

Abstract
The existence of structural intermediates in the processes of microtubule assembly and disassembly, and their relationship with the nucleotide state of tubulin, have been the subject of significant study and recent controversy. The first part of this chapter describes experiments and methods designed to characterize, using cryo-electron microscopy (cryo-EM) and image analysis, the structure of stabilized tubulin assemblies that we propose mimic the growth and shortening states at microtubule ends. We further put forward the idea that these intermediates have important biological functions, especially during cellular processes where the dynamic character of microtubules is essential. One such process is the attachment of spindle microtubules to kinetochores in eukaryotic cell division. The second part of this chapter is consequently dedicated to studies of the yeast Dam1 kinetochore complex and its interaction with microtubules. This complex is essential for accurate chromosome segregation and is an important target of the Aurora B spindle check-point kinase. The Dam1 complex self-assembles in a microtubule-dependent manner into rings and spirals. The rings are able to track microtubule-depolymerizing ends against a load and in a highly processive manner, an essential property for their function in vivo. We describe the experimental in vitro protocols to produce biologically relevant self-assembled structures of Dam1 around microtubules and their structural characterization by cryo-EM.
I. Introduction
A. Microtubule Dynamics: Assembly and Disassembly Intermediates and Nucleotide Versus Lattice Effects
As key cytoskeleton components of eukaryotic cells, microtubules play important roles in many cellular processes, including intracellular transport, cell motility, meiosis, and mitosis. In the microtubule, αβ-tubulin heterodimers bind head to tail into protofilaments, and about 13 protofilaments associate in parallel giving rise to a polar cylindrical polymer. Microtubules can switch stochastically between growing and shrinking phases, a phenomenon known as dynamic instability (Mitchison and Kirschner, 1984). This dynamic character is essential to microtubule function, as evidenced by the large number of natural compounds that bind tubulin, alter dynamics, and result in mitotic arrest (Downing, 2000; Jordan, 2002).
Dynamic instability is an intrinsic property of tubulin observable in purified preparations of this protein and based on the binding, hydrolysis, and exchange of nucleotide. The electron crystallography structure of tubulin assembled into protofilaments (Nogales et al., 1998b; Lowe et al., 2001) has been very useful in explaining the different exchangeability of the nucleotides in α- and β-tubulin, as well as the polymerization-induced hydrolysis of the exchangeable, E-site nucleotide (Nogales, 2000; Nogales et al., 1998a). The N-terminal, nucleotide-binding domain of each tubulin monomer is directly involved in polymerization contacts and is connected through the core H7 helix to a smaller, intermediate domain that is also involved in polymerization. The C-terminal domain includes two antiparallel helices that define the protofilament crest on the outside of the microtubule (Nogales et al., 1998b), while the last, acidic residues are disordered and form a cloud of negative charge around the microtubule. The nonexchangeable, N-site guanosine triphosphate (GTP) in the α-subunit is buried at the monomer–monomer interface within the dimer, while the exchangeable nucleotide at the E-site in β-tubulin sits on the exposed surface of the dimer. The microtubule polymerization process results in the burial of this E-site nucleotide at the newly formed interface within a protofilament, making it nonexchangeable and exposing it to catalytic residues in α-tubulin that promote hydrolysis. Following polymerization the E-site nucleotide is both hydrolyzed and becomes nonexchangeable. This means that the microtubule body is made of guanosine diphosphate (GDP)–tubulin subunits, a structure that is energetically unstable. In the GTP cap model, the microtubule structure is proposed to be stabilized by a layer of GTP–tubulin subunits at the ends that still retain their GTP (Mitchison and Kirschner, 1984; Fig. 1, top center). When this cap is lost the microtubule rapidly depolymerizes (Fig. 1, top left).
Fig. 1.
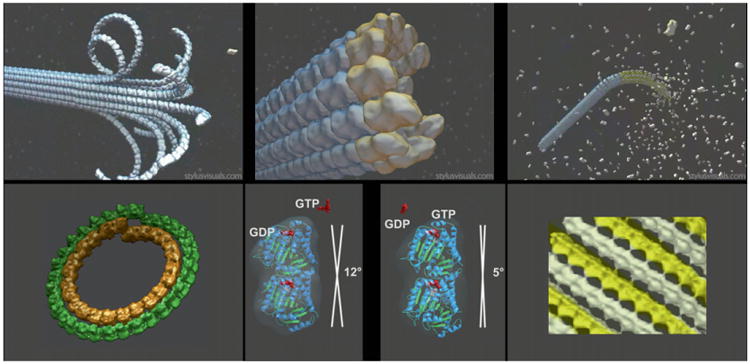
Structural view of microtubule dynamics, disassembly, and assembly intermediates, and nucleotide state of tubulin. Upper panels are schematics of three distinct states for microtubules: shrinkage (left), growth (right), and minimal guanosine triphosphate (GTP) cap (center). Lower panels are the experimental cryo-electron microscopy structures for stabilized mimics of the disassembly (left) and assembly intermediates (right), and the tubulin dimer structures for guanosine diphosphate-bound and GTP-bound tubulin outside the context of the microtubule lattice (center). Adapted from Figs. 1 and 2 in Nogales and Wang (2006). (See Plate no. 5 in the Color Plate Section.)
While the formation of the longitudinal interdimer contact in a protofilament is required for GTP hydrolysis, hydrolysis in turn is likely to affect the longitudinal interface, the strength of lateral contacts between protofilaments, and the overall structure of the protofilaments. Indeed, cryo-electron microscopy (cryo-EM) studies of depolymerizing microtubules show protofilaments peeling from the ends into ring-like structures (Mandelkow et al., 1991), similar to those observed from the self-assembly of GDP-containing tubulin dimers (see Nogales et al., 2003 for a review; Fig. 1, left top). Within the body of the microtubule, GDP subunits are locked in a straight, “GTP-like” conformation due to the structural constraints imposed by the microtubule lattice (Melki et al., 1989), for as long as there is a GTP cap (Fig. 1, top center). Thus, the energy released by GTP hydrolysis is “stored” within the lattice as structural strain (Caplow et al., 1994). The model proposes that when the GTP cap is lost, rapid depolymerization occurs, likely by weakening of lateral contacts at the ends, and the consequent release of the constrained GDP subunits into their curved, lower energy, conformational state (Melki et al., 1989). GDP–tubulin rings can be observed as microtubule depolymerization products (Bordas et al., 1983; Mandelkow et al., 1991) or can be formed from GDP–tubulin as de novo double rings that have been energetically well characterized (Frigon and Timasheff, 1975; Howard and Timasheff, 1986).
Two alternative tubulin structures, corresponding to two different assembly states of the protein, are available at atomic resolution. The first one is that of tubulin in a polymerized, straight protofilament and bound to the stabilizer taxol obtained by electron crystallography of zinc-induced tubulin sheets (Lowe et al., 2001; Nogales et al., 1998b). Docking of this atomic structure of the protofilament into a cryo-EM reconstruction of the microtubule (Li et al., 2002; Nogales et al., 1999) confirmed that the electron crystallography structure corresponds very closely to that in the microtubule and thus represent the bona-fide structure of a “polymerized” tubulin. The second tubulin structure is in a curved conformation, bound to the cellular depolymerizer RB3 (a homologue of stathmin/Op 18) and microtubule-destabilizing ligands (colchicine, podophyllotoxin, or vinblastine) (Gigant et al., 2000, 2005; Ravelli et al., 2004). There is a kink between the two dimers bound to the RB3 fragment, and an indistinguishable kink between monomers within each dimer. The tubulin monomer conformation is different from that in the straight protofilament: there is a rotation of the intermediate domain with respect to the N-terminal domain, as well as a displacement along the dimer axis of the core helix H7.
Depolymerizing factors such as RB3/stathmin are likely to have effects beyond the sequestration of the GDP state of tubulin, so the tubulin structural features observed under those conditions could result from a combination of the nucleotide state and factor binding. To elucidate the structure of GDP–tubulin free of the effect of depolymerizers, we stabilized a polymer of tubulin bound to GDP formed by tight winding of a double spiral where every turn corresponds to a slightly open double-layer ring. While not lacking challenge, this polymer was a good sample for cryo-EM and image reconstruction that allowed us to obtain a structure of the low-energy state of GDP-bound tubulin (Fig. 1, bottom left). Our study showed that irrespective of whether bound or unbound by a depolymerizer, the bending of the intra- and interdimer interfaces in the free GDP–tubulin protofilament is incompatible with the formation of lateral contacts in microtubules. So, how can binding of GTP result in the “straightening” of protofilaments observed in microtubules? An important question to address is whether tubulin structure is defined purely by nucleotide state (now an old concept), purely by lattice contact, or by a combination of both (the case we advocate) (Fig. 1, bottom center).
Andreu and coworkers (Buey et al., 2006), using arguments based mostly on the properties of FtsZ, the bacterial tubulin homolog, whose conformation appears totally insensitive to nucleotide content based on a number of X-ray crystal structures, and more recently Agard and colleagues (Rice et al., 2008) based on SAXS experiments and drug-binding properties, as well as the structure of γ-tubulin (Aldaz et al., 2005), have proposed that nucleotide has no effect on tubulin conformation. The latter explained the different self-assembly properties of the two nucleotide states of tubulin based purely on differences in affinity between subunits due to the presence or absence of the γ-phosphate at the dimer-dimer interface. Incorporation into a microtubule lattice would subsequently result in the straightening of the tubulin dimer. On the other hand, Hyman et al. had provided experimental arguments to show that even out of the context of a microtubule lattice, tubulin bound to GTP analogue guanylyl-(αβ)-methylene-diphosphonate (GMPCPP) (a nonhydrolyzable GTP analog) is “less curved” than its GDP-bound counterpart (Müller-Reichert et al., 1998). In these experiments the depolymerizing products of GDP- and GTP-containing microtubules showed peels of significantly different curvature. Recent data from AFM visualization of individual protofilaments have further supported this idea (Elie-Caille et al., 2007). Our own EM studies of the GMPCPP-bound tubulin polymer described below (Fig. 1, bottom right) agree with the Hyman reports and led us to propose the following model: the exchange of GDP for GTP in the αβ-tubulin dimer results in a change from a more kinked to a smoothly curved conformation and allows for the association of protofilaments into assembly intermediates with lateral contacts that prime closure into a cylinder (Nogales and Wang, 2006; Fig. 1, top right). This closure results in the final straightening of the dimer. The two-step straightening was in fact first suggested by the studies of Chrétien and colleagues about a decade ago. They showed that under conditions of fast tubulin assembly, growth occurs via open sheets at the ends of microtubules that later close into a cylinder (Chrétien et al., 1995). This mechanism, involving both a nucleotide and a lattice effect, may not be present in tubulin isoforms such as γ-tubulin or bacterial homologues that do not form cylindrical polymers, are monomeric, and/or do not exhibit dynamic behavior.
B. Microtubule–Kinetochore Interactions: The Yeast Dam1 Complex
The fundamental property of living systems is their ability to consume resources from their environment in order to reproduce, heritably passing on their genetic program to their offspring. At the level of an individual cell, the irreducible unit of self-replicating life, this propagation occurs through cell division. The problem of delivering exactly one copy of each chromosome to each daughter cell may need to be solved an enormous number of times over the course of an organism's lifetime; errors in this process lead to aneuploidy, which can result in cell transformation (King, 2008).
The kinetochore is a network of protein complexes which assembles on centromeric chromatin to act as the connection point between the chromosomes and the microtubules that segregate them into daughter cells (Cheeseman and Desai, 2008; Westermann et al., 2007). This complex machinery is involved in a multitude of functions during mitosis. A most essential function is to couple chromosome movement to microtubule depolymerization. Once all chromosomes are correctly bioriented, they must be pulled apart into daughter cells. After the spindle checkpoint has been satisfied, the anaphase-promoting complex triggers the destruction of the condensin complexes which tether sister chromatids together (Cheeseman and Desai, 2008; Westermann et al., 2007). Then, by a process that does not require motor activity (Koshland et al., 1988), kinetochores allow chromosomes to track the depolymerizing ends of microtubules, which are the primary site of force generation. Thus, it is thought that the energy stored in the microtubule lattice and released during depolymerization as individual protofilaments splay outward and can be harnessed by the kinetochore machinery to produce movement (Koshland et al., 1988).
The molecular mechanisms by which chromosomes attach to kinetochores, how this attachment is monitored by the cell, and how it is maintained as mitosis progresses have remained a great mystery since the first live movies of the mitotic process. For budding yeast, where this feat has to be achieved with a single microtubule attachment per kinetochore, a full part list of kinetochore proteins has emerged in recent years. The first report on Dam1 complex subunits and their requirement in mitotic spindle function appeared a decade ago. Drubin and coworkers identified Duo1p as a protein that contributed to aspects of spindle function sensed by the spindle checkpoint (Hofmann et al., 1998). Dam1p was identified by two hybrid studies as a Duo1p interacting protein and shown to bind directly to microtubules. Soon after, Winey and colleagues (Jones et al., 1999) reported the identification of the Dam1 gene in a genetic screen that showed it to be involved in spindle integrity and localized to the spindle microtubules and likely to the kinetochore. Major breakthroughs in the next few years were the identification of the rest of the Dam1 complex components (Cheeseman et al., 2001a,b; Enquist-Newman et al., 2001; Janke et al., 2002) and its functional interaction with the checkpoint kinase Ipl1 (Cheeseman et al., 2002; Kang et al., 2001; Shang et al., 2003).
The initial genetic and biochemical studies showed the ten-subunit Dam1 complex to be essential for regulated microtubule–kinetochore attachment. But it has been during the last 4 years, following the expression of the ten Dam1 complex subunits in bacteria by the Harrison Lab, Harvard Medical School (Miranda et al., 2005), that structural and biophysical studies of the interaction of the Dam1 complex with microtubules have become tractable. The first electron microscopy visualization studies of the complex proved most illuminating and exciting by showing the microtubule-induced assembly of Dam1 into rings and spirals (Miranda et al., 2005; Westermann et al., 2005; Fig. 2, left panels). A ring structure seemed an ideal coupler for the energy released during microtubule depolymerization due to protofilament peeling that follows GTP hydrolysis. Indeed, the novel ring–microtubule interaction allows the rings to diffuse on the microtubule and, during microtubule disassembly, to move processively following the depolymerizing end (Westermann et al., 2006). The energy for processive movement comes from the conformational change in tubulin during microtubule depolymerization (Nogales and Wang, 2006). Therefore, this ring structure is able to track the depolymerizing ends of microtubules (as during anaphase), without requiring energy of its own (Fig. 2, right). These data and concepts have captivated the minds of structural biologist and mitosis cell biologists (Salmon, 2005). The last section in this chapter deals with structural studies of the two self-assembly forms of Dam1 so far visualized by electron microscopy.
Fig. 2.
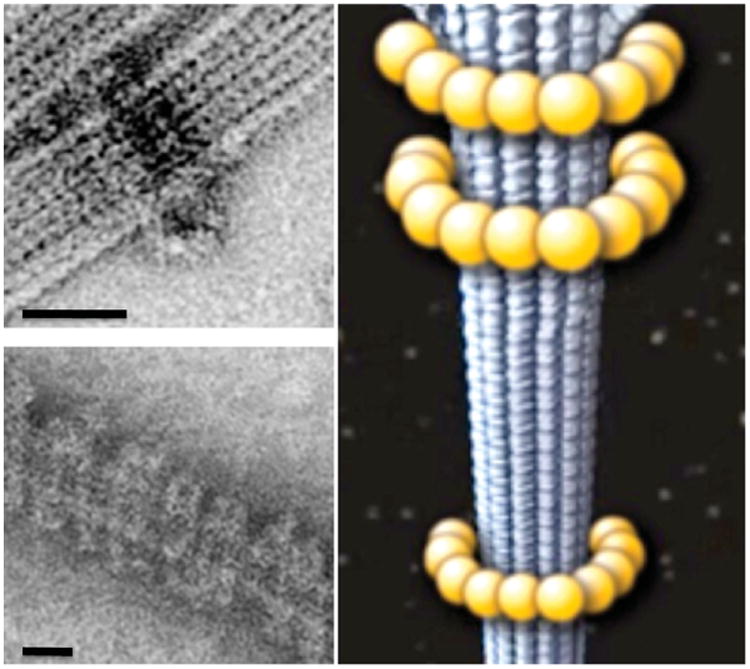
Dam1 complex self-assembly around microtubules. Electron microscopy images of a Dam1 ring (top left) and double spiral (bottom left) around microtubules. Schematic representation of the Dam1 ring around a microtubule (right). Scale bars correspond to 25 nm. (See Plate no. 6 in the Color Plate Section.)
II. Rationale
A. Proposed Structure of the Microtubule Disassembly Intermediate: Depolymerizer-Free GDP–Tubulin
The curved protofilament peels at the end of shortening microtubules constitute a structural intermediate in the disassembly process, where GDP–tubulin is in its relaxed state, clearly distinct from its constraint state in the body of microtubule wall. Such a GDP–tubulin state can be sequestered by microtubule depolymerizers such as RB3/stathmin, colchicine, and vinblastine. The binding of these factors to GDP–tubulin, however, may have additional structural effects that go beyond that determined by the nucleotide state itself. Using the fact that high concentrations of divalent cations (which are known to stabilize tubulin assembly, likely by shielding the negative change in tubulin as microtubule-associated proteins do in the cell), we were able to form stable tubular assemblies of GDP–tubulin in which ring closure does not occur and the protofilaments turn in a tight, double-layer helix (Fig. 3, left). The formation of such polymer does not need any depolymerizer and appears to recapitulate the shape of the horn-like protofilament structures at depolymerizing microtubule ends. Thus, this well-ordered assembly serves as an ideal sample for the structural characterization by cryo-EM of an unconstrained, GDP-bound tubulin in the absence of depolymerizing factors or drugs. Helical reconstruction of such assemblies ultimately allowed us to describe the structure of the protofilaments in this nucleotide state (Fig. 1, bottom left). This structure showed, for the first time, distinctive intra- and interdimer interactions and thus a distinction between the GTP and GDP interfaces. While both interfaces are kinked, the bending angle is clearly different, and one interface is dramatically more flexible than the other (we originally proposed that the GDP interface was more flexible than the GTP-containing intradimer contact, but the alternative possibility cannot be discarded from our data and is supported by recent studies; Bennett et al., 2009). This is in contrast with the RB3-bound tubulin structure where both interfaces were indistinguishable. The small rearrangement seen at the intradimer interface in RB3-bound tubulin may be due to the presence of colchicine at the site (Ravelli et al., 2004). In fact, early studies of colchicine binding interpreted the two-step binding of this drug as due to the induction of a conformational change in the tubulin dimer structure (Andreu et al., 1991). On the other hand, it is possible that the interdimer interface gets locked into an “intradimer-like” state due to the RB3 alpha helix that runs along the surface of both dimers (Ravelli et al., 2004). The EM and X-ray studies showed that, irrespective of the presence of a depolymerizer, the bending of the intra- and interdimer interfaces in GDP–tubulin protofilament is incompatible with the formation of the lateral contacts seen in microtubules (Fig. 1, bottom center). In Section III, we describe in detail the conditions for growth and stabilization of the double-layered helical GDP–tubulin assembly as well as the difficulties encountered and overcome in the image analysis of this tubulin polymer toward its three-dimensional (3D) structure.
Fig. 3.
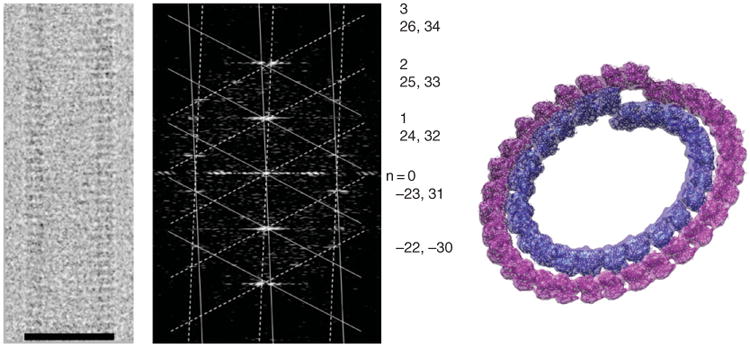
Double-layered guanosine diphosphate (GDP)-tubulin tubes. Cryo-electron microscopy image of a 32/24 double-layered GDP–tubulin tube (left) and its diffraction pattern (center). The pattern was indexed for the near and far side of the helix (solid and dashed lattice, respectively). The indexing indicates severe Bessel overlap of the two layers of helices on all the layer lines (Bessel orders for the inner and outer layer, respectively, are shown on the right). Docking of the tubulin crystal structure into one turn each of the inner and outer layer. Scale bar in left panel corresponds to 50 nm. (See Plate no. 7 in the Color Plate Section.)
B. GTP–Tubulin Structure in an Assembly Intermediate—Before Microtubule Closure and Protofilament Straightening
As has been addressed above, an important question in microtubule polymerization is whether tubulin structure is defined purely by nucleotide state (now an old concept), purely by lattice contact, or by a combination of both (the case we advocate). To reach our conclusions we studied the structure of a tubulin assembly that forms in the presence of a nonhydrolyzable GTP analog (GMPCPP) but low temperatures, where microtubule growth is precluded. At normal assembly temperature (37°) GMPCPP gives rise to structurally normal microtubules but inhibits dynamics: the microtubules are stable. At low temperatures GTP–tubulin forms small oligomers that appear slightly curved. Increasing magnesium concentration, which generally has a stabilizing effect in all forms of tubulin assembly, results in the stabilization of these oligomers and leads to the formation of ring-like structures especially if GDP is used instead of GTP. We were thus very intrigued to see that a new polymer form of tubulin with laterally associated, slightly curved protofilaments appears at low temperatures when hydrolysis is prevented by using GMPCPP. Our initial working hypothesis was that temperature slows down tubulin interactions, without having a significant effect on the rate of hydrolysis upon the formation of interdimer tubulin contact. Thus, under low-temperature conditions little assembly occurs. When it does occur, hydrolysis quickly follows, before tubulin has a chance to make a microtubule closure and store the energy as lattice strain. If the hydrolysis step is eliminated, as in the case of GMPCPP–tubulin, the slow polymerization of GTP–tubulin (GMPCPP) can continue without the conformational change that hydrolysis would bring on tubulin. Under this simple assumption, we proposed that these assembly conditions could shed information on the process of microtubule assembly taking place before microtubule closure. Section III gives details on the process of stabilization and growth of these structures, the image analysis and the structure obtained, and how this structure together with additional experiments supports the idea that this stabilized assembly form corresponds to a GTP-specific intermediate in microtubule growth.
C. Structure of Dam1 Double Spirals to Describe the Oligomeric State of the Complex Around Microtubules
Defining the architecture of the Dam1 complex and its microtubule driven self-assembly is essential to understand the mechanisms by which Dam1 rings are able to couple processive movement to microtubule disassembly and thus contribute to the end-on attachment of chromosomes to the mitotic spindle and their segregation during anaphase. It is also crucial to determine how the assembly of the ring is regulated and how the ring attaches to other components of the kinetochore. Two assembly forms of the Dam1 complex have been observed around microtubules: a closed ring structure (Fig. 2, left top) and a double spiral where each turn of the spiral corresponds to a slightly opened ring (Fig. 2, left bottom; Fig. 6, left and center). While the first is the likely physiological oligomeric form of the complex based on the number of copies of Dam1 estimated from in vivo fluorescence studies (Joglekar et al., 2006), the double spirals offered, in principle, the benefit of a higher ordered arrangement that would make it possible to use conventional helical reconstruction methods for its structural characterization by cryo-EM (Fig. 6). It turned out that the poor helical order of the Dam1 spirals makes this type of analysis more challenging than anticipated. We obtained the cryo-EM structure of the Dam1 spirals both by helical processing of a small, well-ordered segment (Wang et al., 2007) and from poorly ordered assemblies using a new implementation of the iterative real-space helical reconstruction method (Egelman, 2000) that incorporated an intensive image classification step (Ramey et al., 2009). In order to compare the structure of the complex by itself and when oligomerized around microtubules we needed to use two alternative imaging modalities. While the preservation of the cylindrical character of the spiral Dam1 assembly around microtubules requires hydration and cryo-EM procedures, the small size of the unassembled complex makes negative stain studies the method of choice. Interestingly, our 3D reconstruction studies of the Dam1 complex before and after its oligomerization around microtubules identified a large conformational change accompanying self-assembly (Wang et al., 2007). The detailed methods for obtaining the Dam1 spiral assembly around microtubules and the cryo-EM reconstruction procedures of the structure are described in the following section.
Fig. 6.

Self-assembly of Dam1 complexes into double spirals around microtubules. Negative stain image (left). Cryo-electron microscopy image of a small, ordered segment of spiral (center) and its diffraction pattern (right) extending to about 30 Å resolution. The Dam1 lattice is indicated in cyan and magenta. The tubulin 41 Å repeat is indicated. (See Plate no. 9 in the Color Plate Section.)
III. Methods
A. GDP–Tubulin Helical Tubes
SUMMARY: Double-layer tubes of GDP-bound tubulin can be formed when partially subtilisin-cleaved tubulin bound to GDP is incubated at 37°C for a few hours in the presence of high concentrations of manganese. Frozen-hydrated helical tubes can then be imaged by cryo-EM. The images of individual tubes can be classified into distinct helical families containing different number of subunits per turn. We describe how a 24/32 family was selected for further analysis and reconstruction using an iterative Fourier–Bessel algorithm to determine the relative orientation of multiple tube images (19 images used in the final average). Independent reconstructions for the inner and outer layer were then produced by Fourier integration using data up to 10-Å resolution.
1. Assembly of GDP–Tubulin Helical Tubes and Cryo-EM Analysis
We prepare our subtilisin-cleaved GDP–tubulin by closely following the procedures of Correia and coworkers (Lobert and Correia, 1992). Bovine brain tubulin from Cytoskeleton (Denver, CO) at a concentration of 6 mg/ml is incubated at 37°C for 30 min in cytoskeleton buffer II (CBII) buffer (80 mM PIPES, 1 mM ethylene glycol tetraacetic acid, 1 mM MgCl2, and 10% glycerol at pH 6.8) in the presence of 2.5 mM GTP in order to form microtubules. Subtilisin (25 μg/ml) is then added for 10 min to cleave the tubulin C-termini. The reaction is quenched by adding 3.0 mM phenylmethylsulfonyl fluoride. The subtilisin-cleaved microtubules are spun down at 13.5 K rpm for 15 min on a tabletop centrifuge, washed with prewarmed CBII buffer at 37°C, and pelleted by an additional 8 min centrifugation step. The pellet is then resuspended in cold CBI buffer (CBII buffer without glycerol) to a concentration of about 10 mg/ml. After 20 min on ice all the microtubules are fully depolymerized. The solution is centrifuged at 50 K rpm for 15 min at 4°C to remove any insoluble aggregates on a TLA100 rotor using a Beckman Coulter Optima TLX centrifuge. The supernatant can then be characterized by sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting with antibodies against α-tubulin and β-tubulin. Following this procedure about half of β-tubulin C-termini are cleaved, while practically all α-tubulin molecules are intact. Although partial removal of the C-terminus of tubulin by subtilisin treatment is not necessary for the formation of the tubes, it has a positive effect on their crystalline order. More extensive subtilisin digestion results in more triple-layered tubes and was avoided.
To form well-ordered, double-layered tubes the GDP–tubulin prepared as above is diluted to a final concentration of 2–3 mg/ml in CBI buffer. GDP and MnCl2 are added to the tubulin solution to give final concentrations of 2 and 20–50 mM, respectively. The solution is incubated at 37°C to promote GDP–tubulin assembly (although helical tubes can form at any temperature between 4 and 37°C). After 3–5 h incubation, the solution becomes cloudy. For cryo-EM analysis 3.5 μl of this solution is applied to a chloroform pretreated Quantifoil electron microscope grid (R1.2/1.3Cu 400 mesh, SPI, PA) and then the grid is mounted in a Vitrobot (FEI, the Netherlands). The grid is blotted for 1.8 s at room temperature with 100% humidity and plunged into liquid ethane slush cooled by liquid nitrogen at about −170°C. The vitrified grids are then stored in liquid nitrogen until examination. In our studies, cryo-EM grids were transferred to a Gatan 626 cryo-holder (Gatan Inc., PA) and inserted into a CM200 electron microscope with a field emission gun (FEI, the Netherlands). The temperature of the grid in the microscope was maintained below −172°C during the whole process of examination and image recording. Using low-dose protocols, images of the tubular crystals of GDP–tubulin were recorded on Kodak SO-163 films at a magnification of 50,000 and acceleration voltage of 200 kV with a dose lower than 15 e−/Å2 per picture. The defocus of the images ranged between −0.5 and −2.0 μm. In some sessions, focal pairs were taken of the same area to facilitate the classification of the tubes into different families using the higher defocus image. The films were developed and screened both by eye and by optical diffraction to remove those with astigmatism or drift. Selected films were digitized on a Nikon Super Coolscan 8000 ED scanner (Nikon, Japan) at 2000 dpi, resulting in a final pixel size of 2.54 Å. The images were transferred to the processing computer and converted from transmission to optical density using the EMAN image-processing package (Ludtke et al., 1999).
2. Screening and Reconstruction of the Helical Tubes of GDP–Tubulin
In the presence of high concentration of manganese, GDP–tubulin forms well-ordered, multilayered tubular crystals. Most of them are double-layered tubes, while some are triple-layered tubes. Analysis of the diffraction patterns from images of these tubes indicates that each layer is a one-start helix corresponding to a curved tubulin protofilament. Depending on the number of tubulin subunits per turn of these helices, the tubular crystals can be classified into different families. Each tube can be assigned to a certain family based on its number of layers, the diameters of each layer in the tube (and thus the number of tubulin subunits per turn), and the parallel/antiparallel relationship among the protofilaments of different layers (whether they are parallel or antiparallel can be determined by examining the directions of tubulin end-on-view projections at the edge walls of the two layers in filtered images). For our structure analysis we focused on the most abundant family of double-layered tubes with 32/24 subunits in antiparallel protofilaments (Fig. 3, left). The double-layer character of the tubes results in systematic overlap of the Bessel terms from inner and outer layers on all the layer lines and makes it impossible to use traditional helical reconstruction methods (Fig. 3, center). An iterative Fourier–Bessel method developed in our laboratory (Wang and Nogales, 2005a) was used to determine the relative orientation of different tube images and to produce independent 3D reconstructions of the inner and outer layers of the tube.
The image of each tube is cut from the original micrograph within a box about four times wider than that of the tube itself. The in-plane curvature of the tubes is corrected using the straightening algorithm in the Phoelix package (Carragher et al., 1996). After straightening the images are cut into three parts, the tube itself and two images of ice on both sides of the tube. These two images are averaged and used as the estimated background noise for resolution assessment. Each tube image is then padded into 512 × 4096 pixels, its Fourier transform calculated, and the power spectrum is analyzed for indexing. Layer lines are extracted from the Fourier transform of each tube up to a resolution of 8 Å. The helical selection rule for each tube differs slightly from the others because the Z-heights of the layer lines vary in one or two reciprocal units from tube to tube. For the purpose of alignment and average, the layer lines are reassigned to a common selection rule from the average of all the tubes. The same process is followed for the noise images.
Though most of the layer lines of double-layered tubes are composed of overlapping Bessel terms with different Bessel orders from the inner and outer layers, those corresponding to the one-start helical track have the same order (N= 1, 2, 3…). These layer lines, along with the equator, can be used to determine the x-shift and out-of-plane tilt of each tube. Using the same method as Amos and coworkers (Amos and Klug, 1975), the x-shift and out-of-plane tilt are calculated and corrected separately for the inner-layer helix and outer-layer helix of each tube. Tubes with out-of-plane tilt bigger than 5° are discarded. Twenty-nine images of 19 tubes (10 focal pairs) from the antiparallel 32/24 family of double-layered tubes were chosen for alignment in our analysis.
For each tube, only data within a resolution of 23 Å are used for alignment. During the alignment cycle focal paired images are half-weighted in the process of data merging and averaging. The equator is not used for the alignment. The radial scale of each tube is also refined during the process of alignment. The iterative Fourier–Bessel alignment algorithm leads to convergence to a stable solution of the alignment parameters of each layer of each tube after a dozen iterations. The iterative alignment of our 29 images of 19 tubes converged after 10 iterations. To obtain the reconstruction of the two tubulin layers independently, the set of inner-layer and outer-layer alignment parameters are used, respectively, on the Fourier transforms of images for final averaging.
During the averaging, contrast transfer function (CTF) is also corrected. CTF parameters for each image are calculated from the diffraction spectra of the ice within the hole of each micrograph using the SPIDER TF series programs (Zhu et al., 1997). In our analysis, the B factor was set roughly at 100 Å2 based on the above analysis. The amplitude contrast ratio was calculated to be 7% using the methodology of Toyoshima and Unwin (Toyoshima and Unwin, 1988). The alignment parameters are applied to the layer line data and the final average from all the tubes is calculated using the equation:
| (1) |
where M is the total image number, CTFj is the CTF value of the jth image, Fj is the Fourier transform of the jth image, and Favg is the Fourier transform of the final averaged structure.
The noise of the final reconstruction is calculated from the average of CTF-corrected noise data using the same alignment parameters and weighting factors of their corresponding tube image. The signal-to-noise ratio of every point on the layer lines of the final reconstruction is thus calculated. Only those points with signal-to-noise ratio higher than 2.0 are considered to be significant and retained in the final data set. Two final sets of Fourier averages are obtained corresponding to the inner-layer and outer-layer tubes, respectively. After Fourier–Bessel transformation and radial cutting in little g space, the real-space reconstructions of the inner-layer and outer-layer helices are calculated separately by Fourier integration using the averaged layer line data set. We have obtained reconstruction of both layers at a resolution up to 10 Å (one turn of the outer-layer reconstruction is shown in Fig. 1, bottom left panel, and one turn each of the inner and outer layers is shown in Fig. 3, right).
3. Building a Pseudo-atomic Model of GDP–Tubulin-Curved Protofilament
The atomic models of tubulin obtained by electron crystallography in a straight protofilament (1JFF.pdb) and that obtained by X-ray crystallography with tubulin bound to RB3 and colchicine (1SA0.pdb) were used in docking experiments into our 3D reconstructions of the inner and outer layer. Regions missing in the model of one monomer that have a counterpart in another (long loop in the N-terminal domain of β-tubulin; M-loop in the RB3–tubulin structures) were filled in with the available section and energy minimized using molecular dynamic procedures. This procedure is done to be able to account for most of the mass of the proteins and is not intended to produce the real structure. The atomic resolution models were fitted manually as rigid bodies into the OMAP map using the program O (Jones et al., 1991). These manually fitted atomic models of tubulin were then converted into a density map, low-pass filtered to 23 Å, and threshold filtered to masks. The masks are used to segment the tubulin heterodimer densities from the inner-layer and outer-layer reconstructions, respectively. The resulting densities were compared by Fourier shell correlation, indicating that they are practically indistinguishable within a resolution of 15 Å. Helical symmetries of the two layers of tube were then applied to the docked atomic models in the asymmetric unit of the reconstruction to generate a docking of longer protofilaments using HLXBUILD program in Situs (Wriggers et al., 1999; Fig. 3, right).
B. GMPCPP–Tubulin Helical Ribbons and Tubes
SUMMARY: Helical ribbons and tubes of GMPCPP-bound tubulin are obtained by incubating tubulin with this nucleotide at low temperatures (4–15°C) for several hours (up to 4 h) in the presence of high concentration of magnesium ions (8–30 mM). Frozen-hydrated helical tubes can then be imaged by cryo-EM. The images of two tubes corresponding to the same helical family were analyzed using traditional helical methods to obtain a reconstruction at 18-Å resolution.
1. Generating GMPCPP–Tubulin Ribbons and Tubes
The slowly hydrolyzable GTP analogue guanylyl-(αβ)-methylene-diphosphonate (GMPCPP) was purchased from Jena Bioscience Company, Germany. To obtain the GMPCPP-bound tubulin, bovine brain tubulin (Cytoskeleton) is incubated at 37°C for 30 min in CBII buffer supplemented with 2.5 mM GTP to form microtubules. Microtubules are pelleted, then resuspended in cold CBI buffer and allowed to depolymerize in ice for 20 min. Such a polymerization/depolymerization cycle produces pure GDP–tubulin due to the hydrolysis of GTP in the microtubule lattice. The solution is then centrifuged at 4°C to remove insoluble aggregates. GMPCPP is added to the supernatant to a final concentration of 10 mM, and the solution is incubated for 10 min on ice to allow for the exchange of GDP for GMPCPP. The GMPCPP–tubulin is diluted to 2–3 mg/ml in CBI buffer and MgCl2 is added to a final concentration ranging from 4 to 30 mM. The solution is incubated at different temperatures: 4, 15, 25, and 37°C. At 4°C, a temperature at which GTP–tubulin is unable to polymerize into pelletable structures (small oligomers containing a few tubulin dimers are, however, present in solution for most typical tubulin buffers), GMPCPP–tubulin self-assembles slowly into helical ribbons at magnesium concentration higher than 8 mM. When concentrations of MgCl2 as high as 20 mM are used, spiral ribbons are observed at 4°C after just 30 min of incubation. On longer incubation times the spiral ribbons become wider by addition of more protofilaments (Fig. 4, top). At higher temperatures, but lower than 20°C, the formation of spirals is faster. For incubation times longer than a couple of hours, more abundant and fast growing ribbon assemble and ultimately gain enough protofilaments to close into well-ordered helical tubular crystals of GMPCPP–tubulin. At concentration of MgCl2 lower than 6 mM the assemblies become rare. When the incubation temperature is higher than 20°C, GMPCPP–tubulin forms mainly microtubules, irrespective of the concentration of magnesium. But when the magnesium concentration is high (20 mM), some helical ribbons can be seen coexisting with microtubules.
Fig. 4.

Growth of GMPCPP–tubulin ribbon. Time points in the growth of GMPCPP ribbons (top). Early assemblies already show the pairing of protofilaments as shown by the presence of strong 10 nm repeats perpendicular to the protofilament axis (bottom). Scale bars correspond to 50 nm.
The most parsimonious explanation for how divalent cations promote tubulin self-assembly is the shielding of the negative charges in tubulin C-terminal tails by the ions, facilitating the coming together of tubulin subunits (Wolff, 1998). Assemblies that resemble these GMPCPP tubes have been reported for other conditions, both in vitro and in vivo (and generally referred to as “macrotubules”; Unger et al., 1990), although structural details of those assemblies are lacking.
2. Overall Morphology of Ribbons and Tubes
The spiral ribbons and tubes can have various diameters. When we analyzed the curvature of the protofilaments in these assemblies by plotting pitch versus diameter (Fig. 4A), we saw that the curvature of the protofilaments is invariant, that is, the longitudinal bends that exist between tubulin subunits are well defined and of a unique value that do not change as the assemblies grow and eventually close into a tube. The spiral ribbons are composed of parallel, paired tubulin protofilaments just as described for the closed tubes. Pairing is indeed a very strong tendency for protofilaments under these assembly conditions. Even in very early stages of assembly, when protofilament lengthening is limited, small oligomers of about 5–8 dimers interact laterally, robustly, and in a paired fashion (Fig. 4, bottom), similarly to what has been previously proposed by Carlier and coworkers (Carlier et al., 1997).
3. Reconstruction of GMPCPP–Tubulin Tubes and Model Building
Under the conditions described above, GMPCPP–tubulin forms open helical ribbons as well as closed well-ordered helical tubular crystals. Ribbons and tubes have similar ranges of diameter and pitch values and show the pairing of protofilaments. Samples of GMPCPP–tubulin tubes were vitrified and imaged by cryo-EM following the procedures described for the GDP–tubulin double tubes. The GMPCPP tubes have diameters that range from 500 to 600 Å and belong to different helical families. Using the best-ordered tubes belonging to the (11, −37) family, we performed helical reconstruction from two images with defocus values of 1.8 and 0.95 μm, respectively. They were straightened, their layer lines extracted up to 12 Å, and the two Fourier transforms aligned using conventional helical reconstruction methodology. The final average was calculated by merging of the aligned Fourier transforms after CTF correction, just as was done for the GDP–tubulin tubes. This process resulted in a reconstruction of the GMPCPP–tubulin tubes with a resolution of 18 Å (Fig. 5, left). The atomic model of β-tubulin (1JFF.pdb) was manually docked in the monomer densities of two adjacent protofilaments using O. The docked atomic structures were used to generate a full pseudo-atomic model for the whole density by applying helical symmetry (Fig. 5, right).
Fig. 5.
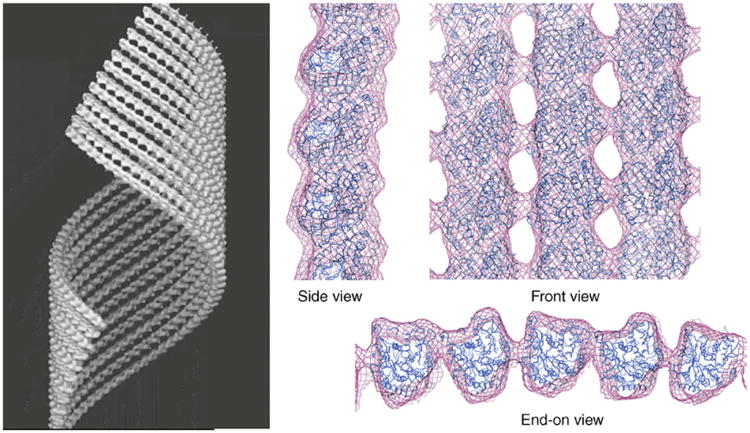
Structure of GMPCPP–tubulin tubes. Three-dimensional cryo-electron microscopy reconstruction where only 10 protofilaments of the tube wall are shown (left). Docking of the crystal structure of a tubulin monomer to reproduce the tubulin lattice in this structure, shown in different views (right). The side view shows the slight outward curvature of the GMPCPP protofilament. The front view shows the striking similarity of the lateral protofilament stagger in this structure and that of microtubules. In the end-on view the pairing of protofilaments is most apparent. Right side adapted from Fig. 3c in Wang and Nogales (2005). (See Plate no. 8 in the Color Plate Section.)
4. Direct Conversion of GMPCPP–Tubulin Tubes into Microtubules
After the formation of GMPCPP–tubulin ribbons at 15°C, increasing the temperature to 37°C resulted in the conversion of the tubes to microtubules. We visualized the samples at different time points during the 37°C incubation by negative stain EM using 2% uranyl acetate and a JEM-1200 EXII transmission microscope (JEOL, Japan). Within half an hour, the ribbons appear more open and coexist with micro-tubules. After incubations longer than an hour, all the ribbons have disappeared and have been substituted by microtubules.
In order to prove that the temperature-driven conversion of GMPCPP tubes into microtubules was direct, without involving a depolymerization step, we devised the following fluorescence microscopy assay. Two populations of GMPCPP–tubulin tubes with different fluorescent labels were prepared by incubation of two types of GMPCPP–tubulin mixtures (5:1 unlabeled tubulin:fluorescein–tubulin and 5:1 unlabeled tubulin:rhodamine–tubulin; fluorescence-labeled tubulins were purchased from Cytoskeleton Inc.). The two GMPCPP tube samples were pelleted by centrifugation at 30,000 rpm for 10 min below room temperature and washed once with CBI buffer containing 15 mM magnesium to remove any free tubulin. The two pellets were resuspended and then mixed in CBI buffer with 15 mM magnesium. The mixture was incubated at 37°C for over 3 h to make sure all the helical ribbons and tubes had converted into microtubules. The incubation solution was then diluted ten times, fixed, and viewed by fluorescence microscopy with a 60× Nikon objective on a Nikon Eclipse fluorescence microscope according to the method described in Westermann et al. (2005). A viewing area was examined using two different filters in order to record the patterns of fluorescein-tubulin (green) and rhodamine-tubulin (red), respectively. The two patterns were then merged to show relative distributions. Images showed no overlap of the two colors, as would have been expected if the GMPCPP tubes had not depolymerized to dimers or small oligomers before microtubule formation. Interestingly, many microtubules showed adjacent green and red regions indicating annealing of the structures. As a control, the two fluorescent tubulins were mixed before any assembly, then incubated at 37°C in the presence of GMPCPP to form microtubules, fixed, and examined in the same way. In this case, the green and red images showed perfect overlap, as expected from assembly of randomly mixed tubulin dimers with the two labels (Wang and Nogales, 2005b).
C. Microtubule-Dam1 Kinetochore Complex Assemblies
SUMMARY: In the presence of microtubules, the yeast kinetochore Dam1 complex assembles into rings or helical spirals around microtubules. EM analysis of the ring revealed it to consist of 16 Dam1 complexes. Segments of well-ordered double spirals of Dam1 helical assemblies were studied by cryo-EM and helical reconstruction methods to provide the 3D structure of the assembly at a resolution of 30 Å. A new algorithm to deal with the disorder and symmetry heterogeneity of this sample has been recently developed, opening new possibilities in the characterization of the Dam1 complex, including the study of mutants or the potential to reach high-resolution reconstructions of the assemblies.
1. Preparing Ring and Spiral Assembly of Dam1 Around Microtubules
The Dam1 complex has been shown to bind more readily to microtubules made up from GMPCPP-bound tubulin (Westermann et al., 2005). GMPCPP microtubules are prepared as described elsewhere (Westermann et al., 2005) at a concentration of about 2 mg/ml by incubation at 37°C. For ring formation the Dam1 complex is diluted to 0.1 mg/ml concentration in microtubule-assembly CBI buffer and incubated with 0.5 mg/ml GMPCPP microtubules at room temperature for about 10 min. The sample is then negatively stained with 2% uranyl acetate and examined by EM. Rings can be readily observed around microtubules. Analysis of some end-on views of the assembly revealed a 16-fold rotational symmetry of the rings (Westermann et al., 2006).
For helical spiral formation the Dam1 complex is first dialyzed against a low-salt buffer (150 mM KCl, 20 mM potassium phosphate, pH6.8, 1 mM ethylenediamine-tetraacetic acid) for 1–2 h at room temperature before addition. This room temperature dialysis in low-salt buffer, along with the high concentration of Dam1 (at least 5 mg/ml), is not necessary for ring formation but appears important in spiral formation, an effect that could be related to the existence of Dam1 oligomers in low salt prior to addition of microtubules (Westermann et al., 2005). After overnight incubation, samples are applied to glow-discharged Quantifoil grids, blotted with filter paper for 1.5–2 s, and plunged into a liquid ethane slush cooled by liquid nitrogen using a Vitrobot®. Microtubules in these conditions are fully decorated with Dam1 spirals and very occasionally fragments with well-ordered assemblies can be observed.
2. Cryo-EM and Helical Reconstruction of the Dam1 Spiral Assembly Around Microtubules
The frozen-hydrated samples were transferred to a Gatan 626 cryo-holder cooled by liquid nitrogen and examined either on a Philips CM200 or on a Tecnai F20 microscope. Both microscopes are equipped with field emission guns and are operated at 200 keV. Micrographs are taken on Kodak SO163 films under low-dose mode with less than 20 e/Å2 at a magnification of 50 K. The defocus range was from −1.5 to −3.0 μm. Films are developed and scanned as described previously for single-particle negatively stained samples. The step size used was 2.54 Å/pixel.
At high concentrations the Dam1 complex decorates microtubules as double spirals that show a tendency to pack tightly with time. Segments of Dam1-wrapped microtubules appearing particularly well ordered were boxed using the program BOXMRC and their power spectrum calculated using the SUPRIM package (Schroeter and Bretaudiere, 1996; Zhou et al., 1996). Out of hundreds of segments examined, two showed clear helical diffraction to resolution better than 40 Å (Fig. 6, right). These two images not only appear particularly tightly wound but also turn out to have a tighter connection with the underlying microtubule, thus maximizing protein contacts into a more crystalline array. Most spirals appear ordered by visual inspection but do not display more than one or two layer lines, likely due to the loss of registry between adjacent turns. The handedness of the spirals was determined to be left-handed using platinum metal shadowing (Wang et al., 2007).
The axial repeats of the microtubule and the Dam1 complex spirals are not a multiple of each other and this required the parallel, independent reconstruction of both structures from the same image. Analysis of two good segments using the SUPRIM image-processing package (Schroeter and Bretaudiere, 1996) and home-modified MRC image-processing software allowed us to index both helical arrangements. While the most common arrangement of a Dam1 ring is a 16-fold symmetric ring around a 13- or 14-protofilament microtubule, the two selected images correspond to a 15-fold Dam1 spiral (14.6 unit numbers per turn, to be precise), around a 14-protofilament microtubule, and a 16-fold spiral (15.7 unit numbers per turn), around a 16-protofilament microtubule. Each particle was processed independently, and the transforms of each showed a clear two-fold symmetry with an axis perpendicular to the helical axis; that is, by changing the azimuthal angle and axial position of the phase origin, about 10 or more layer line peaks for each particle could be brought to within a few degrees of 0° or 180°, thus centering the two-fold axis. Because there are four possible locations for the two-fold axis, it is necessary to choose the same two-fold axis for both data sets. With correct choice of origin, pairs of corresponding peaks in the two data sets are close to 0° or 180° as expected. After out-of-plane tilt correction and origin alignment, the “little g” data of the four sides (near and far sides for each of the two segments) are merged (DeRosier et al., 1999). Only data within a resolution range below the first zero of the CTF (∼35 Å for the first image and ∼28 Å for the second image) are extracted for analysis and averaging. On each layer line, only data points with significant signal for amplitude and phase are maintained. Because the first segment has a larger defocus value, higher resolution layer line data arise solely from the second segment. Most of the densities are within the radii of 150–350 Å, indicating the radial separation of the Dam1 complex density from the microtubule density. After removal in little g space of the low radii data that corresponds to the microtubule density (Wang and Nogales, 2005a), two-fold symmetry is applied on the merged layer lines and a 3D map of the Dam1 complex assembly is obtained by Fourier-Bessel reconstruction of the averaged layer lines. The microtubule layer lines are treated similarly but separately to calculate the 3D map of the 16-protofilament microtubule in the center of the second segment. Summation of the two 3D maps results in a final 3D map of the Dam1 spiral with 15.7 units per turn around a 16-protofilament microtubule (Fig. 7).
Fig. 7.
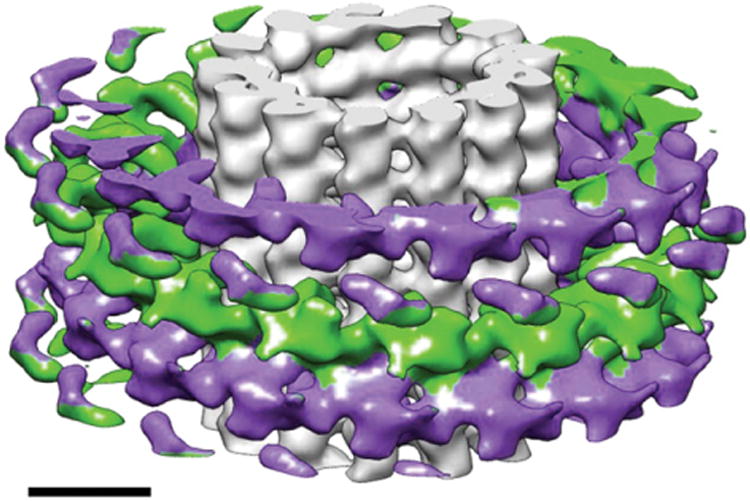
Cryo-electron microscopy reconstruction of the Dam1 double spiral around microtubules. The spirals run antiparallel (each shown in two distinct colors) and the resulting lack of polarity of the Dam1 assembly stands in contrast with the polar character of the underlying microtubule. Scale bar corresponds to 100 nm. (See Plate no. 10 in the Color Plate Section.)
3. Development of New Image Analysis Methodology
At saturating concentrations the Dam1 complex forms tightly packed spirals around microtubules, with each turn corresponding very closely to a slightly skewed ring (or lock-washer). This helical arrangement has the potential to aid structural analysis, by both concentrating the complexes and aligning them. However, as described above, the Dam1 spirals formed around microtubules very rarely sustain the type of long-range helical order required for conventional helical reconstruction methodology (e.g., kinesin; Amos and Hirose, 1997; Arnal et al., 1996; Sosa and Milligan, 1996). Out of hundreds of filaments examined, only two showed clear helical diffraction to a resolution better than 40 Å. A possibility to overcome this pitfall is to use the single-particle helical reconstruction approach developed by Egelman (Egelman, 2000). Ideally we would like to utilize the helical order retained at the level of single turns to avoid this limitation and robustly study this sample. Conventional Fourier–Bessel methods cannot operate on short helical stretches, due to extreme layer line broadening and lack of signal, while the normal single-particle approaches need further modification to deal with the symmetry mismatch between Dam1 and microtubule assemblies to efficiently separate the two parts during the reconstruction.
We have recently developed an approach (Ramey et al., 2009) that overcomes these difficulties in studying Dam1 structure by removing the microtubule from each short segment and then applying two-dimensional image alignment and classification techniques to sort the assembly turn by turn into homogeneous classes, with no assumptions made about the symmetries present or amount of disorder. Inspection of class averages allows us to determine the number of symmetries present and to estimate what helical parameters to use in their reconstruction. Implementation of our method yielded a 30 Å reconstruction of the Dam1 complex in very good agreement with the previous reconstruction by traditional Fourier–Bessel methods (Fig. 8). Importantly, the new structure was obtained using helical segments that were too short for traditional methods and had been discarded in our previous study. This method has general applicability to many other microtubule-associated proteins that assemble around microtubules.
Fig. 8.
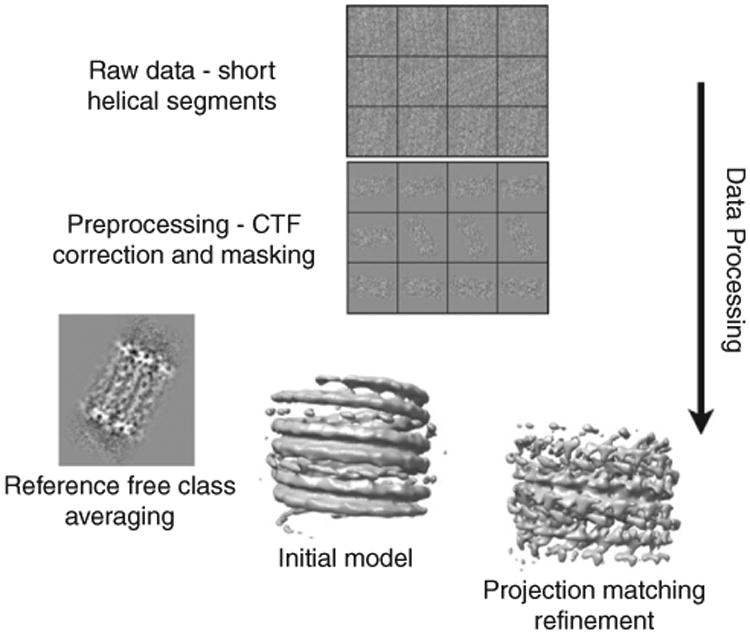
Schematic overview of the single-particle approach utilized for the analysis of Dam1 spirals. Helical segments are boxed, preprocessed to remove the microtubule, and then aligned and classified to reduce heterogeneity. Two-dimensional averages can then be used to estimate helical symmetry and create an initial model for refinement by single-particle methods (adapted from Fig. 5 in Ramey et al., 2009).
IV. Discussion
A. Biological Relevance of Stabilized Tubulin Structures as Intermediates
Our cryo-EM reconstruction of the high-Mg2+, GMPCC-bound, cold-stabilized structures (which we will refer to as “ribbons”) (Fig. 5) showed protofilaments to be slightly and smoothly curved, with apparent, indistinguishable intra- and interdimer kinks between tubulin monomers (Wang and Nogales, 2005b). Most importantly, the structure showed the presence of alternating lateral contacts between protofilaments that otherwise preserved the precise stagger between protofilaments seen in the microtubule. This meant that the structures would be able to convert into microtubules without the need of longitudinal sliding between protofilaments, but by a rotation around a surface of the tubulin dimer. Interestingly, this surface is as or even more conserved than the final interface involved in the stable microtubule lateral contacts (Fygensonm et al., 2004), suggesting its likely relevance in microtubule function. It is important to mention that this type of arrangement, or at least one involving alternative lateral contacts without longitudinal displacements between protofilaments, could have been deduced directly from the extended sheets observed by Chretien and colleagues at the growing end of microtubules, unless extreme deformability is otherwise hypothesized for the tubulin subunit.
Thus, the polymer observed under our conditions has the potential to structurally correspond to the sheet structure proposed as a structural intermediate in microtubule growth. However, many aberrant tubulin polymers have been observed over the years, which reflects tubulin's extreme tendency to polymerize and (importantly) the plasticity of the polymerization process, but which does not necessarily correspond to any structure of functional relevance (see a recent review about the structural plasticity in tubulin and actin; Kueh and Mitchison, 2009). In all those cases, the polymers formed had to be depolymerized down to individual dimers before a microtubule could be formed with the same subunits upon changing polymerization conditions. We proposed that if our structure could directly convert into microtubules, without them breaking down into individual subunits in the process, this behavior would, once again, be in agreement with the idea that our structure corresponds to an early state in microtubule assembly. Our fluorescence microscopy experiments proved that the conversion from ribbons to microtubules is direct. So our ribbon structures have a protofilament arrangement that can explain the sheet structure of Chretien and coworkers, and they convert directly into microtubules, as those sheets do, involving rolling of the lateral interface over a highly conserved region on the protofilament surface. This led us to propose that our ribbon structure is a good model for the early stages of microtubule assembly.
On the other hand, the GDP–tubulin tube represents the structural intermediate during disassembly of microtubules. Within the body of the microtubule, GDP subunits are locked in a straight, “GTP-like” conformation due to the structural constraints imposed by the microtubule lattice (Melki et al., 1989), for as long as there is a GTP cap. Thus, the energy of GTP hydrolysis is “stored” within the lattice as structural strain (Caplow et al., 1994). The model proposes that when the GTP cap is lost, rapid depolymerization occurs by conversion into the low-energy, curved GDP state.
A number of computational simulations have been generated based on existing experimental data (Janosi et al., 2002; Molodtsov et al., 2005; Tuszynski et al., 2005; Vanburen et al., 2005). With some common simplifications, such as assuming GTP–tubulin to be in a straight conformation, GDP–tubulin favoring a curved conformation, and the microtubule wall having elastic properties, they all reach the same conclusion: that the energy released from GTP hydrolysis becomes constrained lattice energy in the microtubule body and that this energy drives microtubule depolymerization and generates force that may perform work, like pulling a kinetochore along the microtubule. Simulations suggest that the mechanical stress is at a minimum at the connection between the GTP cap and the body of the microtubule and that only a couple layers of GTP–tubulin are sufficient to stabilize the microtubule (Molodtsov et al., 2005). A metastable intermediate state is also predicted and suggests a mechanism for rescue (the process of switching from a shrinking to a growing phase; Janosi et al., 2002; Vanburen et al., 2005). More elaborate simulation studies will now be possible following new knowledge of the structure of depolymerized GDP–tubulin and the process of GTP–tubulin assembly.
The elastic energy stored in the microtubule wall is released during its disassembly. A question is where this energy goes and what it is for. Work from McIntosh group showed that the frayed end of a depolymerizing microtubule can generate enough force to push a load to the opposite end of the microtubule (Grishchuk et al., 2005). The discovery of the Dam1 ring complex has given further biological relevance to the curved GDP–tubulin protofilaments at the microtubule end as the potential generators of the processive and directional movement of Dam1 with depolymerizing microtubules (Nogales and Wang, 2006; Westermann et al., 2006).
B. Biological Relevance of Dam1 Spirals and Rings
The flexible, electrostatic interaction between the mismatched symmetries of Dam1 and the microtubule suggested that a Dam1 ring would be able to diffuse along the microtubule lattice by switching stochastically from one set of E-hooks to another. The implications for kinetochore function are considerable and have led to experiments that showed how the rings were capable, not only of one-dimensional diffusion on the microtubule lattice but also of tracking depolymerizing microtubule ends, with high processivity and without energy consumption of their own (Westermann et al., 2006). In the model put forward, Dam1 ring structures interact with the microtubule via flexible elements and lack a footprint on the microtubule lattice, allowing for diffusion that becomes biased and unidirectional when the microtubule depolymerizes (Nogales and Wang, 2006; Westermann et al., 2006). Robust connection of the depolymerizing spindle microtubule to the kinetochore requires a highly processive and mobile attachment. There is currently disagreement about whether this is accomplished by the Dam1 complex through a biased diffusion or forced walk mechanism (Grishchuk et al., 2008; Westermann et al., 2006).
An encircling ring is clearly an attractive model for processivity and a solution evolution has found for tracking another linear substrate, DNA. However, some of the required properties of Dam1 at the kinetochore have been recapitulated in vitro by nonencircling oligomers or single complexes, pointing to the idea that a ring may not be necessary for function (Gestaut et al., 2008; Grishchuk et al., 2008; see Chapter 33 by Grishchuk and Ataullakhanov, this volume). On the other hand, fluorescence data have put the number of Dam1 complexes at the budding kinetochore at 16–20 (Joglekar et al., 2006), in agreement with the number seen in a single ring. Using Dam1-coated beads, Asbury et al. (2006) showed that the complex could follow both growing and shrinking microtubule ends. The attachments made between the beads and the microtubule were load-bearing, and sliding experiments indicated that Dam1 was forming a ring that saw growing and shrinking ends as barriers. Interestingly, the presence of the bead at microtubule ends had no apparent effect on microtubule dynamics. However, Ashbury and coworkers went on to show that applying tension to the bead against the direction of microtubule depolymerization decreased catastrophe frequency, slowed shortening, and increased rescue frequency. The authors noted the compatibility of a ring model with the force-dependent changes in dynamic parameters they observed: the ring would transmit tension to the peels at the end of depolymerizing microtubules and tend to straighten them, resulting in a microtubule-stabilizing effect. The dependency of microtubule stabilization on tension also agrees well with the idea of multiple weak binding sites for the ring: weak attachments would allow for diffusion and by themselves have little effect on disassembly parameters. However, the ring would be able to affect the stability of the microtubule when force is exerted through it against the depolymerizing direction. These concepts, however, had already been put into question. Davis and coworkers developed a fluorescence-based assay to measure Dam1–microtubule affinity with high sensitivity at extremely low concentrations of tubulin (Gestaut et al., 2008). They reported values 30-fold greater than previously described (in the nanomolar range). Furthermore, single-molecule experiments showed that at low concentrations of Dam1, where only short oligomers, 1–4 copies in size are formed, this complex is able to follow both assembling and disassembling microtubule ends, although with reduced processivity.
The issue of the oligomerization state of Dam1 and its functional relevance has been further analyzed by McIntosh and coworkers (Grishchuk et al., 2008). Based on fluorescence assays they proposed that Dam1 ring oligomers are unable to diffuse but able to track depolymerizing ends. Smaller oligomers diffuse with rates inversely proportional to their sizes. Interestingly, they also found that single rings are able to track microtubule ends, but not larger assemblies. While the presence of one tracking ring reduces the depolymerization rate, its encountering a second ring results in halting of the depolymerization process until one of the rings disassembles, most likely the one in contact with the microtubule edge. These results are reminiscent of the effect of force on depolymerization speed and rescue rates in the bead experiments of Asbury and coworkers. Experiments by McIntosh and coworkers using Dam1-coated beads show two very different behaviors depending on whether soluble Dam1 complexes were also available in solution. Without soluble Dam1 the beads bound only to the GMPCCP section of microtubules (a region that would mimic the stabilizing GTP cap at growing microtubule ends). These beads increased the depolymerization rate with increasing Dam1 bead decoration and tracked depolymerizing ends by rolling, rather than sliding of the beads. The authors interpret these results as a lack of ring formation and thus postulate two distinct mechanisms for end tracking by rings and small oligomers.
It is interesting to note that phosphomimetic mutants, which are impaired for ring formation but are able to track depolymerizing microtubule ends in vitro (Gestaut et al., 2008), are, however, deleterious in vivo, where they result in chromosome loss (Cheeseman et al., 2002). It is thus likely that smaller oligomers, while retaining some of the functional properties of the rings, are deficient in a cellular context.
V. Summary
Dynamic instability is essential for microtubule function, specially during mitosis. This chapter deals with studies aimed at understanding the coupling of microtubule assembly and disassembly to the nucleotide state of tubulin, and the existence and relevance of structural intermediates important in both processes. We describe the details of both the stabilization of assemblies that mimic these intermediates and the structural characterization of these assemblies using cryo-EM and image analysis. The robust and regulated engagement of highly dynamic spindle microtubules with chromosomes during mitosis involves the interaction of microtubules with kinetochore components. To begin to shed light on this important process we have studied the yeast Dam1 kinetochore complex. Using electron microscopy and image analysis we have defined the structure of this complex and its microtubule-induced self-assembly into rings and spirals. Our structural studies of these two self-assembly systems, which are exquisitely coupled during mitosis, are important for our molecular understanding of the mitotic process, and together with other functional assays, are giving us a first glimpse at a molecular mechanism essential in the life cycle of all cells.
Acknowledgments
We are in debt with all our collaborators in the studies of microtubule structure and dynamics and kinetochore complexes over the years. We are thankful to Greg Alushin for his insight into the mitotic process and to members of the Nogales lab for inspiration and technical know how. This work was funded by grants from the National Institutes of Health and the Agouron Foundation. EN is a Howard Hughes Medical Institute Investigator.
References
- Aldaz H, Rice LM, Stearns T, Agard DA. Insights into microtubule nucleation from the crystal structure of human gamma-tubulin. Nature. 2005;435:523–527. doi: 10.1038/nature03586. [DOI] [PubMed] [Google Scholar]
- Amos L, Hirose K. The structure of microtubule-motor complexes. Curr Biol. 1997;9:4–11. doi: 10.1016/s0955-0674(97)80145-7. [DOI] [PubMed] [Google Scholar]
- Amos LA, Klug A. Three-dimensional image reconstructions of the contractile tail of T4 bacteriophage. J Mol Biol. 1975;99:51–64. doi: 10.1016/s0022-2836(75)80158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu JM, Gorbunoff MJ, Medrano FJ, Rossi M, Timasheff SN. Mechanism of colchicine binding to tubulin. Tolerance of substituents in ring C′ of biphenyl analogues. Biochemistry. 1991;30:3777–3786. doi: 10.1021/bi00229a027. [DOI] [PubMed] [Google Scholar]
- Arnal I, Metoz F, DeBonis S, Wade RH. Three-dimensional structure of functional motor proteins on microtubules. Curr Biol. 1996;6:1265–1270. doi: 10.1016/s0960-9822(02)70712-4. [DOI] [PubMed] [Google Scholar]
- Asbury CL, Gestaut DR, Powers AF, Franck AD, Davis TN. The Dam1 kinetochore complex harnesses microtubule dynamics to produce force and movement. Proc Natl Acad Sci USA. 2006;103:9873–9878. doi: 10.1073/pnas.0602249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Chik J, Slysz GW, Luchko T, Tuszynski J, Sackett DL, Schriemer DC. Structural mass spectrometry of the alpha beta-tubulin dimer supports a revised model of microtubule assembly. Biochemistry. 2009;48:4858–4870. doi: 10.1021/bi900200q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordas J, Mandelkow EM, Mandelkow E. Stages of tubulin assembly and disassembly studied by time-resolved synchrtron X-ray scattering. J Mol Biol. 1983;164:89–135. doi: 10.1016/0022-2836(83)90089-x. [DOI] [PubMed] [Google Scholar]
- Buey RM, Diaz JF, Andreu JM. The nucleotide switch of tubulin and microtubule assembly: A polymerization-driven structural change. Biochemistry. 2006;45:5933–5938. doi: 10.1021/bi060334m. [DOI] [PubMed] [Google Scholar]
- Caplow M, Ruhlen RL, Shanks J. The free energy of hydrolysis of a microtubule-bound nucleotide triphosphate is near zero: All of the free energy for hydrolysis is stored in the microtubule lattice. J Cell Biol. 1994;127:779–788. doi: 10.1083/jcb.127.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier MF, Didry D, Pantaloni D. Hydrolysis of GTP associated with the formation of tubulin oligomers is involved in microtubule nucleation. Biophys J. 1997;73:418–427. doi: 10.1016/S0006-3495(97)78081-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carragher B, Whittaker M, Milligan RA. Helical processing using PHOELIX. J Struct Biol. 1996;116:107–112. doi: 10.1006/jsbi.1996.0018. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J, Yates JR, III, Chan CS, Drubin DG, Barnes G. Phospho-regulation of kinetochore-microtubule attachments by the Aurora kinase Ipl1p. Cell. 2002;111:163–172. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Brew C, Wolyniak M, Desai A, Anderson S, Muster N, Yates JR, Huffaker TC, Drubin DG, Barnes G. Implication of a novel multiprotein Dam1p complex in outer kinetochore function. J Cell Biol. 2001a;155:1137–1145. doi: 10.1083/jcb.200109063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Enquist-Newman M, Muller-Reichert T, Drubin DG, Barnes G. Mitotic spindle integrity and kinetochore function linked by the Duo1p/Dam1p complex. J Cell Biol. 2001b;152:197–212. doi: 10.1083/jcb.152.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrétien D, Fuller SD, Karsenti E. Structure of growing microtubule ends: Two-dimensional sheets close into tubes at variable rates. J Cell Biol. 1995;129:1311–1328. doi: 10.1083/jcb.129.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier D, Stokes DL, Darst SA. Averaging data derived from images of helical structures with different symmetries. J Mol Biol. 1999;289:159–165. doi: 10.1006/jmbi.1999.2677. [DOI] [PubMed] [Google Scholar]
- Downing KH. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu Rev Cell Dev Biol. 2000;16:89–111. doi: 10.1146/annurev.cellbio.16.1.89. [DOI] [PubMed] [Google Scholar]
- Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- Elie-Caille C, Severin F, Helenius J, Howard J, Muller DJ, Hyman AA. Straight GDP-tubulin protofilaments form in the presence of taxol. Curr Biol. 2007;17:1765–1770. doi: 10.1016/j.cub.2007.08.063. [DOI] [PubMed] [Google Scholar]
- Enquist-Newman M, Cheeseman IM, Van Goor D, Drubin DG, Meluh PB, Barnes G. Dad1p, third component of the Duo1p/Dam1p complex involved in kinetochore function and mitotic spindle integrity. Mol Biol Cell. 2001;12:2601–2613. doi: 10.1091/mbc.12.9.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigon RP, Timasheff SN. Magnesium-induced self-association of calf brain tubulin. I. Stoichiometry. Biochemistry. 1975;14:4559–4566. doi: 10.1021/bi00692a001. [DOI] [PubMed] [Google Scholar]
- Fygensonm D, Needleman D, Sneppen K. Variability-based sequence alignment identifies residues responsible for functional differences in α and β tubulin. Protein Sci. 2004;13:25–31. doi: 10.1110/ps.03225304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestaut DR, Graczyk B, Cooper J, Widlund PO, Zelter A, Wordeman L, Asbury CL, Davis TN. Phosphoregulation and depolymerization-driven movement of the Dam1 complex do not require ring formation. Nat Cell Biol. 2008;10:407–414. doi: 10.1038/ncb1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Curmi PA, Martin-Barbey C, Charbaut E, Lachkar S, Lebeau L, Siavoshian S, Sobel A, Knossow M. The 4 angstrom X-ray structure of a tubulin: Stathmin-like domain complex. Cell. 2000;102:809–816. doi: 10.1016/s0092-8674(00)00069-6. [DOI] [PubMed] [Google Scholar]
- Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435:519–522. doi: 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- Grishchuk EL, Molodtsov MI, Ataullakhanov FI, McIntosh JR. Force production by disassembling microtubules. Nature. 2005;438:384–388. doi: 10.1038/nature04132. [DOI] [PubMed] [Google Scholar]
- Grishchuk EL, Spiridonov IS, Volkov VA, Efremov A, Westermann S, Drubin D, Barnes G, Ataullakhanov FI, McIntosh JR. Different assemblies of the DAM1 complex follow shortening microtubules by distinct mechanisms. Proc Natl Acad Sci USA. 2008;105:6918–6923. doi: 10.1073/pnas.0801811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann C, Cheeseman IM, Goode BL, McDonald KL, Barnes G, Drubin DG. Saccharomyces cerevisiae Duo1p and Dam1p, novel proteins involved in mitotic spindle function. J Cell Biol. 1998;143:1029–1040. doi: 10.1083/jcb.143.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard WD, Timasheff SN. GDP state of tubulin: Stabilization of double rings. Biochemistry. 1986;25:8292–8300. doi: 10.1021/bi00373a025. [DOI] [PubMed] [Google Scholar]
- Janke C, Ortiz J, Tanaka TU, Lechner J, Schiebel E. Four new subunits of the Dam1-Duo1 complex reveal novel functions in sister kinetochore biorientation. Embo J. 2002;21:181–193. doi: 10.1093/emboj/21.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janosi IM, Chretien D, Flyvbjerg H. Structural microtubule cap: Stability, catastrophe, rescue, and third state. Biophys J. 2002;83:1317–1330. doi: 10.1016/S0006-3495(02)73902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AP, Bouck DC, Molk JN, Bloom KS, Salmon ED. Molecular architecture of a kinetochore-microtubule attachment site. Nat Cell Biol. 2006;8:581–585. doi: 10.1038/ncb1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MH, Bachant JB, Castillo AR, Giddings TH, Jr, Winey M. Yeast Dam1p is required to maintain spindle integrity during mitosis and interacts with the Mps1p kinase. Mol Biol Cell. 1999;10:2377–2391. doi: 10.1091/mbc.10.7.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjelgaard M. Improved methods of building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anti-Cancer Agents. 2002;2:1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- Kang JS, Cheeseman IM, Kallstrom G, Velmurugan S, Barnes G, Chan CSM. Functional cooperation of Dam1, Ipl1, and the inner centromere protein (INCENP) related protein Sli15 during chromosome segregation. J Cell Biol. 2001;155:763–774. doi: 10.1083/jcb.200105029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW. When 2+2=5: The origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE, Mitchison TJ, Kirschner MW. Polewards chromosome movement driven by microtubule depolymerization in vitro. Nature. 1988;331:499–504. doi: 10.1038/331499a0. [DOI] [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, DeRosier D, Nogales E, Downing KH. Structure of the microtubule at 8Å resolution. Structure. 2002;10:1317–1328. doi: 10.1016/s0969-2126(02)00827-4. [DOI] [PubMed] [Google Scholar]
- Lobert S, Correia J. Subtilisin cleavage of tubulin heterodimers and polymers. Arch Bioch Biophy. 1992;296:152–160. doi: 10.1016/0003-9861(92)90557-d. [DOI] [PubMed] [Google Scholar]
- Lowe J, Li H, Downing KH, Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E, Milligan RA. Microtubules dynamics and microtubules caps: A time-resolved cryo-electron microscopy study. J Cell Biol. 1991;114:977–991. doi: 10.1083/jcb.114.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki R, Carlier MF, Pantaloni D, Timasheff SN. Cold depolymerization of micro-tubules to double rings: Geometric stabilization of assemblies. Biochemistry. 1989;28:9143–9152. doi: 10.1021/bi00449a028. [DOI] [PubMed] [Google Scholar]
- Miranda JJ, De Wulf P, Sorger PK, Harrison SC. The yeast DASH complex forms closed rings on microtubules. Nat Struct Mol Biol. 2005;12:138–143. doi: 10.1038/nsmb896. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Molodtsov MI, Ermakova EA, Shnol EE, Grishchuk EL, McIntosh JR, Ataullakhanov FI. A molecular-mechanical model of the microtubule. Biophys J. 2005;88:3167–3179. doi: 10.1529/biophysj.104.051789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Reichert T, Chrétien D, Severin F, Hyman AA. Structural changes at microtubule ends accompanying GTP hydrolysis: Information from a slowly hydrolyzable analogue of GTP, guanylyl (α,β)methylenediphosphonate. Proc Natl Acad Sci USA. 1998;95:3661–3666. doi: 10.1073/pnas.95.7.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E. Structural insights into microtubule function. Ann Rev Biochem. 2000;69:277–302. doi: 10.1146/annurev.biochem.69.1.277. [DOI] [PubMed] [Google Scholar]
- Nogales E, Downing KH, Amos LA, Löwe J. Tubulin and FtsZ form a distinct family of GTPases. Nat Struc Biol. 1998a;5:451–458. doi: 10.1038/nsb0698-451. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wang HW. Structural intermediates in microtubule assembly and disassembly: How and why? Curr Opin Cell Biol. 2006;18:179–184. doi: 10.1016/j.ceb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wang HW, Niederstrasser H. Tubulin rings: Which way do they curve? Curr Opin Struct Biol. 2003;13:256–261. doi: 10.1016/s0959-440x(03)00029-0. [DOI] [PubMed] [Google Scholar]
- Nogales E, Whittaker M, Milligan RA, Downing KH. High resolution structure of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the αβ tubulin dimer by electron crystallography. Nature. 1998b;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Ramey VH, Wang HW, Nogales E. Ab initio reconstruction of helical samples with heterogeneity, disorder and coexisting symmetries. J Struct Biol. 2009;167(2):97–105. doi: 10.1016/j.jsb.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- Rice LM, Montabana EA, Agard DA. The lattice as allosteric effector: Structural studies of alphabeta- and gamma-tubulin clarify the role of GTP in microtubule assembly. Proc Natl Acad Sci USA. 2008;105:5378–5383. doi: 10.1073/pnas.0801155105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon ED. Microtubules: A ring for the depolymerization motor. Curr Biol. 2005;15:R299–R302. doi: 10.1016/j.cub.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Schroeter JP, Bretaudiere JP. SUPRIM: Easily modified image processing software. J Struct Biol. 1996;116:131–137. doi: 10.1006/jsbi.1996.0021. [DOI] [PubMed] [Google Scholar]
- Shang C, Hazbun TR, Cheeseman IM, Aranda J, Fields S, Drubin DG, Barnes G. Kinetochore protein interactions and their regulation by the Aurora kinase Ipl1p. Mol Biol Cell. 2003;14:3342–3355. doi: 10.1091/mbc.E02-11-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa H, Milligan RA. Three-dimensional structure of ncd-decorated microtubules obtained by a back-projection method. J Mol Biol. 1996;260:743–755. doi: 10.1006/jmbi.1996.0434. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Unwin N. Contrast transfer for frozen-hydrated specimens: Determination from pairs of defocused images. Ultramicroscopy. 1988;25:279–291. doi: 10.1016/0304-3991(88)90003-4. [DOI] [PubMed] [Google Scholar]
- Tuszynski JA, Luchko T, Portet S, Dixon JM. Anisotropic elastic properties of microtubules. Eur Phys J E Soft Matter. 2005;17:29–35. doi: 10.1140/epje/i2004-10102-5. [DOI] [PubMed] [Google Scholar]
- Unger E, Böhm JK, Vater W. Structural diversity and dynamics of microtubules and polymorphic tubulin assemblies. Electron Microsc Rev. 1990;3:355–395. doi: 10.1016/0892-0354(90)90007-f. [DOI] [PubMed] [Google Scholar]
- Vanburen V, Cassimeris L, Odde DJ. A mechanochemical model of microtubule structure and self-assembly kinetics. Biophys J. 2005;89:2911–2926. doi: 10.1529/biophysj.105.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Nogales E. An iterative Fourier-Bessel algorithm for reconstruction of helical structures with severe Bessel overlap. J Struct Biol. 2005a;149:65–78. doi: 10.1016/j.jsb.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Wang HW, Nogales E. Nucleotide-dependent bending flexibility of tubulin regulates microtubule assembly. Nature. 2005b;435:911–915. doi: 10.1038/nature03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Ramey VH, Westermann S, Leschziner AE, Welburn JP, Nakajima Y, Drubin DG, Barnes G, Nogales E. Architecture of the Dam1 kinetochore ring complex and implications for microtubule-driven assembly and force-coupling mechanisms. Nat Struct Mol Biol. 2007;14:721–726. doi: 10.1038/nsmb1274. [DOI] [PubMed] [Google Scholar]
- Westermann S, Avila-Sakar A, Wang HW, Niederstrasser H, Wong J, Drubin DG, Nogales E, Barnes G. Formation of a dynamic kinetochore- microtubule interface through assembly of the Dam1 ring complex. Mol Cell. 2005;17:277–290. doi: 10.1016/j.molcel.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Westermann S, Drubin DG, Barnes G. Structures and functions of yeast kinetochore complexes. Annu Rev Biochem. 2007;76:563–591. doi: 10.1146/annurev.biochem.76.052705.160607. [DOI] [PubMed] [Google Scholar]
- Westermann S, Wang HW, Avila-Sakar A, Drubin DG, Nogales E, Barnes G. The Dam1 kinetochore ring complex moves processively on depolymerizing microtubule ends. Nature. 2006;440:565–569. doi: 10.1038/nature04409. [DOI] [PubMed] [Google Scholar]
- Wolff J. Promotion of microtubule assembly by oligocations: Cooperativity between charged groups. Biochemistry. 1998;37:10722–10729. doi: 10.1021/bi980400n. [DOI] [PubMed] [Google Scholar]
- Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Zhou ZH, Hardt S, Wang B, Sherman MB, Jakana J, Chiu W. CTF determination of images of ice-embedded single particles using a graphics interface. J Struct Biol. 1996;116:216–222. doi: 10.1006/jsbi.1996.0033. [DOI] [PubMed] [Google Scholar]
- Zhu J, Penczek PA, Schröder R, Frank J. Three-dimensional reconstruction with contrast transfer function correction from energy-filtered cryoelectron micrographs: Procedure and application to the 70S Escherichia coli ribosome. J Struct Biol. 1997;118:197–219. doi: 10.1006/jsbi.1997.3845. [DOI] [PubMed] [Google Scholar]