

Abstract

Harnessing the biotechnological potential of the large number of proteins available in sequence databases requires scalable methods for functional characterization. Here we propose a workflow to address this challenge by combining phylogenomic guided DNA synthesis with high-throughput mass spectrometry and apply it to the systematic characterization of GH1 β-glucosidases, a family of enzymes necessary for biomass hydrolysis, an important step in the conversion of lignocellulosic feedstocks to fuels and chemicals. We synthesized and expressed 175 GH1s, selected from over 2000 candidate sequences to cover maximum sequence diversity. These enzymes were functionally characterized over a range of temperatures and pHs using nanostructure-initiator mass spectrometry (NIMS), generating over 10,000 data points. When combined with HPLC-based sugar profiling, we observed GH1 enzymes active over a broad temperature range and toward many different β-linked disaccharides. For some GH1s we also observed activity toward laminarin, a more complex oligosaccharide present as a major component of macroalgae. An area of particular interest was the identification of GH1 enzymes compatible with the ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]), a next-generation biomass pretreatment technology. We thus searched for GH1 enzymes active at 70 °C and 20% (v/v) [C2mim][OAc] over the course of a 24-h saccharification reaction. Using our unbiased approach, we identified multiple enzymes of different phylogentic origin with such activities. Our approach of characterizing sequence diversity through targeted gene synthesis coupled to high-throughput screening technologies is a broadly applicable paradigm for a wide range of biological problems.
A grand challenge in genomics is to accurately assign function to the ever-growing numbers of genes found in sequence databases. Until recently much of the sequence data was generated from isolated organisms, but with the advent of metagenomics and next-generation sequencing technologies,1 the sequence space that can be surveyed has increased dramatically, thereby providing new opportunities for the discovery and characterization of novel biological functions including biocatalysts.
Presently, the economical conversion of cellulosic biomass to biofuels is limited by the lack of sufficiently active and inexpensive enzymes that can operate at conditions compatible with currently used biomass pretreatment methods. Necessary enzymatic activities for biomass deconstruction include endoglucanases (EC 3.2.1.4), cellobiohydrolases (EC 3.2.1.91), and β-glucosidases (EC 3.2.1.21).2
β-Glucosidases are ubiquitous enzymes that catalyze the hydrolysis of β1–4 bonds in short-chain oligosaccharides into glucose.3 These enzymes were selected for study because adequate β-glucosidase levels are critical for efficient biological deconstruction of lignocellulosic materials, as accumulation of cellobiose in reactors has been shown to inhibit the activity of endoglucanases and cellobiohydrolases.4,5 β-Glucosidase activity has been demonstrated across multiple enzyme families within the glycoside hydrolase (GH) superfamily, including GH1, GH3, GH5, GH9, and GH30 (see CAZy database www.cazy.org) but has been most extensively characterized within the GH1 group of glycoside hydrolases.6 This family of enzymes have a well conserved structure defined by a (β/α)8-barrel fold and conserved active-site residues comprising a proton donor glutamate residue located in the fourth β-sheet and a nucleophile glutamate residue in the seventh β-sheet.6 Whereas the structures and the catalytic mechanism of GH1s have been extensively investigated,7−11 there has been no systematic characterization of the functional diversity within this enzyme group in terms of substrate specificity and activity at different operating conditions.
Relevant GH1 properties that define their suitability for industrial purposes vary according to the biomass pretreatment method used. As such, industrially useful β-glucosidases need to be stable at pretreatment-specific conditions, have high activity throughout the course of saccharification, and ideally hydrolyze multiple oligosaccharides present in the reaction mix. Commonly used pretreatment methods include dilute acid, concentrated acid, organic solvent, and ammonia fiber explosion (AFEX), all of which have advantages and disadvantages related to their costs, energy inputs, sugar yields, feedstock flexibility, generation of inhibitory side products, and safety.12,13 Recently, ionic liquids (ILs) have emerged as a promising new approach for the thermo-chemical pretreatment of biomass, based on a class of RT molten salts capable of dissolving and fractionating lignocellulose at relatively low temperatures (<100 °C). One important challenge for the large-scale implementation of IL technology for biomass pretreatment is the paucity of known enzymes that can tolerate even low concentrations of these solvents.
Although substantial effort has been devoted to understanding GH activities, structures, and mechanism of action,6 past efforts have been focused mostly on studying one or few enzymes at a time. As such, functional characterization is sparse relative to the vast number of sequences available and not uniform in terms of assays, conditions, and substrates used.
Here we describe a phylogenomic approach to systematically characterize the GH1 sequence space through DNA synthesis coupled to high-throughput functional screening, to broadly define their activity, thermal stability, and substrate specificity. To this end we identified ∼2000 putative GH1s from sequence databases and selected 175 representatives that capture maximum phylogenetic space. The reduced but highly diverse set of GH1 genes was synthesized and heterologously expressed in E. coli. High-throughput functional characterization of proteins was performed using nanostructure-initiator mass spectrometry (NIMS) coupled to acoustic printing.14,15 This combination of technologies facilitated the rapid screening of enzymatic activity and substrate specificity, over a range of temperature and pH conditions. The most promising candidates from the NIMS screening were further characterized by tracking sugar profiles using HPLC to determine specific activities against a number of native substrates as well as performance at 70 °C and 20% (v/v) of the ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]), conditions relevant for the deconstruction of IL-pretreated biomass. This study demonstrates the power of phylogenomic guided DNA synthesis coupled to high-throughput screening technologies for the rapid identification of enzymes or other functional activities with properties relevant to a wide range of industrial applications.
Results and Discussion
Phylogenetic Sampling
As a first step to functionally characterize the GH1 sequence space, we retrieved all full-length GH1 enzymes present in the CAZy database. This resulted in the identification of 2319 putative GH1 genes, defined by the presence of the pfam domain PF00232.
To reduce redundancy in the set, we applied a 95% identity threshold, so that for any pair of sequences with >95% identity only the longest sequence was retained, reducing our data set to 928 candidates. In addition, we mined a recently sequenced and assembled metagenome data set16 for potential GH1 enzymes and identified 39 additional candidates. In total, our starting data set comprised 967 putative GH1 encoding genes.
Our strategy was to sample and characterize a set of representatives that would capture a substantial fraction of the sequence diversity in the GH1 phylogenetic space. To this end, we aligned all 967 genes and built a phylogenetic tree (Figure 1). The alignment was visually inspected revealing a high degree of conservation within this family, as we did not observe sequences with additional domains or large unexpected tracts of sequence that could result from unspliced introns (in the case of eukaryotic genes). With few exceptions, sequences clustered well according to kingdom and phyla. The predicted metagenomic GH1 genes from cow rumen data16 contributed to expanding the GH1 sequence space, adding several new clades to the tree. For example, genes CR34 and CR35 formed a new sister clade to a group of mostly actinobacterial GH1s (Figure 1: orange rectangles). This illustrates the value of incorporating novel metagenome sequences into systematic bioprospecting efforts, and as more metagenomics data is generated and assembled, this approach is likely to become a major driver for expanding protein sequence space available for enzyme characterization.
Figure 1.
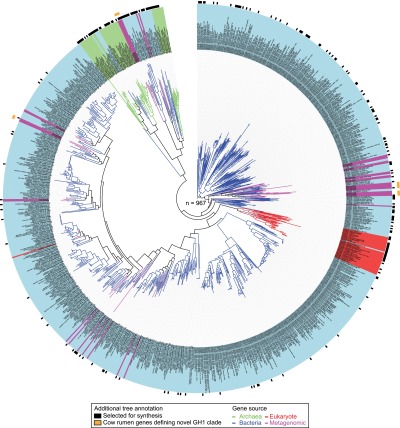
Phylogenetic tree of GH1 enzymes (n = 967) retrieved from sequence databases and metagenomic data. Branch and background color of the leaf labels indicate the phylogenetic origin of each GH1 sequence representative by kingdom (blue = bacteria, light green = archaea, red = eukaryota, purple = metagenome). Black rectangles in first outer circle show the GH1 representatives selected for synthesis on the basis of our criteria for maximizing phylogenetic space covered. Orange rectangles on second outer circle show examples of metagenome-derived representatives that define new clades.
To select a representative set of genes for functional studies, we sampled from our compiled phylogenetic tree using an algorithm that maximizes the phylogenetic distance covered for any given number of representatives.17 Initially we selected 175 candidates solely on the basis of maximizing phylogenetic distance and further curated the list to include, where possible, matched pairs of proteins from thermophilic and mesophilic organisms, as well as genes encoding proteins with known crystal structures, as this could be informative when looking for sequence to function correlations (Supplementary Table 1). Sampling was not uniformly spread across the tree, as for example a large area comprising bacterial mesophiles was undersampled due to their lower sequence diversity relative to Archaea or deep branching bacteria (including bacterial extremophiles, marine bacteria, and plant-associated bacteria) where most of the diversity resides (Figure 1). The final set for synthesis included 130 bacterial, 19 archaeal, 16 eukaryotic, and 10 metagenome-derived genes (Supplementary Table 1).
High-Throughput GH1 Activity Screens Using NIMS
Candidate GH1s were synthesized, cloned, and sequence verified in-house. All constructs were heterologously overexpressed in E. coli, and soluble proteins were purified and analyzed by SDS-PAGE. For 105 candidates a distinct band at around the expected 50 kDa was observed (Supplementary Figure 1), with eukaryotic genes showing markedly lower expression success rates (only 3/16 expressed in soluble form) likely due to the lack of adequate post-translational modification systems or to the formation of inclusion bodies in the bacterial host.
The set of 105 candidate GH1 enzymes for which soluble expression was detected was screened for activity using nanostructure-initiator mass spectrometry (NIMS),15 a scalable and versatile platform that allows screening of enzyme activities on many different substrates and reaction conditions in much higher throughput than more traditional carbohydrate analytical techniques such as HPLC–UV, HPLC–MS, and GC–MS. We tested GH1 activity against four disaccharide substrates: cellobiose, lactose, xylobiose, and maltose. A mass tagging strategy18 was used to enable all four substrates to be assayed in a single multiplex reaction, by attaching fluorous tags of discriminating mass to the reducing ends of each disaccharide (Supplementary Figure 2). Cellobiose and xylobiose were chosen because of their relevance as high abundance oligosaccharides present in lignocellulosic biomass. Lactose and maltose were chosen to determine enzyme specificity toward different structural (gluco-/galacto-) and variously bond-linked (α/β) sugars.
To assess temperature stability in our set of candidate GH1s, enzymes were preincubated at 40, 60, 80, and 90 °C and residual activity at each temperature was measured upon substrate addition. These enzyme–substrate reaction solutions were transferred onto a nanostructured solid surface, referred to hereafter as the “NIMS chip”, using acoustic deposition followed by imaging using a time-of-flight mass spectrometer as described previously14 (Figure 2a). In-house analysis software was used to determine mass spectrum and the conversion-rate for each substrate for individual “spots” on the NIMS chip (Figure 2b). All assays were performed in triplicate for a total of 10,080 experimental conditions. The average coefficient of variation for the entire data set was around 14%.
Figure 2.

Overview of acoustic deposition process and NIMS data. (a) Samples are acoustically transferred from the assay plate to the NIMS chip. Individual reactions on the NIMS chip are ionized by a laser and detected by a time-of-flight mass spectrometer. (b) A representative mass spectrum from a single sample derived from the mean of 12 laser shots. The text above the peaks denotes the multiple substrates and products that can be resolved on the basis of mass tags that encode the identity of the substrate (CB = cellobiose, Lac = lactose, XB = xylobiose, Mal = maltose). (c) Enzyme assay results (n = 10,080) for all synthesized GH1 representatives (n = 175) displayed on a phylogenetic tree. Branches are colored according to kingdom. Four concentric outer circles indicate soluble expression and maximum temperature at which enzyme activity was observed for the cellobiose, lactose, and xylobiose NIMS substrates, respectively.
We found that under at least one condition 59/105 enzymes were active toward cellobiose and 50/105 were active against lactose with an almost perfect correlation observed between these two activities (Figure 2c). We also observed that 7/105 enzymes had activity toward xylobiose, a β1–4 linked five-carbon substrate not commonly associated with GH1 hydrolysis. No activity toward maltose (a glucose dimer with an α1–4 bond) was observed, showing, as expected, that GH1 activity is specific to the β bond configuration. In addition, for about 1/3 of the expressed GH1s no activity was detected toward any of the tested substrates.
Temperature, but not pH, was observed to have a major impact on GH1 enzyme activities. We found that 21/105 enzymes were stable at either 80 or 90 °C, 41/105 enzymes were active up to 60 °C, and 59/105 enzymes were active at 40 °C (Figure 2c). As anticipated, most enzymes stable at the highest temperatures originated from thermophilic bacteria and archaea. Thermostable enzymes were found scattered throughout the phylogenetic tree. For example, enzymes ABW01253 from the archaea Caldivirga maquilingensis and ACM22958 from bacterium Thermotoga neapolitana are both active at 90 °C, and yet these proteins exhibit only 27% sequence identity. The presence of multiple clusters of thermostable enzyme suggests that this property arose multiple times in the evolution of the GH1 protein family. In contrast to the temperature-dependent spread in enzyme activities, we did not see significant changes in the number of enzymes active between pH 5 and 8, although conversion rates were highly variable between these conditions (Supplementary Table 2).
Because most previous efforts to characterize GH1 enzymes have relied on the use of the chromogenic substrate p-nitrophenyl β-d-glucoside (pNPβG) as a model compound, we assayed the activity of our GH1 set using this substrate at 40 °C (pH 7) to compare with the NIMS results. We observed a concordance rate of 88% between the two methods (Supplementary Table 3).
HPLC-Based Sugar Profiling
To validate the NIMS data and to further characterize GH1 functional diversity in terms of substrate specificity, we selected 28 diverse enzymes for detailed HPLC profiling based on the NIMS results. This set included thermophilic (n = 10), mesophilic (n = 9), and expressed enzymes for which no activity against NIMS substrates was detected (n = 9).
We measured specific activity toward a set of 8 natural substrates (Table 1), mostly disaccharides produced during the biomass deconstruction of various feedstocks. In addition, we included laminarin, a glucose oligomer (7–10mer) with β1–3 and β1–6 bonds, which is the main storage hydrocarbon found in brown algae, a potential source of biomass for biofuel production.19
Table 1. HPLC-Based Sugar Profiling of Selected GH1s toward a Panel of Relevant Carbohydrate Substratesa.
| specific
activity (U mg–1) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| enzyme | reaction temp (°C) | sophorose (β1,2)-Glc-Glc | laminaribiose (β1,3)-Glc-Glc | cellobiose (β1,4)-Glc-Glc | gentiobiose (β,6)-Glc-Glc | lactose (β1,4)-Gal-Glc | glucomannan (β1,4)-Glc-Man | galactobiose (β1,4)-Gal-Gal | mannobiose (β1,4)-Man-Man | laminarin (β1,3) and (β1,6) |
| Mesophillic | ||||||||||
| CR_14_Cow_Rumen | 30 | 35.1 | 54.6 | 8.6 | 0.2 | 0.4 | 1.8 | 0.1 | nd | 0.1 |
| CAA82733.1_Streptomyces-sp. | 40 | 8.3 | 8.5 | 6.9 | 1.0 | 4.9 | 2.6 | 0.1 | nd | 0.1 |
| CR_19_Cow_Rumen | 40 | 25.1 | 20.0 | 3.1 | 0.7 | nd | 2.5 | nd | nd | 0.4 |
| AAF37730.1_Thermobifida-fusca | 50 | 21.4 | 42.6 | 34.8 | 0.6 | 7.9 | 22.2 | 0.1 | 0.4 | 0.1 |
| AAZ81839.1_Alicyclobacillus-acidocaldarius | 55 | 86.6 | 95.1 | 70.8 | 6.9 | 16.9 | 34.0 | 0.9 | nd | 1.1 |
| ACJ34717.1_Anoxybacillus-flavithermus-WK1 | 60 | 52.0 | 71.6 | 4.2 | 0.6 | 0.6 | 3.3 | 0.1 | nd | 0.7 |
| ACL70277.1_Halothermothrix-orenii-H-168 | 60 | 74.9 | 62.1 | 29.4 | 7.1 | 7.9 | 18.2 | 1.9 | nd | 1.0 |
| ADD27066.1_Meiothermus-ruber-DSM-1279 | 60 | 5.1 | 4.6 | 2.8 | 1.8 | 0.4 | 0.5 | nd | nd | 3.7 |
| CAA42814.1_Clostridium-thermocellum-ATCC-27405 | 65 | 78.8 | 127.7 | 3.2 | 1.0 | 7.3 | 5.2 | 0.9 | 0.2 | 0.4 |
| Thermophillic | ||||||||||
| ABW01253.1_Caldivirga-maquilingensis-IC-167 | 70 | 53.9 | 53.2 | 28.0 | 13.3 | 23.9 | 17.6 | 2.0 | nd | 0.3 |
| ACJ75238.1_Thermosipho-africanus-TCF52B | 70 | 95.6 | 94.0 | 59.1 | 2.7 | 7.3 | 37.2 | 0.4 | 0.3 | 0.2 |
| ACJ76349.1_Thermosipho-africanus-TCF52B | 70 | 98.5 | 100.6 | 38.2 | 13.7 | 58.7 | 27.7 | 2.4 | nd | 0.8 |
| ACZ42845.1_Thermobaculum-terrenum-ATCC-BAA-798 | 75 | 48.8 | 75.9 | 49.8 | 15.9 | 61.7 | 46.1 | 10.2 | nd | 9.2 |
| AAA72843.1_Sulfolobus-solfataricus | 80 | 19.7 | 19.2 | 16.9 | 30.9 | 16.6 | 11.9 | 5.1 | nd | 8.5 |
| AAY81155.1_Sulfolobus-acidocaldarius-DSM-639 | 80 | 38.1 | 48.4 | 8.4 | 3.6 | 1.9 | 4.5 | 1.9 | nd | 0.1 |
| AAF36392.1_Thermus-nonproteolyticus | 85 | 21.8 | 32.6 | 25.9 | 14.4 | 21.9 | 15.3 | 1.5 | 0.3 | 10.7 |
| ABS61401.1_Fervidobacterium-nodosum-Rt17-B1 | 85 | 43.6 | 44.3 | 22.2 | 1.3 | 9.2 | 14.9 | 0.1 | 1.9 | 0.1 |
| AAL81332.1_Pyrococcus-furiosus-DSM-3638 | 95 | 75.9 | 45.3 | 28.6 | 1.6 | 9.5 | 20.9 | 1.1 | 15.9 | 0.2 |
| ACM22958.1_Thermotoga-neapolitana-DSM-4359 | 95 | 147.9 | 163.8 | 26.8 | 4.9 | 19.1 | 23.2 | 5.6 | nd | 5.1 |
A unit (U) is defined as 1 μmol of total sugar monomers produced in 1 min. Values represent the average of three measurements with a coefficient of variation less than 10%. nd = not detectable. β1,2 refers to a β1–2 glycosidic bond. Glc = glucose, Gal = galactose, Man = mannose.
We first looked at concordance between the NIMS and HPLC results. All 19 enzymes that showed activity toward NIMS substrates were found to be active against the natural substrates at the conditions identified from the NIMS screen, confirming the biochemical relevance of the NIMS data accrued (Table 1). However, enzymes identified to have xylobiase activity showed only minimal hydrolysis against natural xylobiose after extended incubation times (specific activity less than 0.03 U mg–1). This illustrates that the high sensitivity of the NIMS technology allowed detection of GH1 enzymes with trace xylobiase activity, but the specific activity of this reaction is too low to be of relevance in an industrial setting. For the 9 enzymes that were not active against NIMS substrates, no detectable activity against any of the natural substrates was observed, again confirming the NIMS results.
HPLC data on the natural substrate panel showed that apart from the expected activity toward cellobiose (glucose dimer with β1–4 bond), widespread activity for substrates containing β1–2 and β1–3 linkages was also observed. Surprisingly, activities against sophorose (β1–2) and laminaribiose (β1–3) were consistently higher than those against cellobiose (β1–4). This finding likely reflects the large number of natural products conjugated to glucose by beta linkages that are found in natural environments and that are substrates to these enzymes in addition to their role in biomass breakdown.6 Previous studies using automated computational docking20 had predicted potentially higher affinities against disaccharides with alternative linkages, but this had not been experimentally validated across the GH1 family.
Enzymes with high activity toward gentiobiose (β1–6) were less common and largely confined to thermophiles from different phylogenetic origin (Table 1). We also confirmed widespread activity against lactose (as expected from the NIMS data) and glucomannan, although the specific activity values were generally lower than those observed toward the glucose–glucose dimers. We also noted rare instances of enzymes active toward galactobiose and mannobiose, with no clear correlation to their position on the phylogenetic tree (Table 1).
An interesting and unexpected result was the observation that some GH1 enzymes exhibited activity toward the more complex oligosaccharide laminarin, the principal storage carbohydrate in a number of brown macroalgae (Table 1). This activity had not been previously reported and is of relevance given that brown algae have been proposed as an economically viable source of biomass for biofuel production.19
Overall, we noted that for the subset of enzymes that were characterized by HPLC, thermophilic enzymes had significantly broader substrate specificity compared to their mesophilic counterparts (pANOVA = 2 × 10–5).
GH1 Specificity for Phosphorylated Substrates
An intriguing observation from the NIMS (Figure 2c) and HPLC data was the lack of activity toward any of the tested substrates for a number of well-expressed bacterial GH1s that co-localize on the GH1 phylogenetic tree. To ascertain whether these enzymes have different substrate specificities, we searched the CAZy database for previously reported activities for the synthesized GH1s that cluster to this area of the tree. In two cases (BAD76141 and BAD76622 both from Geobacillus kaustophilus), 6-phospho-β-glucosidase (EC 3.2.1.86) activity had been reported, suggesting that this subgroup of GH1s might specifically catalyze the hydrolysis of phosphorylated β-glucosides. Inspection of the multiple sequence alignment (Figure 3a) revealed a glutamic acid (E) to serine (S) substitution at position 428 (L. lactis numbering), a lysine (K) or asparagine (N) at position 435, and a tyrosine (Y) at position 437; this SKY/SNY motif was found in all GH1 proteins clustered in this region of the tree but not elsewhere. Recently published crystal structures of GH1 6-phospho-β-glucosidases from lactic acid bacteria,21Streptococcus pneumoniae,22 and S. pyogenes(23) show that the three SKY residues coordinate the phosphoryl moiety of the substrate via hydrogen bonds. To validate the correlation between GH1 enzymes containing the SKY/SNY motif and activity toward phosphorylated substrates, all (n = 26) of the expressed GH1s from this area of the tree were tested for their capacity to hydrolyze the chromogenic analogue, p-nitrophenol-β-d-glucopyranoside 6-phosphate (pNPβG6P). Twenty-three out of the 26 enzymes tested (previously shown to be inactive against the NIMS and native substrates) readily hydrolyzed pNPβG6P to form G6P and the yellow p-nitrophenolate anion (Figure 3b), but 19 enzymes from other areas of the tree that do not contain the SKY or SNY motif did not show activity against this substrate.
Figure 3.

Activity of subgroup of GH1s toward phosphorylated substrates. (a) Multiple sequence alignment highlighting the SKY/SNY motif found in a subgroup of GH1s that showed no activity toward the NIMS substrates, along with the sequence from a 6-phospho-β-glucosidase GH1 from L. lactis with a known crystal structure (top). Four representative GH1s from this study that showed activity toward the NIMS and real substrates are shown for comparison (bottom). (b) A phylogenetic tree highlighting the subgroup of GH1s with the SKY/SNY motif; the three outer concentric rings show (i) expression, (ii) SKY/SNY motif identity, and (iii) their enzyme activity toward the phosphorylated chromogenic substrate, pNPβG6P.
Identification of Ionic Liquid ([C2mim][OAc])-Tolerant GH1s
A specific goal of our study was the identification of enzymes that could efficiently function under conditions compatible with the use of ILs as the biomass pretreatment method. In particular, we were searching for GH1 enzymes with sustained activity at 70 °C and in the presence of 20% (v/v) 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]) for use in combination with similarly tolerant endoglucanases and exoglucanases. Commercially available enzyme cocktails are inactive under these conditions.
Our initial candidate set for IL-tolerance screening included all 21 enzymes that showed activity at 80 or 90 °C (Figure 2c). To simulate the time scale of a typical saccharification reaction, enzymes were first preincubated at 70 °C with 20% (v/v) [C2mim][OAc] for 24 h. Cellobiose was then added, and the residual specific activity of the β-glucosidase was determined. A reaction under the same conditions but without [C2mim][OAc] was used as a control. Following the prolonged heat treatment, 16/21 enzymes showed some level of activity in the absence of [C2mim][OAc], but only 5 of those 16 enzymes retained activity in the presence of [C2mim][OAc], with residual activity for those enzymes ranging from 74% to 3% relative to the control (Figure 4a).
Figure 4.

Activity of thermophilic GH1s in [C2mim][OAc]. (a) Specific activities of thermophilic GH1s toward cellobiose after a 24-h preincubation at 70 °C in the presence (orange) or absence (black) of 20% (v/v) [C2mim][OAc]. Text labels refer to the accession numbers for the tested enzymes. (b) Glucose and cellobiose release from [C2mim][OAc]-pretreated switchgrass after 24 h saccharification at 70 °C and 20% (v/v) [C2mim][OAc] in the presence of accessory glycosyl hydrolases. Numbers to the right denote % conversion of biomass into glucose for each enzyme.
To test whether our results were relevant for the deconstruction of “real-world” biomass, we then performed a saccharification with IL-pretreated switchgrass in which the activity of each thermophilic GH1 was assayed in a cocktail containing an endoglucanase and a cellobiohydrolase. We measured glucose production from [C2mim][OAc]-pretreated switchgrass (diluted to 20% (v/v) IL) in the presence of CelB and Cel5A, two enzymes that have been previously shown to function (albeit not optimally) under these conditions.24 Whereas very little glucose was observed in mixtures containing only CelB and Cel5A, addition of any one of the five thermostable and IL-tolerant enzymes identified in the previous experiment resulted in complete or near-complete conversion of cellobiose into glucose, with up to 66% of theoretically maximum glucose yield observed (Figure 4b). We thus identified 5 GH1 enzymes that function at our target process conditions that are compelling candidates for use in larger scale saccharification reactions. These enzymes are spread throughout the GH1 phylogenetic tree but originate from archaeal and bacterial lineages identified mostly from volcanic saline environments at various pHs (Supplementary Figure 3).
In summary, we have described a widely applicable methodology for sampling sequence diversity through phylogenomically guided DNA synthesis coupled to high-throughput NIMS screening to rapidly determine proteins with desirable biochemical properties. This approach allowed us to successfully identify GH1 enzymes with sought-after specifications, notably activity at high temperatures in the presence of IL, broad substrate specificity, and activity against more complex substrates such as laminarin, that are potentially useful in a biotechnological setting. Our approach offers a powerful paradigm for rapidly characterizing the expanding protein sequence space in a systematic and unbiased manner that can be applied to a broad range of biological functions.
Methods
Phylogenetic Sampling
Putative GH1 enzymes were retrieved from CAZy (http://www.cazy.org/) characterized by pfam domain PF00232, and amino acid sequences were retrieved from NCBI using the Batch Entrez tool. In addition, we searched an assembled cow rumen metagenome data set for additional potential GH1s as described in ref (16). In total, we identified 2358 GH1 representatives. These sequences were filtered for uniqueness using the UCLUST algorithm.25 We aligned the filtered sequences using Muscle26 and constructed a phylogentic tree using FastTree27 using default parameters. The resulting tree was generated using the ITOL software.28 To extract a highly informative set of representatives that cover maximal phylogenetic distance, we used the MaxPD algorithm as described in ref (17). This yielded our set of 175 GH1 representatives for synthesis.
Gene Synthesis and Cloning
Amino acid sequences were codon optimized for expression in E. coli using an empirically derived codon usage table.29 Optimized DNA, gene partitioning, and construction oligos were generated by GeneDesign.30 Oligonucleotides were pooled for synthesis using a Hamilton STAR liquid handler (Hamilton Robotics). Assembly proceeded in a two-step PCA reaction in 25 μL final volume using KOD polymerase (EMD Millipore). Assembled products were cloned into pET45b(+) vector containing an N-terminal His-Tag (EMD Millipore) by In-Fusion cloning (Clontech) and transformed into BL21 cells in 96-well plates. Plating and picking was performed using a QPix 400 system (Molecular Devices). Eight colonies per construct were sequenced verified using PACBIO RSII system (Pacific Biosciences).
Protein Expression and Purification
GH1 proteins were expressed in 5 mL Overnight Express Instant TB medium (EMD Millipore) containing 50 μg mL–1 carbenicillin for 16 h at 37 °C with 300 rpm shaking. Cells were pelleted and lysed at RT with 1 mL of BugBuster Master Mix (EMD Millipore) containing protease inhibitor for 20 min. Soluble proteins were collected after centrifugation of lysed cells at ∼20,000g for 15 min at 4 °C. His-tagged GH1 proteins were purified from the soluble fraction using Ni-NTA spin columns (Qiagen) according to the manufacturer’s protocol. The eluted pure proteins were dialyzed in HN buffer (50 mM HEPES, 100 mM NaCl, pH 7.4) using 96 D-tube dialyzers (EMD Millipore). Glycerol (10% v/v) was added to the dialyzed pure protein and stored at −80 °C. Purified protein (7.5 μL) was electrophoresed on 10–20% polyacrylamide gel (Bio Rad) under denaturing condition. The gel was stained with Bio-Safe Coomassie Stain (Bio Rad) and destained with water. The protein concentration was determined by densitometry using a number of protein standards of known concentration using Image Lab software (Bio Rad).
NIMS
The NIMS chips were produced as described elsewhere.31
The NIMS assay substrates were a mixture of four separate probes (cellobiose, lactose, xylobiose, and maltose), synthesized as described in ref (18) (Supplementary Figure 2) . Each probe was at a concentration of 40 μM, dissolved in 100 mM acetate (pH 5) or 100 mM HEPES (pH 8) buffer.
Purified enzymes were diluted to a concentration of 40 ng μL–1 in 5 mM MES buffer pH 6.5; then 5 μL samples of the diluted enzymes were transferred to 384-well PCR plates (Bio Rad). The PCR plate was sealed with Microseal B film (Bio Rad) and heated to the reaction temperature (a single plate was heated to one of the following temperatures: 40, 60, 80, or 90 °C) for 10 min and then cooled to 4 °C. Immediately after reaching 4 °C, 5 μL of NIMS substrate was added to each well. Final reaction concentrations were 20 ng μL–1 enzyme, 50 mM buffer, and 20 μM NIMS substrate. The plate was resealed and heated back to the original incubation temperature for an additional 10 min and then cooled back to 4 °C. After cooling, 90 μL of an ice-cold 5:4 methanol/water mixture was added to each well to quench the enzymatic reaction. Samples were centrifuged at 3000 rpm for 1 min to remove solid debris, and then 10 μL of the supernatant was transferred to 384-well acoustic plates (Greiner Bio-one, Germany) for printing.
The assay mixture was acoustically printed onto a NIMS chip using EDC ATS-100 acoustic transfer system (EDC Biosystems) with a sample deposition volume of 1 nL. Samples were printed with the microarray spot pitch (center-to-center distance) set at 450 μm. This format allowed ∼2 samples per mm2.
NIMS was performed using a 5800 MALDI TOF/TOF (AB/Sciex) mass spectrum with laser intensity of 2500 over a mass range of 700–1500 Da. The data collection was controlled using MALDI MSI 4800 imaging tool, each position on a NIMS chip accumulated 12 laser shots, and scanning step size was set at 75 μm step both vertically and horizontally. The total array acquisition time was 1.3 h per 384 samples. The enzymatic activity was determined by calculating the fractional conversion for each reaction. First, the signal intensities for probe and product were determined from the acquired spectrum; next the fractional conversion (i.e., activity) was calculated using the following equation: product/(probe + product). Each pixel of the image was analyzed in this manner using an in-house analysis algorithm written in Matlab and was plotted as a false color image. Negative control of nonenzymatic hydrolysis was subtracted to correct the calculated activities; this approach minimizes the effects of intensity across the surface. All experiments were done in triplicate.
HPLC Sugar-Profiling
To determine specific activities toward sophorose, cellobiose, lactose, gentibiose, galactobiose, xylobiose (Sigma), glucomannan, laminaribiose, laminarin, and mannobiose (Megazyme), we first determined the maximum temperature at which the enzymes retained >90% activity for at least 1 h using standard colorimetric assays. For all colorimetric assays, purified enzymes were diluted in 5 mM MES buffer pH 6.5 to a concentration of 40 ng μL–1. We heated the enzymes at a range of temperatures from 40 to 90 °C for 60 min and cooled them to 4 °C and added 45 μL of a 1 mM p-nitrophenol-β-d-glucoside (pNPβG, Sigma), 25 mM acetate buffer pH 5 solution. The plate was resealed, and the mixture was heated to 40 °C for 5 min, cooled to 4 °C, and quenched using 50 μL of 0.5 M NaOH. Enzyme activity was determined by comparing the absorbance at 400 nm to those of known standards of p-nitrophenol also treated with 0.5 M NaOH.
Based on the activity toward pNPβG, the purified enzymes were diluted to a concentration between 1–40 ng μL–1 in 5 mM MES buffer pH 6.5. We then transferred 10 μL of diluted enzymes to a 96-well plate and mixed them with 10 μL of each substrate at a concentration of 20 mM in 100 mM acetate buffer pH 5, except for laminarin, which was at a stock concentration of 6 mg mg–1 in the same buffer. The well plate was sealed and heated to the appropriate reaction temperature for 10 min and then cooled to 4 °C. We quenched the reactions by adding 20 μL of ice-cold 100 mM glycine buffer pH 1.3. Substrate conversion was determined by HPLC analysis using an Aminex-HPX-87H column (Bio Rad) and appropriate monomer standards. Specific activities were calculated at enzyme concentrations where <10% substrate conversion occurred.
Colorimetric Assays for Phospho-glycosidase Activity
The activity of 6-phospho-β-glucosidases was tested using the chromogenic sugar analogue p-nitrophenol-β-d-glucopyranoside 6-phosphate (pNPβG6P, provided by Dr. Jack Thompson). Purified enzymes were diluted to a concentration of 40 ng μL–1 in 5 mM MES buffer pH 6.5; then 5 μL of diluted enzyme was mixed with 45 μL of a solution of 1 mM pNPβG6P and 25 mM acetate buffer pH 5. The mixture was heated at 40 °C for 5 min and then cooled to 4 °C. The reaction was quenched by adding 50 μL of 0.5 M NaOH. Enzyme activity was determined by comparing the absorbance at 400 nm to those of known standards of p-nitrophenol also treated with 0.5 M NaOH.
IL ([C2mim][OAc])-Tolerance Testing
1-Ethyl-3-methylimidazolium acetate, abbreviated hereafter as [C2mim][OAc], was purchased from BASF (lot no. 08-0010, purity >95%, Basionics BC-01, BASF). Purified enzymes were diluted in 0.5 mM HEPES, 1.5 mM NaCl pH 7.4 to a concentration of 40 ng μL–1, and then 10 μL samples of diluted enzymes were transferred to a 96-well PCR plate and mixed with either 10 μL of 40% (v/v) [C2mim][OAc]/water or 10 μL of 100 mM MES buffer pH 7.2 (MES buffer pH changed to 6.65 at 70 °C, matching the pH of the IL/water mixture). The plate was sealed, heated for 24 h at 70 °C, and then cooled to 4 °C. Immediately after reaching 4 °C, 20 μL of either a 60 mM cellobiose solution in 20% (v/v) [C2mim][OAc]/water or a 60 mM cellobiose solution in 50 mM MES pH 7.2 was added to the appropriate enzyme mixture (i.e., cellobiose-IL to the enzyme-IL wells; cellobiose buffer to the enzyme-buffer wells). Final reaction conditions were 10 ng μL–1 enzyme, 30 mM cellobiose, and either 20% (v/v) [C2mim][OAc] or 50 mM MES pH 7.2 (pH 6.6 at 70 °C). This mixture was heated to 70 °C for 10 min and then cooled to 4 °C. Immediately after cooling, 10 μL of the reaction mixture was removed and quenched with 90 μL of an ice-cold 5:4 methanol/water mixture. Glucose production was determined by an Agilent 1100 series HPLC equipped with a Aminex HPX-87H ion exchange column (Bio Rad) and a refractive index detector, using 4 mM H2SO4 as the mobile phase at a flow rate of 0.6 mL min–1 and a column temperature of 60 °C. All experiments were done in three replicates.
IL-Pretreated Biomass Saccharifications
Switchgrass (Panicum virgatum) was provided by Dr. Daniel Putnam, University of California at Davis and was subsequently ground by a Wiley Mill through a 2 mm screen. The switchgrass contains 34.6% cellulose, 20.2% xylan, 19.0% lignin, and 26.2% of other compounds remaining unidentified, on dry basis. Dry switchgrass was mixed with [C2mim][OAc] at a 3% (w/w) biomass loading and pretreated at 160 °C for 3 h. After pretreatment, the slurry was cooled to 100 °C, and 100 μL of slurry (determined to contain 1.08 mg glucan) was aliquoted into conical 1.5 mL screw top tubes (VWR no. 211-0090) and stored at 4 °C.
The recombinant cellobiohydrolase (CBH) from Caldicellulosiruptor saccharolyticus was a truncated construct of CelB (Uniprot ID: A4XIF7) containing only the CBM3 and GH5 domains, residues 374–1039; it was prepared from E. coli as described in ref (32). The recombinant endoglucanse (Cel5A) from Thermotoga maritima (Uniprot ID: Q9X273) was prepared from E. coli as described in ref (33). Both enzymes were stored at −80 °C in 25 mM HEPES, 75 mM NaCl, 25% (w/v) glycerol pH 7.5.
Three-part enzyme mixtures were prepared for each GH1 by combining 10.8 μg CBH, 5.4 μg Cel5A, and 5.4 μg GH1 in a volume of 400 μL deionized water. Enzyme mixtures were then added to the screw top tubes containing 100 μL biomass slurry for a final volume of 500 μL, 20% (v/v) [C2mim][OAc] and a total enzyme loading of 20 mg g–1 glucan. Tubes were sealed and incubated at 70 °C while shaking at 900 rpm for 24 h, after which the filtered supernatant was analyzed for glucose and cellobiose via HPLC as described above.
Acknowledgments
We thank EDC biosystems for providing instrumentation and support regarding acoustic printing.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ These authors contributed equally to this work.
The work conducted by the U.S. Department of Energy Joint Genome Institute and Joint BioEnergy Institute is supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH1123. In addition, this work was supported by the Swiss National Science Foundation [PA0033-121414 to S.D.] and the Intramural Research Program of the National Institute of Dental and Craniofacial Research to J.T.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tringe S. G.; Rubin E. M. (2005) Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Genet. 6, 805–814. [DOI] [PubMed] [Google Scholar]
- Kubicek C. P.; Mikus M.; Schuster A.; Schmoll M.; Seiboth B. (2009) Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol. Biofuels 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B. (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280(Pt 2), 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andric P.; Meyer A. S.; Jensen P. A.; Dam-Johansen K. (2010) Reactor design for minimizing product inhibition during enzymatic lignocellulose hydrolysis: II. Quantification of inhibition and suitability of membrane reactors. Biotechnol. Adv. 28, 407–425. [DOI] [PubMed] [Google Scholar]
- Gruno M.; Valjamae P.; Pettersson G.; Johansson G. (2004) Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol. Bioeng. 86, 503–511. [DOI] [PubMed] [Google Scholar]
- Ketudat Cairns J. R.; Esen A. (2010) beta-Glucosidases. Cell. Mol. Life Sci. 67, 3389–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Aparicio J.; Hermoso J. A.; Martinez-Ripoll M.; Lequerica J. L.; Polaina J. (1998) Crystal structure of beta-glucosidase A from Bacillus polymyxa: insights into the catalytic activity in family 1 glycosyl hydrolases. J. Mol. Biol. 275, 491–502. [DOI] [PubMed] [Google Scholar]
- Hakulinen N.; Paavilainen S.; Korpela T.; Rouvinen J. (2000) The crystal structure of beta-glucosidase from Bacillus circulans sp. alkalophilus: ability to form long polymeric assemblies. J. Struct. Biol. 129, 69–79. [DOI] [PubMed] [Google Scholar]
- Jeng W. Y.; Wang N. C.; Lin M. H.; Lin C. T.; Liaw Y. C.; Chang W. J.; Liu C. I.; Liang P. H.; Wang A. H. (2011) Structural and functional analysis of three beta-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J. Struct. Biol. 173, 46–56. [DOI] [PubMed] [Google Scholar]
- Kempton J. B.; Withers S. G. (1992) Mechanism of Agrobacterium beta-glucosidase: kinetic studies. Biochemistry 31, 9961–9969. [DOI] [PubMed] [Google Scholar]
- Vocadlo D. J.; Davies G. J. (2008) Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 12, 539–555. [DOI] [PubMed] [Google Scholar]
- Agbor V. B.; Cicek N.; Sparling R.; Berlin A.; Levin D. B. (2011) Biomass pretreatment: fundamentals toward application. Biotechnol Adv. 29, 675–685. [DOI] [PubMed] [Google Scholar]
- Blanch H. W.; Simmons B. A.; Klein-Marcuschamer D. (2011) Biomass deconstruction to sugars. Biotechnol. J. 6, 1086–1102. [DOI] [PubMed] [Google Scholar]
- Greving M.; Cheng X.; Reindl W.; Bowen B.; Deng K.; Louie K.; Nyman M.; Cohen J.; Singh A.; Simmons B.; Adams P.; Siuzdak G.; Northen T. (2012) Acoustic deposition with NIMS as a high-throughput enzyme activity assay. Anal. Bioanal. Chem. 403, 707–711. [DOI] [PubMed] [Google Scholar]
- Northen T. R.; Lee J. C.; Hoang L.; Raymond J.; Hwang D. R.; Yannone S. M.; Wong C. H.; Siuzdak G. (2008) A nanostructure-initiator mass spectrometry-based enzyme activity assay. Proc. Natl. Acad. Sci. U.S.A. 105, 3678–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M.; Sczyrba A.; Egan R.; Kim T. W.; Chokhawala H.; Schroth G.; Luo S.; Clark D. S.; Chen F.; Zhang T.; Mackie R. I.; Pennacchio L. A.; Tringe S. G.; Visel A.; Woyke T.; Wang Z.; Rubin E. M. (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. [DOI] [PubMed] [Google Scholar]
- Wu D.; Hugenholtz P.; Mavromatis K.; Pukall R.; Dalin E.; Ivanova N. N.; Kunin V.; Goodwin L.; Wu M.; Tindall B. J.; Hooper S. D.; Pati A.; Lykidis A.; Spring S.; Anderson I. J.; D’Haeseleer P.; Zemla A.; Singer M.; Lapidus A.; Nolan M.; Copeland A.; Han C.; Chen F.; Cheng J. F.; Lucas S.; Kerfeld C.; Lang E.; Gronow S.; Chain P.; Bruce D.; Rubin E. M.; Kyrpides N. C.; Klenk H. P.; Eisen J. A. (2009) A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng K.; George K. W.; Reindl W.; Keasling J. D.; Adams P. D.; Lee T. S.; Singh A. K.; Northen T. R. (2012) Encoding substrates with mass tags to resolve stereospecific reactions using Nimzyme. Rapid Commun. Mass Spectrom. 26, 611–615. [DOI] [PubMed] [Google Scholar]
- Wargacki A. J.; Leonard E.; Win M. N.; Regitsky D. D.; Santos C. N.; Kim P. B.; Cooper S. R.; Raisner R. M.; Herman A.; Sivitz A. B.; Lakshmanaswamy A.; Kashiyama Y.; Baker D.; Yoshikuni Y. (2012) An engineered microbial platform for direct biofuel production from brown macroalgae. Science 335, 308–313. [DOI] [PubMed] [Google Scholar]
- Hill A. D.; Reilly P. J. (2008) Computational analysis of glycoside hydrolase family 1 specificities. Biopolymers 89, 1021–1031. [DOI] [PubMed] [Google Scholar]
- Michalska K.; Tan K.; Li H.; Hatzos-Skintges C.; Bearden J.; Babnigg G.; Joachimiak A. (2013) GH1-family 6-P-beta-glucosidases from human microbiome lactic acid bacteria. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W. L.; Jiang Y. L.; Pikis A.; Cheng W.; Bai X. H.; Ren Y. M.; Thompson J.; Zhou C. Z.; Chen Y. (2013) Structural insights into the substrate specificity of a 6-phospho-beta-glucosidase BglA-2 from Streptococcus pneumoniae TIGR4. J. Biol. Chem. 288, 14949–14958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepper J.; Dabin J.; Eklof J. M.; Thongpoo P.; Kongsaeree P.; Taylor E. J.; Turkenburg J. P.; Brumer H.; Davies G. J. (2013) Structure and activity of the Streptococcus pyogenes family GH1 6-phospho-beta-glucosidase SPy1599. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 16–23. [DOI] [PubMed] [Google Scholar]
- Park J. I.; Steen E. J.; Burd H.; Evans S. S.; Redding-Johnson A. M.; Batth T.; Benke P. I.; D’Haeseleer P.; Sun N.; Sale K. L.; Keasling J. D.; Lee T. S.; Petzold C. J.; Mukhopadhyay A.; Singer S. W.; Simmons B. A.; Gladden J. M. (2012) A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels. PLoS One 7, e37010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N.; Dehal P. S.; Arkin A. P. (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I.; Bork P. (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan J.; Saaem I.; Tang N.; Ma S.; Negre N.; Gong H.; White K. P.; Tian J. (2011) Parallel on-chip gene synthesis and application to optimization of protein expression. Nat. Biotechnol. 29, 449–452. [DOI] [PubMed] [Google Scholar]
- Richardson S. M.; Nunley P. W.; Yarrington R. M.; Boeke J. D.; Bader J. S. (2010) GeneDesign 3.0 is an updated synthetic biology toolkit. Nucleic Acids Res. 38, 2603–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reindl W.; Deng K.; Gladden J. M.; Cheng G.; Wong A.; Singer S. W.; Singh S.; Lee J. C.; Yao C. H.; Hazen T. C.; Singh A. K.; Simmons B. A.; Adams P. D.; Northen T. R. (2011) Colloid-based multiplexed screening for plant biomass-degrading glycoside hydrolase activities in microbial communities. Energy Environ. Sci. 4, 2884–2893. [Google Scholar]
- Shi J.; Gladden J. M.; Sathitsuksanoh N.; Kambam P.; Sandoval L.; Mitra D.; Zhang S.; George A.; Singer S. W.; Simmons B. A.; Singh S. (2013) One-pot ionic liquid pretreatment and saccharification of switchgrass. Green Chem. 15, 2579–2589. [Google Scholar]
- Pereira J. H.; Chen Z. W.; McAndrew R. P.; Sapra R.; Chhabra S. R.; Sale K. L.; Simmons B. A.; Adams P. D. (2010) Biochemical characterization and crystal structure of endoglucanase Cel5A from the hyperthermophilic Thermotoga maritima. J. Struct. Biol. 172, 372–379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.