was statistically significant, but the spreading rates were not significantly different from control for the other molecular labels. Thus, generally speaking, the macroscopic contact area increased from a few square microns for cells gently resting on the substrate to ∼80 μm2 in the first minute after the start of spreading, increasing further to ∼150 μm2 over the next 30 s.
Figure 2.
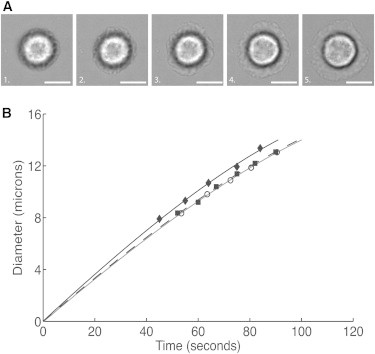
Spreading velocity was determined from brightfield images where the edge of the lamellipodium could be distinguished clearly. (A) Brightfield images of a neutrophil spreading on an IL-8-coated surface. The diameter as a function of time was fit to an empirical relationship (Eq. 6). Extrapolation to zero diameter enabled the determination of the start time for spreading, and the slope of the fitted curve at a diameter of 10 μm was used as the characteristic spreading velocity. (Scale bars, 5.0 μm.) (B) Three examples of diameter as a function of time for neutrophils spreading on an IL-8-coated coverslip. Curves were extrapolated to zero diameter to determine the start of spreading (time 0) and then replotted from a common origin.
Table 1.
Spreading rates
| Label | Characteristic velocity (μm/s) | SD (μm/s) | n |
|---|---|---|---|
| Control | 0.150 | 0.023 | 17 |
| CXCR1 | 0.135 | 0.026 | 34 |
| CXCR2 | 0.176 | 0.023 | 36 |
| L-selectin | 0.150 | 0.027 | 28 |
| LFA-1 | 0.16 | 0.022 | 28 |
Interfacial receptor redistribution during neutrophil spreading
During neutrophil spreading onto IL8, the normalized fluorescence intensity under TIRF illumination provided a measure of the change in the proximity of the molecules to the substrate as the cells spread (see Movies S1–S5 in the Supporting Material). Shown in Fig. 3 are representative examples of the first and last images for the four different molecules (L-selectin, LFA-1, CXCR1, and CXCR2) analyzed in this study. Before spreading, when the neutrophil was freely resting on the glass, the TIRF/Epi ratio indicated that L-selectin is located much closer to the coverslip compared to LFA-1 and CXCR1/2 (Fig. 3, A and C). In contrast, after 60 s of spreading, L-selectin localization relative to the substrate changed very little, whereas LFA-1 and CXCR1 redistributed closer to the cell-substrate interface (Fig. 3, B and C). The nonspecific label of the cell surface showed an intermediate change. Note the rapid increase in surface proximity for both integrins and chemokine receptors, reflected in a roughly 10-fold increase in TIRF intensity over 40 s.
Figure 3.
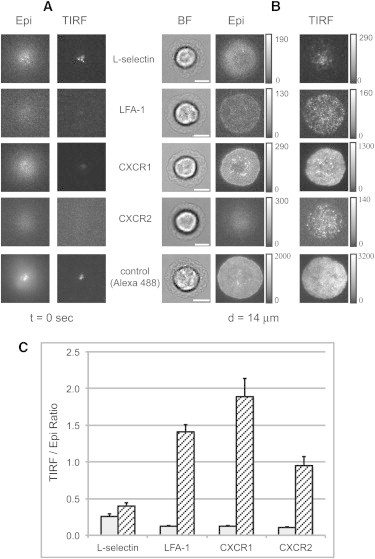
Human neutrophils labeled for L-selectin, LFA-1, CXCR1, or CXCR2 spreading on IL8 coated substrate. (A and B) Images acquired at the start of spreading (A) and after spreading to a diameter of 14 μm (B). Contrast and brightness have been adjusted for visibility, but the original gray values are indicated in the scale bars to the right of each image. All images in the same row are at the same magnification. Scale bars, 5.0 μm. (C) Column graph showing the TIRF/epifluorescence intensity ratios at the center of the contact zone before spreading (open bars) and when the spreading diameter reaches 10 μm (hatched bars). Each bar represents the average of 25–34 cells measured, and error bars represent the mean ± SE. Before spreading, the mean ratio for L-selectin was significantly greater than those for the other three, which were not significantly different from each other (ANOVA, p < 0.05). Note the large increases in TIRF intensity for LFA-1, CXCR1, and CXCR2 that accompany spreading. When the spreading diameter reached 10 μm, the ratio for L-selectin was significantly less than those for CXCR1 and LFA-1, which were not different from each other (ANOVA, p < 0.05).
Measurements of the local intensity of molecules at the interface as a function of radial position in the contact zone provide additional insights into the evolution of molecules in close contact with the substrate. In Fig. 4 are shown the radial distributions of normalized TIRF intensity for each of the four molecules and the nonspecific membrane label. LFA-1 and the two chemokine receptors exhibited similar behavior. The intensity of the TIRF signal near the center of the contact zone increased with time, indicating either that new molecules are diffusing into that region or that the surface of the cell is being drawn into closer contact with the substrate. Two pieces of evidence point to the latter explanation. First, the epifluorescence signal at the center of the contact zone was also monitored over time but showed little change in intensity over that time period (Fig. 4 F). Second, the nonspecific membrane label, which is expected to be uniform on the cell surface, also showed an increase in TIRF signal at the center of the contact zone with time (Fig. 4 A). At larger radii, the intensity is lower near the periphery of the cell than at the center but also increases with time, indicating that here, too, there is a progressive remodeling of the cell topography, drawing molecules into closer contact with the substrate. In contrast, L-selectin (Fig. 4 B) showed decreasing intensity at the center over time and much lower intensity in the newly formed regions of contact nearer the periphery. This difference in behavior appears to be due to the lateral redistribution of L-selectin during spreading (see below).
Figure 4.
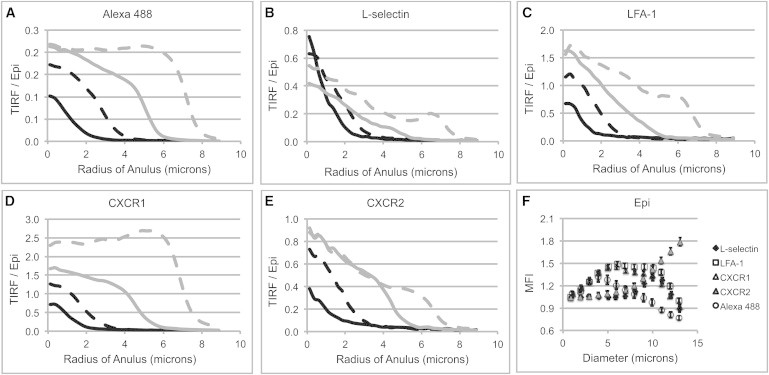
(A–E) Variation in TIRF signal as a function of radial distance from the center of the contact region. Each curve shows the intensity profile (averaged over 10–34 cells) at a different stage of spreading corresponding to lamellipodial diameters of 1.0 μm (solid black curves), 5.0 μm (dashed black curves), 9.0 μm (solid gray curves), and 13.0 μm (dashed gray curves). Results for the nonspecific membrane label are shown in A, and the corresponding curves for the four molecular labels L-selectin, LFA-1, CXCR1, and CXCR2 in B–E. (F) Epifluorescence intensity (normalized by the epiintensity at the start of spreading) that was used to correct the TIRF signal for possible bleaching as a function of increasing lamellipodium diameter.
Comparison with model predictions
We used model calculations to understand the implications of the increase in TIRF intensity in terms of the number of molecules that are within sufficient proximity to the surface to form bonds. In these calculations, we focused on the region at the center of the contact zone and compared the model predictions with the observed changes in TIRF/epifluorescence intensity ratios (Fig. 5 A). We compared the predictions for a uniform distribution of label to the data obtained using Alexa 488 label to determine the maximum microvillus height and the time constant for the rate of microvillus height decrease. With these parameters fixed, we adjusted the molecular distribution parameter (σf) to match the observed changes in TIRF signal for each molecular label (see the Supporting Material for details). The best-fit distributions for LFA-1 and CXCR1 are shown in Fig. 5, B and C. These fitted results enable us to estimate how many molecules of each different type are within range of bond formation at any time during spreading. For example, if the ligand on the surface is extended ∼70 nm from the glass surface (as is estimated for the surface-bound IL8 in these experiments) then ∼0.1% of the LFA-1 or CXCR2 molecules on the cell should be capable of interacting with the surface-bound ligand initially, and ∼0.02% of CXCR1 would be available for binding. This implies that the possible number of bonds per unit membrane area that can be formed by these molecules at the interface increases by >1000-fold as a result of topographical remodeling during cell spreading.
Figure 5.
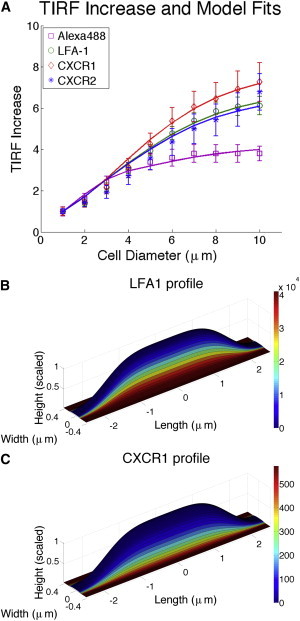
Correlation of model calculations with changes in TIRF intensity. (A) Measurements were taken over a 2.0 μm radius at the center of the contact region. The data for the nonspecific Alexa label were used to calculate the characteristic height of the microvilli and the rate of change in the height of the microvilli. A single coefficient characterizing the distributions of molecules relative to the topography was adjusted to match the data for individual molecular labels. All curves show good agreement. The distribution of CXCR1 (B) and LFA-1 (C) overlaid onto the shape of a model microvillus. Scale bars map colors to molecular density in number/μm2.
Lateral receptor redistribution on the cell surface during spreading
Lateral receptor redistribution over the surface of the cell during chemokine-induced spreading was measured in live labeled cells interacting with IL8-coated beads, and the redistribution of fluorescence was observed in cross section to see how molecules were distributed over the cell body (Fig. 6 A). In this case, too, L-selectin exhibited behavior distinct from those of the other molecules tested. LFA-1 and the chemokine receptor CXCR1 remained more or less uniformly distributed over the cell surface as the cell spread onto the bead, but L-selectin was observed first to gather on the cell body near the contact zone and to redistribute away from the contact zone at later times (Fig. 6 A and Movies S6–S8). In a second approach to evaluate this redistribution, neutrophils labeled with an antibody linked to a quantum dot were fixed with 2.5% glutaraldehyde during their spreading on IL8 substrate, and the images of fluorescently labeled L-selectin or CXCR1 were acquired as serial Z-stacks and displayed as 3D reconstructions (Fig. 6, B and C). Although the resolution is lower using this approach, the two approaches revealed similar behavior of the different molecules.
Figure 6.
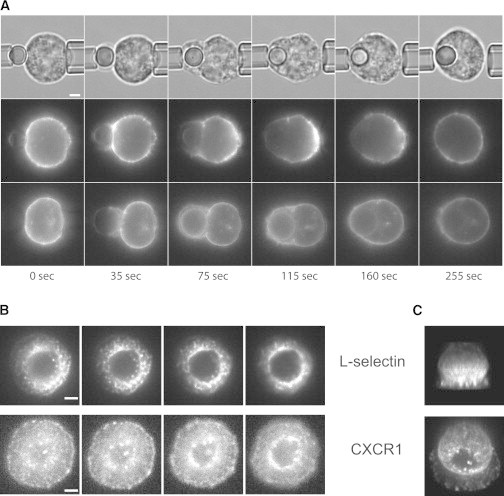
Lateral redistribution of the molecules during neutrophil spreading. (A) A series of images shows the progressive redistribution of L-selectin (middle row) and CXCR1 (lower row) as cells spread onto, then engulf, a glass bead coated with IL8. The upper row shows the brightfield images corresponding to the L-selectin distribution (middle row). LFA-1 (not shown) exhibited behavior similar to that of CXCR1. (B and C) Reconstruction of through-focus fluorescence images of a fixed cell spreading onto an IL8 coverslip. (B) Four of the 29 through-focus images (0.4 μm spacing) used in the 3D reconstructions shown in C. Scale bars, 2.0 μm.
These fixed cells were also observed using SEM. A silver enhancement procedure was used to visualize the quantum dots on different regions of the cell and in relation to the surface topography (Fig. 7). Consistent with our observations in fluorescence images, L-selectin was observed during cell spreading to be concentrated on the upper surface of the spreading lamellipodium, and it appeared to be depleted on the upper portions of the cell body away from the contact zone. As expected, based on observations and evidence from previous fluorescence measurements (11–13) (Fig. 7), the L-selectin remained concentrated at the tips of microvilli. In contrast, CXCR1 appeared to be less concentrated in the upper part of the lamellipodium and was found principally on the body of the cell in the valleys between microvilli.
Figure 7.
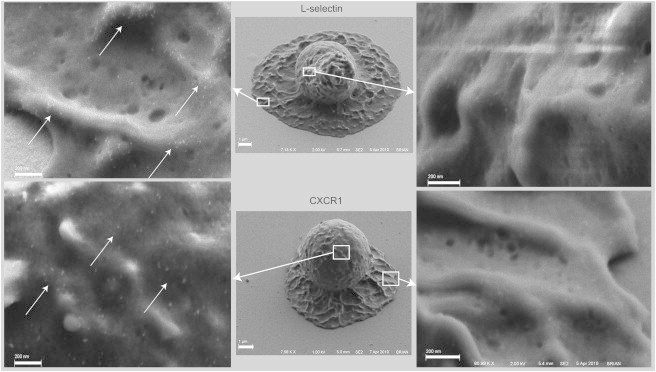
Scanning electron micrographs of neutrophils spreading on immobilized IL8 at the 60-s time point. (Upper) L-selectin redistribution upon spreading. Most L-selectin is located on the microvilli tips along the top of the lamellipodium (left, arrows), and no detectable L-selectin can be seen in the valleys between the ridges or on the cell body far from the surface (right). (Lower) Distribution of CXCR1 upon spreading on IL8. Most CXCR1 is found in the valleys between microvilli (left, arrows), and no detectable CXCR1 was found on the top of the lamellipodium (right).
Discussion
Irregularities in the cell surface affect the ability of the cell to form bonds with a substrate. This was first demonstrated experimentally by Williams et al. (8) who showed a 50-fold increase in the rate of bond formation for the same molecular pair located on a smooth rather than a ruffled cell surface. The effects of topography are due in part simply to a limitation on how much of the cell membrane can come close enough to the surface to form bonds. However, the simple limitation of cell contact area may be compounded by a nonuniform distribution of molecules relative to the surface features on the cell. For example, both electron micrographic studies on fixed cells (11) and TIRF microscopy studies on live cells (14) have shown that L-selectin tends to be distributed near the tips of microvilli on neutrophils, whereas integrins (and PSGL1) tend to be located more in the valleys, away from the microvillus tips (14). Results shown here demonstrate that the chemokine receptors CXCR1 and CXCR2 are also located far from microvillus tips in the resting cell. This distribution relative to the surface topography substantially limits the ability of these molecules to form bonds with the substrate before cell spreading. This is likely to have direct relevance to cell recruitment in vivo, where it has been demonstrated that IL8 can be localized on endothelial cell surfaces (17,18) via association with the glycocalyx (19,20).
The effects that confinement to a surface may have on bond formation and breakage have been examined in previous work. Much of this work has been focused on the important effect of force on both the formation and breakage of bonds between surface-attached molecules (21–25). Although the process of bond breakage under force has proven to be amenable to precise physical analysis and characterization, modifying physical influences on bond formation have been less easy to characterize. Largely, the confinement of reacting molecules to surfaces affects bond formation by restricting the ability of molecules to come within sufficient proximity to interact. For example, a theoretical model suggests that lateral convection of surface-bound receptors and ligands on surfaces moving relative to each other can enhance the probability of bond formation (26). Others have proposed that the time of interaction, rather than an activation energy, determines the kinetics of bond formation, resulting in novel functional forms that more accurately describe bond formation kinetics on smooth surfaces (27,28). This work is significant in that it extends these fundamental considerations to explore how mesoscopic factors (surface topography and nonuniform molecular distribution) can affect bond formation for a living cell. Indeed, our measurements and calculations suggest that these are major factors in determining bond formation and can result in changes in effective surface concentrations of >1000-fold.
Our estimation of a 1000-fold increase in receptor availability is based on the calculated ratio of the number of molecules within 70 nm of the substrate in the resting cell to the total number of molecules on the cell surface. As such, it is not a direct measure of receptor engagement but an estimate based on a model calculation of the distribution of molecules on the cell surface. Details of the calculation method, assumptions, and rationale are given in the Supporting Material. Principal assumptions and approximations of the model include a mathematical description of the shape of the microvillus, the relative distribution of microvillus heights, and the functional forms describing the nonuniform distribution of molecular density. Although the modeling approach is not unique, we have taken pains to ensure that model assumptions are consistent with all that we know about the topography of the cell surface, and we have used our experimental measurements to constrain the model assumptions and determine the key parameters that lead to our conclusion. Although there may be subtle differences in the calculated molecular distributions with the use of different models, we do not expect different models to result in significantly different conclusions. For example, we have shown that the use of two completely different functional forms to describe the nonuniform distribution of molecules on the surface does not significantly affect the estimation of the fold increase in available molecules. This is the case because our measurements of changes in fluorescence intensity during cell spreading provide a strong constraint on the distribution of the distances of molecules away from the contacting substrate, a constraint that is independent of the modeling approach. In real-world situations, other factors may affect the availability of molecules for receptor ligation. For example, compressive force between the cell and the substrate will deform the microvilli and bring additional molecules on the cell membrane into range (14). In addition, extension of the counterligand on the substrate to different distances from the surface (for example, localization of the molecules in the glycocalyx layer above the endothelial surface) will also affect the fold increase that might be seen. Nevertheless, our measurements and calculations make it clear that changing surface topography can have a dramatic effect (three orders of magnitude is a reasonable expectation) on the availability of molecules for surface ligation.
Given that such a large proportion of chemokine receptors are kept away from ligand engagement by surface topography, one might question how spreading and signaling could be initiated at all. In a parallel study (M. T. Beste, E. B. Lomakina, D. A. Hammer, and R. E. Waugh, unpublished), we show that impingement of a neutrophil onto an IL8-coated bead does in fact result in adhesion, although the number of bonds formed may be quite small (<10). We also demonstrate that occupation of this small a number of receptors is sufficient to trigger cell spreading and that the consequent increase in occupied receptors is required to initiate robust signaling responses within the cell (leading to a burst of intracellular calcium). Thus, the initial occupation of a small number of receptors leads to topographical remodeling, which results in an increase in receptor engagement of up to 1000-fold, causing a robust signaling response within the cell. Although our experimental system is much simpler than the situation observed in vivo, our results suggest a scenario in which initial contact and adhesion via selectins causes a compressive impingement of the leading edge of the cell, enabling occupation of a small number of integrins and chemokine receptors in the contact zone. This in turn leads to cell arrest and the initiation of cell spreading, allowing further integrin and chemokine receptor engagement.
In addition to the smoothing of the surface in the contact zone, we also observe the lateral redistribution of L-selectin away from the region of substrate interaction. Similar redistribution to the uropod of migrating neutrophils has been observed for PSGL1 (29), leukosialin (CD43), and the hyaluronic acid receptor (CD44) (30,31), and in T cells, a number of membrane proteins have been identified that localize to the rear of migrating cells (32). A common feature of many of these proteins is an association with ezrin-radixin-moesin (ERM) proteins linking them to the actin cytoskeleton, and several groups have shown that these ERM proteins are localized at the rear of migrating cells (33,34). Indeed, L-selectin is also known to interact with ERM proteins (35). Our approach enables us to observe L-selectin redistribution over the entire process, from stimulus to spreading to bead engulfment, and this has led to a more detailed understanding of the dynamics of L-selectin redistribution, first toward the edge of the region of stimulus and only subsequently to the rear of the cell, before regaining a uniform distribution after a bead is engulfed. This concentration of L-selectin near, but not on, the thin lamellipodium during neutrophil spreading is reminiscent of the distribution of myosin II between the cell body and the lamellipodium in T cells forming an immune synapse (36). Others have shown that myosin IIA is required for proper formation and stabilization of the immune synapse (37). These observations invite speculation that a similar mechanism might be at work in the neutrophil, where a myosin-II-based contraction might lead to concentration of ERM-linked proteins and stabilization of the cell body shape adjacent to the lamellipodial extension.
The velocity of lamellipodial extension observed in our work (∼10.0 μm/min) is comparable to neutrophil migration speeds in chemotactic gradients (38). It is interesting that blocking CXCR1 caused a significant reduction in spreading velocity. This indicates that signal transduction plays a significant role in determining the velocity of neutrophil spreading in these initial stages. This contrasts with results obtained in other cell types, where it has been observed that initial cell spreading behavior is independent of cell signaling and can be attributed simply to the balance of adhesive and viscous forces (39,40). The spreading regime over which those conclusions were drawn is confined to a diameter of close contact that is less than the cell equatorial diameter. In our studies, we also have observed a nearly linear increase in the diameter of close contact, but for the neutrophil, the regime extends to diameters significantly greater than the cell equatorial diameter. Thus, although there is a similar dependence of contact area on time, it does not appear that these two cases are comparable mechanistically.
Conclusion
Changes in surface topography during neutrophil spreading lead to dramatic increases in the number of chemokine receptors and integrins in close proximity to the substrate on which the cell spreads. Model calculations based on measurements of molecular proximity using TIRF microscopy indicate that the effective concentration of receptors at the surface can increase by >1000-fold. This is equivalent to a change in an apparent association constant of roughly three orders of magnitude and thus represents a potentially dominant mechanism by which cells may regulate adhesion and contact-mediated cell-cell and cell-substrate communication.
Acknowledgments
The authors thank Douglas Clift and Richard Bauserman for obtaining the images of labeled neutrophils engulfing glass beads.
This work was supported by the U.S. Public Health Service under National Institutes of Health grant P01 HL018208.
Supporting Material
Brightfield (left), epiflourescence (center), and TIRF (right) views of a neutrophil with surface labeled nonspecifically using Alexa Fluor 488 carboxylic-acid-TFP spreading onto a glass slide coated with IL 8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-LFA-1 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-CXCR1 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-CXCR2 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-L-selectin spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-LFA-1 spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-CXCR-1 spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-L-selectin spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.
Supporting Citations
References
- 1.Addison W. On inflammation. BMJ. 1873;2:513–514. doi: 10.1136/bmj.2.670.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohnheim J. The New Sydenham Society; London: 1889. Lectures on General Pathology: A Handbook for Practitioners and Students. [Google Scholar]
- 3.Virchow R. August Hirschwald; Berlin: 1871. Die Cellularpathologie in Ihrer Begrundung auf Physiologische und Pathologische Gewebelehre. [PubMed] [Google Scholar]
- 4.Dana N., Todd R.F., 3rd, Arnaout M.A. Deficiency of a surface membrane glycoprotein (Mo1) in man. J. Clin. Invest. 1984;73:153–159. doi: 10.1172/JCI111186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallatin W.M., Weissman I.L., Butcher E.C. A cell-surface molecule involved in organ-specific homing of lymphocytes. Nature. 1983;304:30–34. doi: 10.1038/304030a0. [DOI] [PubMed] [Google Scholar]
- 6.Snyderman R., Pike M.C. Chemoattractant receptors on phagocytic cells. Annu. Rev. Immunol. 1984;2:257–281. doi: 10.1146/annurev.iy.02.040184.001353. [DOI] [PubMed] [Google Scholar]
- 7.Ley K., Laudanna C., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 8.Williams T.E., Nagarajan S., Zhu C. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J. Biol. Chem. 2001;276:13283–13288. doi: 10.1074/jbc.M010427200. [DOI] [PubMed] [Google Scholar]
- 9.Anderson A.O., Anderson N.D. Lymphocyte emigration from high endothelial venules in rat lymph nodes. Immunology. 1976;31:731–748. [PMC free article] [PubMed] [Google Scholar]
- 10.Warfel A.H., Elberg S.S. Macrophage membranes viewed through a scanning electron microscope. Science. 1970;170:446–447. doi: 10.1126/science.170.3956.446. [DOI] [PubMed] [Google Scholar]
- 11.Bruehl R.E., Springer T.A., Bainton D.F. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J. Histochem. Cytochem. 1996;44:835–844. doi: 10.1177/44.8.8756756. [DOI] [PubMed] [Google Scholar]
- 12.Erlandsen S.L., Hasslen S.R., Nelson R.D. Detection and spatial distribution of the β2 integrin (Mac-1) and L-selectin (LECAM-1) adherence receptors on human neutrophils by high-resolution field emission SEM. J. Histochem. Cytochem. 1993;41:327–333. doi: 10.1177/41.3.7679125. [DOI] [PubMed] [Google Scholar]
- 13.Hasslen S.R., Burns A.R., Erlandsen S.L. Preservation of spatial organization and antigenicity of leukocyte surface molecules by aldehyde fixation: flow cytometry and high-resolution FESEM studies of CD62L, CD11b, and Thy-1. J. Histochem. Cytochem. 1996;44:1115–1122. doi: 10.1177/44.10.8813076. [DOI] [PubMed] [Google Scholar]
- 14.Hocdé S.A., Hyrien O., Waugh R.E. Cell adhesion molecule distribution relative to neutrophil surface topography assessed by TIRFM. Biophys. J. 2009;97:379–387. doi: 10.1016/j.bpj.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillipson M., Heit B., Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sumagin R., Prizant H., Sarelius I.H. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J. Immunol. 2010;185:7057–7066. doi: 10.4049/jimmunol.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Middleton J., Neil S., Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 18.Rot A., Hub E., Dukor P. Some aspects of IL-8 pathophysiology. III: chemokine interaction with endothelial cells. J. Leukoc. Biol. 1996;59:39–44. doi: 10.1002/jlb.59.1.39. [DOI] [PubMed] [Google Scholar]
- 19.Kuschert G.S., Hoogewerf A.J., Sanderson P.N. Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry. 1998;37:11193–11201. doi: 10.1021/bi972867o. [DOI] [PubMed] [Google Scholar]
- 20.Pichert A., Samsonov S.A., Pisabarro M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology. 2012;22:134–145. doi: 10.1093/glycob/cwr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 22.Dembo M., Torney D.C., Hammer D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B Biol. Sci. 1988;234:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]
- 23.Evans E., Ritchie K., Merkel R. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophys. J. 1995;68:2580–2587. doi: 10.1016/S0006-3495(95)80441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammer D.A., Apte S.M. Simulation of cell rolling and adhesion on surfaces in shear flow: general results and analysis of selectin-mediated neutrophil adhesion. Biophys. J. 1992;63:35–57. doi: 10.1016/S0006-3495(92)81577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spillmann C.M., Lomakina E., Waugh R.E. Neutrophil adhesive contact dependence on impingement force. Biophys. J. 2004;87:4237–4245. doi: 10.1529/biophysj.103.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang K.-C., Hammer D.A. The forward rate of binding of surface-tethered reactants: effect of relative motion between two surfaces. Biophys. J. 1999;76:1280–1292. doi: 10.1016/S0006-3495(99)77291-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robert P., Limozin L., Bongrand P. Biomolecule association rates do not provide a complete description of bond formation. Biophys. J. 2009;96:4642–4650. doi: 10.1016/j.bpj.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robert P., Nicolas A., Limozin L. Minimal encounter time and separation determine ligand-receptor binding in cell adhesion. Biophys. J. 2011;100:2642–2651. doi: 10.1016/j.bpj.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruehl R.E., Moore K.L., Bainton D.F. Leukocyte activation induces surface redistribution of P-selectin glycoprotein ligand-1. J. Leukoc. Biol. 1997;61:489–499. doi: 10.1002/jlb.61.4.489. [DOI] [PubMed] [Google Scholar]
- 30.Seveau S., Eddy R.J., Pierini L.M. Cytoskeleton-dependent membrane domain segregation during neutrophil polarization. Mol. Biol. Cell. 2001;12:3550–3562. doi: 10.1091/mbc.12.11.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seveau S., Keller H., Halbwachs-Mecarelli L. Neutrophil polarity and locomotion are associated with surface redistribution of leukosialin (CD43), an antiadhesive membrane molecule. Blood. 2000;95:2462–2470. [PubMed] [Google Scholar]
- 32.Sánchez-Madrid F., del Pozo M.A. Leukocyte polarization in cell migration and immune interactions. EMBO J. 1999;18:501–511. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alonso-Lebrero J.L., Serrador J.M., Sánchez-Madrid F. Polarization and interaction of adhesion molecules P-selectin glycoprotein ligand 1 and intercellular adhesion molecule 3 moesin and ezrin in myeloid cells. Blood. 2000;95:2413–2419. [PubMed] [Google Scholar]
- 34.Serrador J.M., Nieto M., Sánchez-Madrid F. CD43 interacts with moesin and ezrin and regulates its redistribution to the uropods of T lymphocytes at the cell-cell contacts. Blood. 1998;91:4632–4644. [PubMed] [Google Scholar]
- 35.Ivetic A., Deka J., Ager A. The cytoplasmic tail of L-selectin interacts with members of the Ezrin-Radixin-Moesin (ERM) family of proteins: cell activation-dependent binding of Moesin but not Ezrin. J. Biol. Chem. 2002;277:2321–2329. doi: 10.1074/jbc.M109460200. [DOI] [PubMed] [Google Scholar]
- 36.Babich A., Li S., Burkhardt J.K. F-actin polymerization and retrograde flow drive sustained PLCγ1 signaling during T cell activation. J. Cell Biol. 2012;197:775–787. doi: 10.1083/jcb.201201018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumari S., Vardhana S., Dustin M.L. T lymphocyte myosin IIA is required for maturation of the immunological synapse. Front. Immunol. 2012;3:230. doi: 10.3389/fimmu.2012.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aomatsu K., Kato T., Kitagawa S. Toll-like receptor agonists stimulate human neutrophil migration via activation of mitogen-activated protein kinases. Immunology. 2008;123:171–180. doi: 10.1111/j.1365-2567.2007.02684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuvelier D., Théry M., Mahadevan L. The universal dynamics of cell spreading. Curr. Biol. 2007;17:694–699. doi: 10.1016/j.cub.2007.02.058. [DOI] [PubMed] [Google Scholar]
- 40.Etienne J., Duperray A. Initial dynamics of cell spreading are governed by dissipation in the actin cortex. Biophys. J. 2011;101:611–621. doi: 10.1016/j.bpj.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattheyses A.L., Axelrod D. Direct measurement of the evanescent field profile produced by objective-based total internal reflection fluorescence. J. Biomed. Opt. 2006;11:014006. doi: 10.1117/1.2161018. [DOI] [PubMed] [Google Scholar]
- 42.Hocdé S.A., Hyrien O., Waugh R.E. Analysis of molecular accessibility in relation to cell surface topography and compression against a flat substrate. Biophys. J. 2009;97:369–378. doi: 10.1016/j.bpj.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Brightfield (left), epiflourescence (center), and TIRF (right) views of a neutrophil with surface labeled nonspecifically using Alexa Fluor 488 carboxylic-acid-TFP spreading onto a glass slide coated with IL 8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-LFA-1 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-CXCR1 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-CXCR2 spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left), epifluorescence (center), and TIRF (right) views of a neutrophil with surface labeled with Alexa Fluor 488-conjugated anti-L-selectin spreading onto a glass slide coated with IL8-fractalkine stalk chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-LFA-1 spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-CXCR-1 spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.
Brightfield (left) and fluorescence (right) images of a neutrophil labeled with Alexa 488-conjugated anti-L-selectin spreading onto and engulfing a glass bead coated with IL-8-fractalkine chimera. Movie plays at ∼30× actual speed.