

Abstract
Objective:
The present study was designed to elucidate the impact of oral administration of aluminium chloride for 28 days with respect to oxidative stress in the cerebral cortex of female rats. Further, to investigate the potentials of Coenzyme (Co) Q10 (4, 8, and 12 mg/kg, i.p.) in mitigating the detrimental changes.
Materials and Methods:
Biochemical estimations of cerebral lipid peroxidation (LPO), reduced glutathione (GSH), vitamin E and activities of superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) were carried out after 28 days of aluminium chloride (AlCl3) and Co Q10 exposures along with histopathological examination of cerebral cortex of the rats.
Results:
Subacute exposure to AlCl3(5 mg/kg) led to significant decrease in levels of GSH, vitamin E and activities of SOD, CAT, GPx, and an increase in LPO of cerebral cortex. These aberrations were restored by Co Q10 (12 mg/kg, i.p.). This protection offered was comparable to that of L-deprenyl (1 mg/kg, i.p.) which served as a reference standard. Histopathological evaluations confirmed that the normal cerebral morphology was maintained by Co Q10.
Conclusion:
Thus, AlCl3 exposure hampers the activities of various antioxidant enzymes and induces oxidative stress in cerebral cortex of female Wistar rats. Supplementation with intraperitoneal Co Q10 abrogated these deleterious effects of AlCl3.
Keywords: Aluminium chloride, coenzyme Q10, L-deprenyl, neurotoxicity, oxidative stress
INTRODUCTION
Oxidative stress ensues in brain due to imbalance between cellular pro-oxidants and anti-oxidants, and consequential unwarranted generation of reactive free radicals.[1] Oxidative stress and mitochondrial dysfunction is an important cause and consequence of neurological disorders.[2] Many chemicals used by humans are dispersed in the ecosystem. Environmental toxicants alter the normal functioning of the neurons and induce toxicity which leads to cell death.[3] Environmental toxicants impart toxicity by formation of free radicals and alteration of cellular components.[4] Many of them have easy access to the brain due to lipophilic nature that enables them to cross the blood brain barrier rapidly.[3] The metals like lead, mercury and methyl mercury are potential neurotoxicants which increases the formation of reactive radicals and accelerates free radical reactions eventually leading to oxidative stress.[5,6,7] Some produce disturbances in brain antioxidant enzyme capacity.[8]
Aluminium (Al) is the most abundant metal in the earth's crust and ubiquitously found in every food product. Al is found in corn, yellow cheese, salt, herbs, spices, tea, cosmetics, aluminium ware, and containers. Although drinking water standard for Al of the World Health Organization (1984) is 0.2 mg/l, Al content in water is extensively studied and only 1% of the total daily Al intake in human.[9] Slanina et al., has reported that Al absorption is enhanced up to four- to five-fold in combination with food or beverage.[10] Al is a very strong oxygen acceptor and favors binding to other oxygen donors such as citrate, phosphate, lactic acid, oxalic acid, citric acid and catecholamines.[11] The in vivo Al intoxication induces oxidative damage due to the formation of Reactive Oxygen Species (ROS), which further potentiates with continued Al administration. In addition, Al has been shown to facilitate the oxidant activity of ROS.[12] This increase in ROS in brain causes disruption of the neurons.[2] Nitric oxide (NO) plays an important role in the normal functioning of cell. However, in Al induced-toxicity, NO forms complex with superoxide anions to form peroxynitrite anions which is more potent than hydroxyl radical to induce oxidative stress.[13] Al causes hypo-metabolism of glucose in brain mainly due to the disruption of tricarboxylic acid (TCA) cycle. Further, decrease in glucose levels in brain induces the amyloidogenic amyloid precursor protein processing which increase the oxidative stress.[14] Al salt is relevant to the study of biologically occurring Al compounds that may be absorbed through the gastrointestinal (GI) tract.[15] Aluminum salts in biological tissue do not have any direct pro-oxidant properties; in actuality it potentiates the ROS formation via iron salts. Aluminum chloride (AlCl3) has the potential to get accumulated in specific brain regions, which was earlier correlated with the degenerative changes.[16] Some clinical reports demonstrated an increase in Al absorption during aging and certain pathologies, leading to Al accumulation in the body.[17,18] Since Al can get across the blood/brain barrier,[19,20] its accumulation represents an enhanced neurotoxicological risk.
Coenzyme Q10 (Co Q10), also called as ubiquinone, is a fat-soluble vitamin-like substance present in every cell of the body. Co Q10 serves up as a coenzyme for several of the key enzymatic steps in the production of energy within the cell. Co Q10 has a wide variety of functions and applications in the body. It is the coenzyme for at least three mitochondrial enzymes (complexes I, II, and III) as well as enzymes in other parts of the cell.[21] Mitochondrial enzymes of the oxidative phosphorylation pathway are essential for the production of adenosine triphosphate (ATP), upon which all cellular functions depend.[22,23] Co Q10 supplementation is stated to be beneficial in the treatment of several health problems, primarily cardiac conditions and diseases, breast cancer, diabetes mellitus, immune deficiency, muscular dystrophy, and periodontal disease.[24] Co Q10 levels has been found to be decreased in various diseases both in animals and humans.[25] Especially in diseases linked to oxidative stress like Alzheimer's disease, diabetes, prion disease, and carcinogenesis.[26] Further, there is strong evidences of the absorption and distribution of Co Q10 to neural tissues as well as preliminary evidence for its role as a neuroprotective in animal models.[27,28] So, in the light of the existing knowledge, the present study was designed to study the impact of Co Q10 on the oxidative stress induced by oral 28 days exposure of AlCl3 on the cerebral cortex of female Wistar rats. L- deprenyl was used as a reference standard as it possesses the property to increase survival of neurons.[29]
MATERIALS AND METHODS
Drugs and chemicals
All the chemicals required for biochemical assays were obtained from S.D. Fine Chemicals Ltd., Thomas Baker Ltd., and Hi-media Laboratories Ltd. Co Q10 was procured and authenticated from M/s. Sterling Biotech, Vadodara, Gujarat, India. Selegiline HCl (L-deprenyl) was obtained from Cipla Ltd., Mumbai, India. All chemicals used were of analytical grade.
Animals and treatments
Thirty-six female Wistar rats (160-220 g) were used in the study. Animals were housed in standard polypropylene cages with wire mesh top and maintained at 23 ± 2ºC and relative humidity 60 ± 5% with 12 h light-dark cycle. Animals were fed with commercially available standard rodent pellet diet (Amrut rat and mice feed manufactured by Navi Maharashtra Chakan Oil Mill Ltd. India). Water was provided to the animals ad libitum. For all animal experimentation protocols prior approval was obtained from ‘Institutional Animal Ethics Committee’ of Bombay College of Pharmacy, Mumbai, India and all studies were performed in accordance with ‘Committee for the Purpose of Control and Supervision on Experiments on Animals’ (CPCSEA) guidelines. Dose of AlCl3 exposure was decided based on a previous study by Jyoti et al., 2007; AlCl3 was dissolved in distilled water (50 mg/ml), while the dose range of Coenzyme Q10 doses was decided on the basis of the investigations by Binukumar et al.[30] Co Q10 was dissolved in 25 μl corn oil. Standard drug L-deprenyl was dissolved in 0.9% saline. AlCl3 was given orally and Co Q10 and L-deprenyl were administered intraperitoneally. Rats were randomized and equally segregated into six groups. The Group 1 consisted of vehicle (distilled water, p.o.) control animals, Group 2 was of animals exposed to AlCl3 (50 mg/kg, p.o.), Group 3 consisted of animals treated with Co Q10 (4 mg/kg, i.p.) daily followed by AlCl3 (50 mg/kg, p.o.) daily, Group 4 consisted of animals treated with Co Q10 (8 mg/kg, i.p.) daily, followed by AlCl3 (50 mg/kg, p.o.), Group 5 consisted of animals treated with Co Q10 (12 mg/kg, i.p.) daily, followed by AlCl3 (50 mg/kg, p.o.), and Group 6 was of L-deprenyl (1 mg/kg, i.p.) daily, followed by AlCl3 (50 mg/kg, p.o.). All the administrations were carried out for a period of 28 days. The last dose was administered 24 h before sacrifice.
Preparation of homogenate
After treatment period, experimental and control animals were sacrificed using CO2 chamber. Brains were immediately taken out and washed thoroughly with ice-cold saline to remove blood. The cerebral cortex was rapidly micro-dissected from intact brain carefully on ice plate. The left and right cerebral cortices were pooled to make one sample of the tissue. Cerebral cortex was homogenized in 50 mM phosphate buffer pH 7.4 with homogenizer fitted with teflon plunger. Ten percent tissue homogenate (w/v) was made. Supernatant was made by centrifuging homogenate at 10,000 rpm for 20 min. Homogenate was used for the assay of LPO, GSH, and Vitamin E, while supernatant was used for the assay of CAT, SOD, and GPx activities. For total protein estimation, Erba Mannheim diagnostics kit (Biuret method) was used.
Thiobarbituric acid reactive substance
The brain tissue homogenate (0.1 ml) was mixed with acetic acid (20%, 1.5 ml), sodium lauryl sulfate (0.2 ml) and thiobarbituric acid (1.5 ml). After incubating at 95ºC for 1 h, the mixture was cooled and 5 ml of n-butanol and pyridine (15:1) were added, shaken vigorously and centrifuged. The absorbance of the supernatant was read at 532 nm against the blank containing all reagents except the homogenate. The inhibition of lipid peroxidation was determined by the procedure previously described.[31] The values were expressed as nmol of malondialdehyde (MDA)/gm tissue.
Reduced glutathione
To the brain tissue homogenate (0.25 ml), 0.5 ml of 5% TCA in 1 mM EDTA was added. The sample was centrifuged at 2000 g for 10 min. The supernatant was mixed with 2.5 ml of 0.1 M phosphate buffer (pH 8.0) and 100 μl of (0.001%) 5, 5’-dithiobis-2-nitrobenzoic acid (DTNB). Absorbance was read at 412 nm. Values were calculated from a standard graph of GSH treated with same reagents.[32] The amount of reduced glutathione was expressed as μg of GSH/mg protein.
Assay of Vitamin E
Reaction mixture containing 1.5 ml of brain tissue homogenate, 1 ml ethanol and 0.5 ml of 25% ascorbic acid was pre-incubated for 5 min at 70°C, followed by addition of 1 ml 10 N KOH and again incubation for 30 min. Mixture was extracted with hexane and the supernatant evaporated to dryness and the residue dissolved in ethanol. To each tube 0.4 ml each of 0.2% bathophenanthroline, ferric chloride reagent, and 0.001 M of ortho-phosphoric acid was added and mixed thoroughly. Absorbance was read at 536 nm. Values were calculated from a standard plot of α-tocopherol treated with the same reagents. The amount of vitamin E was expressed as μg of vitamin E/gm tissue.[33]
Catalase activity
The procedure was followed as discussed by Beers (1952).[34] The reaction mixture consisted of 2 ml phosphate buffer (pH 7.0), 0.95 ml of hydrogen peroxide (0.019 M), and 0.05 ml of supernatant in a final volume of 3 ml. Absorbance were recorded at 240 nm every 10 sec for 1 min. The results were expressed as units of CAT activity/mg protein.
Superoxide dismutase activity
The reaction mixture consisted of 0.05 ml of supernatant, 2 ml of carbonate buffer, and 0.5 ml of EDTA solution. The reaction was initiated by the addition of 0.5 ml of epinephrine and the autoxidation of adrenaline to adrenochrome at pH 10.2 was measured by following the change in optical density at 480 nm. Change in optical density every minute was measured at 480 nm against reagent blank. The results were expressed as units of SOD activity/mg protein.[35]
Glutathione peroxidase activity
The reaction mixture contained 0.5 ml of phosphate buffer (20 mM, pH 7.0), 0.1 ml of glutathione reductase (0.24 U), and 0.1 ml of 10 mM GSH and 0.05 ml of supernatant. Sodium azide (1 mM) was added to the reaction mixture in order to inhibit remnant CAT activity. Thereafter 0.1 ml NADPH solution (0.118 mM) was added and the hydroperoxides independent consumption of NADPH was monitored for about 3 min. Addition of 0.1 ml hydrogen peroxide (12.5 mM) started the overall reaction and the decrease in absorption at 240 nm was monitored for 5 min. The results were expressed as units of GPx activity/mg protein.[36]
Histopathological evaluations
The cerebral cortex was fixed in 10% formalin. The specimens were then processed for standard procedure and were embedded in paraffin wax. The blocks were then sectioned according to hematoxylin and eosin method.[37] Five-micrometer thick histological sections were obtained from the paraffin blocks. The sections were examined under the light microscope and photographs were taken under 100x using Motic camera.
Statistical analysis
All results are expressed as mean ± standard error of mean (n = 6). Statistical analysis was carried out using one-way analysis of variance (ANOVA), followed by Bonferroni's multiple comparison test, # indicates level of significance in comparison to vehicle control group. * indicates level of significance in comparison to AlCl3 treated group. Differences were considered significant at # P < 0.05, ## P < 0.001, and * P < 0.05, ** P < 0.001 compared to the vehicle control and AlCl3 treated groups, respectively. All analysis was done with Graph pad PRISM Version 4 software (Graphpad Software, Inc).
RESULTS
Effect of AlCl3 and Co Q10 on antioxidant status of rat cerebral cortex
Thiobarbituric acid reactive substances
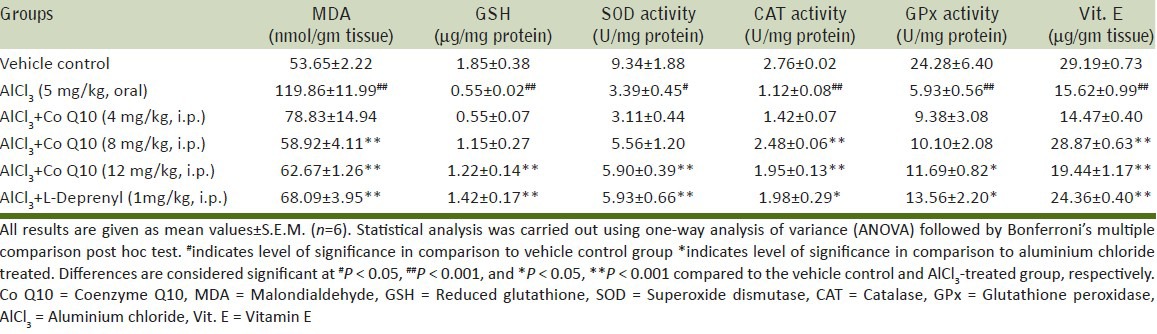
The levels of malondialdehyde (MDA), an indicator of lipid peroxidation were increased in the cerebral cortex of rats treated with AlCl3 (119.86 nmol/gm tissue) as compared to vehicle control group (53.65 nmol/gm tissue). This increase was countered by Co Q10 at the doses of 8 and 12 mg/kg i.p. (58.92 and 62.69 nmol/gm tissue, respectively) as compared to that of L-deprenyl treated group (68.09 nmol/gm tissue) [Table 1].
Table 1.
Effect of Co Q10 on antioxidant markers in aluminium chloride induced oxidative stress in rat cerebral cortex
Reduced glutathione
Reduced glutathione plays an important role in protection of cells against damage from endogenous and exogenous free radicals and oxidants. Subacute exposure to AlCl3 led to decrease in GSH content of cerebral cortex (0.55 μg/mg protein) verses vehicle (1.85 μg/mg protein). Intraperitoneal administration of Co Q10 (12 mg/kg) increased the average level of GSH to 1.22 μg/mg protein. The increase in GSH content by Co Q10 was comparable to the increase obtained by L-Deprenyl (1.42 μg/mg protein) [Table 1].
Vitamin E
Vitamin E, a cell membrane antioxidant has been demonstrated to be capable of scavenging lipid peroxyl radicals, thereby preventing propagation of chain reaction during lipid peroxidation. The subacute exposure of AlCl3 (50 mg/kg, p.o.) for 28 days resulted in a significant decrease in vitamin E levels of cerebral cortex to 15.62 μg/gm tissue verses vehicle control (29.19 μg/gm tissue). Supplementation with intraperitoneal Co Q10 at the doses of 8 and 12 mg/kg increases the average vitamin E content to 22.87 and 19.44 μg/gm tissue, respectively, which is comparable to L-Deprenyl treated group (24.36 μg/gm tissue) [Table 1].
Catalase activity
The ability of catalase to induce reduction in levels of hydrogen peroxidase was evaluated. Hydrogen peroxidase gives rise to free radicals which are congeners of oxidative stress in the body. AlCl3(50 mg/kg, p.o.) exposure for 28 days resulted in a significant decrease in catalase activity to 1.12 U/mg protein as compared to vehicle control group (2.76 U/mg protein). Supplementation with intraperitoneal Co Q10 at the doses 8 and 12 mg/kg increases this activity to 2.48 and 1.95 U/mg protein, respectively, which is comparable to L-Deprenyl-treated group (1.98 units/mg protein) [Table 1].
Superoxide dismutase activity
SOD is a measure of the oxidative stress produced in the organ as it dismutase O2 anions. Decrease in its activity directly corresponds to the increase in oxidative stress. There was significant reduction of SOD activity in cerebral cortex of rats to 3.39 U/mg protein on the AlCl3 exposure as compared to the control group (9.34 U/mg protein) Co Q10 (12 mg/kg, i.p.) increased the activity of SOD (5.90 U/mg protein) which is comparable to that of L-Deprenyl (5.93 U/mg protein) [Table 1].
Glutathione peroxidase activity
GPx is mainly responsible for alleviating the effects of free radicals in brain. The decrease in its activity in AlCl3 exposed group (5.93 U/mg protein) as compared to vehicle control group (24.26 U/mg protein) bears testimony to the oxidative stress inducing effects of AlCl3 on the cerebral cortex of rats. Co administration of Co Q10 intraperitoneally moderately prevented the dramatic decrease in activity (11.69 U/mg protein). The L-deprenyl group also showed an improvement in the enzyme activity (13.56 U/mg protein) [Table 1].
Effect of AlCl3 and Co Q10 on histopathology of rat cerebral cortex
Histopathological evaluation of sections of cerebral cortex of rat brains exhibits degeneration, congestion, and infiltration in AlCl3 exposed group (B), as compared to vehicle control group with normal morphology (A). Significant abatement of AlCl3 induced morphological changes was seen in Co Q10 treated group at the doses 8 and 12 mg/kg, i.p. (D and E, respectively) which was comparable to the standard L-deprenyl treated group (F)as shown in Figure 1.
Figure 1.
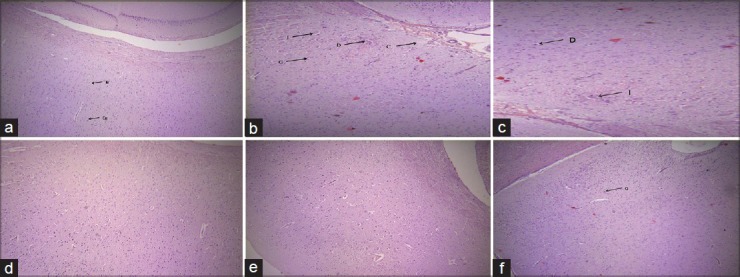
Photomicrographs of sections of rat cerebral cortex (a) Vehicle control: Shows normal architecture (N- Nerve cell, Ca- capillary), (b) AlCl3 exposed: Shows degeneration, congestion and infiltration (D: Degeneration G: Gliosis, I: MNC Infiltrate), (c) Coenzyme Q10 (4 mg/kg, i.p.) treated: Fails to repeal the detrimental outcomes of AlCl3, (d) and (e) Co Q10 (8 and 12 mg/kg, i.p.) treated: Significantly reverses the lesions due to AlCl3, (f) L- deprenyl (1 mg/kg, i.p.) treated: Demonstrates mild congestion (×100, H and E)
DISCUSSION
Cerebral cortex of brain is more susceptible to oxidative stress.[38] The cerebral cortex and hippocampus regions that control the cognitive and motor functions appear to depend on several antioxidants for additional protection.[39] Oxidative damage of the cerebral cortex in rats during Al exposure increases oxidative stress and is thought to contribute to impairment of cognitive functions, for example learning and memory deficits.[40] Cerebral dysfunction was reported in people exposed to drinking water that had been contaminated with Al sulphate.[14]
Several studies have shown that oral administration of Co Q10 can produce protection in experimental models of cerebral ischemia or against mitochondrial toxins.[41,42,43] According to literature, Co Q10 exerts neuroprotective effects in several in vivo and in vitro models of neurodegenerative disorders.[42,44,45] It is reported that oral administration of Co Q10 at the dose of 200 mg/kg/day for 2 months significantly increases the content of Co Q10 and mitochondrial concentrations in cerebral cortex of rats.[41] Raucher et al. (2001) demonstrated increase in oxidized glutathione and reduction of lipid peroxidation in brains of normal and streptozotocin induced diabetic rats treated with Co Q10 at the dose of 10 mg/kg/day, i.p. for 14 days, signifying its antioxidant potential.[46]
The present study was designed to investigate the potentials of Co Q10 to protect the cerebral cortex from AlCl3 induced neurotoxicity. In order to confirm the neuroprotective potential of Co Q10, we compared its effects with that of the standard L-deprenyl (selegiline) with established neuroprotective action in Al neurotoxicity.[47] L-deprenyl is a monoamino oxidase-B inhibitor that protects against the effects of neurotoxins and excitatory amino acids.[48] It is used in for Parkinson's disease with underlying pathogenesis involving oxidative stress. L-deprenyl is also a well known antioxidant and is proven to have antiaging effects. It is known to extend life expectancy.[49] It has been established that the effect of L-deprenyl given i.p. is greater than oral administration; hence it was deemed appropriate to administer it intraperitoneally.
The Thiobarbituric acid reactive substance (TBARS) is the secondary products of lipid peroxidation (LPO) and have been shown to further catalyze the process of oxidative insult to membranes. LPO is an important cause of neuronal damage, as encountered in ischemic injuries, neurotrauma, and neurodegenerative disorders, such as Parkinson's disease and Alzheimer's disease.[50] LPO leads to a decrease in Co Q10 content and inactivation of respiratory chain enzymes in mitochondria.[51] Al administration causes significant increase in brain lipid peroxidation. The increased accumulation of this by product strongly reflects Al inflicted oxidative damage.[49,52] In the present study, synchronous administration of Co Q10 along with AlCl3 prevents increase in TBARS which strongly suggests its neuroprotective abilities. Co Q10 at dose 4 mg/kg has anti-lipid peroxidation activity, this indicates that Co Q10 can cross blood brain barrier at this dose. In normal physiology neurons have the capacity of de novo Co Q10 synthesis.[53] So, exogenous supply of Co Q10 does not elevate its levels in the brain. But in some mitochondrial disorders or in chemical toxicity the Co Q10 content was known to be lowered in brain.[43] This explains the supply and uptake of exogenous Co Q10 due to increase in oxidative stress in neurons.
GSH plays multiple roles in the protection of cells from ROS, electrophiles, and xenobiotics. In the present study, the GSH concentration was reduced in AlCl3 treated group reflecting its utilization in combating the oxidative stress. In vitro study carried out by Satoh et al. (2005) showed that exposure of rat pheochromocytoma cells to Al increases ROS and decreases GSH concentration intracellularly.[54] The current study confirms that Al mediates cellular toxic effect by decreasing GSH. Co Q10 at the dose of 12 mg/kg and L-deprenyl prevented the depletion of GSH levels, suggesting that they are efficacious in countering the oxidative stressors and associated alterations.
The results of the investigations are in agreement with the finding of Rauscher and coworkers (2001) implicating increased glutathione peroxidase in brain upon treatment with Co Q10.[46] GPx catalyses the reduction of H2O2 and alkyl hydroperoxides at the expense of GSH. Although catalase can also remove H2O2, the relative levels of GPx and catalase vary greatly from tissue to tissue.[55] In particular, the brain has very low levels of catalase activity and relatively higher levels of GPx activity. Furthermore, GPx but not catalase (except in the heart) is found in the mitochondria and so play a critical role in removing H2O2 produced in the mitochondria.[56] The study by Jyoti et al. (2007) showed reduction of GSH levels and GPx activity in cerebral cortex of Al-treated rats.[49] Our results are in concurrence with this report, Co Q10 at doses of 12 mg/kg and L-deprenyl prevented the fall in GPx activity.
Superoxide dismutase catalytically scavenges superoxide anion (O2-) and catalase scavenges hydrogen peroxide (H2O2). Both enzymes are essential components of the biological defense against toxicity induced by Al. SOD plays an important role to inactivate free radical generated in electron transport chain reaction. However, the loss of SOD activity does not occur in the presence of GPx and CAT, suggesting role of H2O2 in inactivation of superoxide dismutase. Catalase and peroxidase function to protect superoxide dismutase from inactivation by H2O2.[57] So we can postulate that the increase in the CAT and GPx are key for the enhanced SOD action. Our findings suggests that AlCl3 increases the oxidative stress in cerebral cortex by inhibiting the enzyme SOD and CAT and Co Q10 at the dose of 12 mg/kg prevented this decrease in the activity of these enzymes. This depicts that the elevated activity of SOD, GPx, and CAT act interchangeably or complementarily supporting each other to reduce the deleterious effect of AlCl3 and undoubtedly indicate the neuroprotective potential of Co Q10.
Neural tissue has high fat content and vitamin E is a fat soluble vitamin and plays an instrumental role as membrane antioxidant. The inner mitochondrial membrane contains Co Q10 as well as vitamin E, both of which have antioxidant properties. Vitamin E has been demonstrated to be capable of scavenging lipid peroxyl radicals, thereby preventing the propagation of chain reactions during lipid peroxidation.[58] Several studies seem to confirm the regenerative effect of Co Q10 on vitamin E in vivo.[59,60] In the present study, vitamin E content was enhanced by Co Q10 at the doses of 8 and 12 mg/kg while AlCl3 depleted the levels of vitamin E. Further, the histopathological evidences of the cerebral cortex of rat brains corroborate the protection offered by Co Q10 in countering the AlCl3 induced neurotoxicity by maintenance of the normal morphology.
CONCLUSION
In summary, our results clearly showed that L-deprenyl when co-administered with AlCl3 in rat inhibited TBARS formation while elevating the SOD, CAT, and GPx activities in rat brain cerebral cortex. It was also found to prevent decrease in GSH and vitamin E levels. Moreover, Co Q10 at dose 12 mg/kg i.p. shows anti-oxidant effect which appears to be comparable to that of L-deprenyl. Our investigation leads us to conclude that Co Q10 like L-deprenyl has potential to prevent AlCl3 neurotoxicity in brain region like cerebral cortex. Since this region is responsive to Co Q10, this may be used as a protective drug in preventing neurodegenerative diseases precipitated by the oxidative insults.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Al Ahmad A, Gassmann M, Ogunshola OO. Involvement of oxidative stress in hypoxia induced blood brain barrier break down. Microvasc Res. 2012;84:222–5. doi: 10.1016/j.mvr.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmcol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci. 2011;124:225–50. doi: 10.1093/toxsci/kfr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright DA, Welbourn PM. 1st ed. Cambridge University Press series publication; 2002. Environmental Toxicology; p. 70. [Google Scholar]
- 5.Marrugo-Negrete J, Verbel JO, Ceballos EL, Benitez LN. Total mercury and methylmercury concentrations in fish from the Mojana region of Colombia. Environ Geochem Health. 2008;30:21–30. doi: 10.1007/s10653-007-9104-2. [DOI] [PubMed] [Google Scholar]
- 6.Monnet-Tschudi F, Zurich MG, Boschat C, Corbaz A, Honegger P. Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev Environ Health. 2006;21:105–18. doi: 10.1515/reveh.2006.21.2.105. [DOI] [PubMed] [Google Scholar]
- 7.Wormser U, Brodsky B, Milatovic D, Finkelstein Y, Farina M, Rocha JB, et al. Protective effect of a novel peptide against methylmercury-induced toxicity in rat primary astrocytes. Neurotoxicology. 2012;33:763–68. doi: 10.1016/j.neuro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lobo Torres LH, Moreira WL, Tamborelli Garcia RC, Annoni R, Nicoletti Carvalho AL, Teixeira SA, et al. Environmental tobacco smoke induces oxidative stress in distinct brain regions of infant mice. J Toxicol Environ Health A. 2012;75:971–80. doi: 10.1080/15287394.2012.695985. [DOI] [PubMed] [Google Scholar]
- 9.Yokel RA, McNamara PJ. Aluminium toxicokinetics: An updated minireview. Pharmacol Toxicol. 2001;88:159–67. doi: 10.1034/j.1600-0773.2001.d01-98.x. [DOI] [PubMed] [Google Scholar]
- 10.Slanina P, French W, Ekstrom LG, Lööf L, Slorach S, Cedergren A. Dietary citric acid enhances absorption of aluminum in antacids. Clin Chem. 1986;32:539–41. [PubMed] [Google Scholar]
- 11.Harris WR, Berthon G, Day JP, Exley C, Flaten TP, Forbes WF, et al. Speciation of aluminium in biological systems. J Toxicol Environ Health. 1996;48:543–68. [PubMed] [Google Scholar]
- 12.Kong S, Liochev S, Fridovich I. Aluminum (III) facilitates the oxidation of NADH by the superoxide anion. Free Radic Biol Med. 1992;13:79–81. doi: 10.1016/0891-5849(92)90168-g. [DOI] [PubMed] [Google Scholar]
- 13.Szutowicz A. Aluminium NO, and nerve growth factor neurotoxicity in cholinergic neurons. J Neurosci Res. 2001;66:1009–18. doi: 10.1002/jnr.10040. [DOI] [PubMed] [Google Scholar]
- 14.Nayak P. Aluminum: Impacts and disease. Environ Res. 2002;89:101–15. doi: 10.1006/enrs.2002.4352. [DOI] [PubMed] [Google Scholar]
- 15.Powell AK, Heath SL. X-ray structural analysis of biological relevant aluminium (III) complexes. Coordination Chem Rev. 1996;149:59–80. [Google Scholar]
- 16.Bihagi SW, Sharma M, Singh AP, Tiwari M. Neuroprotective role of Convolvulus pluricaulis on aluminium induced neurotoxicity in rat brain. J Ethnopharmacol. 2009;124:409–15. doi: 10.1016/j.jep.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 17.McDermott JR, Smith AI, Iqbal K, Wisniewski HM. Brain aluminium in aging and Alzheimer disease. Neurology. 1979;29:809–14. doi: 10.1212/wnl.29.6.809. [DOI] [PubMed] [Google Scholar]
- 18.Yokel RA. Blood-brain barrier flux of aluminum, manganese, iron and other metals suspected to contribute to metal-induced neurodegeneration. J Alzheimers Dis. 2006;10:223–53. doi: 10.3233/jad-2006-102-309. [DOI] [PubMed] [Google Scholar]
- 19.Ackley DC, Yokel RA. Aluminium citrate is transported from brain into blood via the monocarboxylic acid transporter located at the blood/brain barrier. Toxicology. 1997;120:89–97. doi: 10.1016/s0300-483x(97)03640-8. [DOI] [PubMed] [Google Scholar]
- 20.Yokel RA, Allen DD, Ackley DC. The distribution of aluminium into and out of the brain. J Inorg Biochem. 199;6:127–32. doi: 10.1016/s0162-0134(99)00124-5. [DOI] [PubMed] [Google Scholar]
- 21.Kumar NS, Agrawal P, Sujata AS, Bhavana BK. Fermentation, media optimization studies for Coenzyme Q10 production by Saccharomyces cerevisiae. Int Res J Pharm. 2012;3:132–8. [Google Scholar]
- 22.Beal F. Therapeutic effects of coenzyme Q10 in neurodegenerative diseases. Methods Enzymol. 2006;382:473–87. doi: 10.1016/S0076-6879(04)82026-3. [DOI] [PubMed] [Google Scholar]
- 23.Bentiger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7:S41–50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Crane FL. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7:S2–7. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Borekova M, Hojerova J, Koprda V, Bauerova K. Nourishing and health benefits of coenzyme Q10-a review. Czech J Food Sci. 2008;26:229–41. [Google Scholar]
- 26.Turunen M, Olssan J, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660:171–99. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Santos GC, Antunes LM, Santos AC, Maria LP. Coenzyme Q10 and its effects in the treatment of neurodegenerative diseases. Braz J Pharm Sci. 2009;45:607–18. [Google Scholar]
- 28.Tina K. Coenzyme Q10 in Parkinson's disease: Ready for first line use? Nat Med J. 2010;2:3–7. [Google Scholar]
- 29.Todd KG, Butterworth RF. Increased neuronal cell survival after L-deprenyl treatment in experimental thiamine deficiency. J Neurosci Res. 1998;52:240–6. doi: 10.1002/(SICI)1097-4547(19980415)52:2<240::AID-JNR12>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 30.Binukumar BK, Gupta N, Bal A, Gill KD. Protection of dichlorvos induced oxidative stress and nigrostriatal neuronal death by chronic Coenzyme Q10 pretreatment. Toxicol Appl Pharmacol. 2011;256:73–82. doi: 10.1016/j.taap.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 31.Ohkawa H, Onishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–8. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 32.Moron MS, Depierre JW, Mannervik B. Levels of glutathione, glutathione reductase and glutathione S-transferase activities in rat lung and liver. Biochim Biophys Acta. 1979;582:67–78. doi: 10.1016/0304-4165(79)90289-7. [DOI] [PubMed] [Google Scholar]
- 33.Desai ID. Vitamin E analysis methods for animal tissues. Methods Enzymol. 1984;105:138–47. doi: 10.1016/s0076-6879(84)05019-9. [DOI] [PubMed] [Google Scholar]
- 34.Beers RF, Jr, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–40. [PubMed] [Google Scholar]
- 35.Sun M, Zigman S. An improved spectrophotometric assay for superoxide dismutase based on epinephrine autoxidation. Anal Biochem. 1978;90:81–9. doi: 10.1016/0003-2697(78)90010-6. [DOI] [PubMed] [Google Scholar]
- 36.Carrillo MC, Kanai S, Nokubo M, Kitani K. Deprenyl induces activities of both superoxide dismutase and catalase but not of glutathione peroxidase in the striatum of young male rats. Life Sci. 1991;48:517–21. doi: 10.1016/0024-3205(91)90466-o. [DOI] [PubMed] [Google Scholar]
- 37.Culling CF. 3rd ed. 1974. Handbook of histopathological and histochemical techniques: (including museum techniques): Butterworth; p. 361. [Google Scholar]
- 38.Sathyasaikumar KV, Swapna I, Reddy PV, Murthy CR, Dutta GA, Senthilkumaran B, et al. Fulminant hepatic failure in rats induces oxidative stress differentially in cerebral cortex, cerebellum and pons medulla. Neurochem Res. 2007;32:517–24. doi: 10.1007/s11064-006-9265-x. [DOI] [PubMed] [Google Scholar]
- 39.Onodera K, Omoi NO, Fukui K, Hayasaka T, Shinkai T, Suzuki S, et al. Oxidative damage of rat cerebral cortex and hippocampus, and changes in antioxidative defense systems caused by hyperoxia. Free Radic Res. 2003;37:367–72. doi: 10.1080/1071576031000090019. [DOI] [PubMed] [Google Scholar]
- 40.Fukui K, Onodera K, Shinkai T, Suzuki S, Urano S. Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann N Y Acad Sci. 2000;928:169–73. doi: 10.1111/j.1749-6632.2001.tb05646.x. [DOI] [PubMed] [Google Scholar]
- 41.Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Nati Acad Sci U S A. 1998;95:8892–7. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mounsey RB, Teismann P. Mitochondrial dysfunction in Parkinson's disease: Pathogenesis and neuroprotection. Parkinson's Dis 2010. 2011:617472. doi: 10.4061/2011/617472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koppula S, Kumar H, More SV, Kim BW, Kim IS, Choi DK. Recent advances on the neuroprotective potential of antioxidants in experimental models of Parkinson's disease. Int J Mol Sci. 2012;13:10608–29. doi: 10.3390/ijms130810608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elipenahli C, Stack C, Jainuddin S, Gerges M, Yang L, Starkov A, et al. Behavioral improvement after chronic administration of coenzyme Q10 in P301S transgenic mice. J Alzheimers Dis. 2012;28:173–82. doi: 10.3233/JAD-2011-111190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rauscher FM, Sanders RA, Watkins JB., 3rd Effects of coenzyme Q10 treatment on antioxidant pathways in normal and streptozotocin-induced diabetic rats. J Biochem Mol Toxicol. 2011;15:41–6. doi: 10.1002/1099-0461(2001)15:1<41::aid-jbt5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 47.Muralikrishnan D, Samantaray S, Mohanakumar KP. D-deprenyl protects nigrostriatal neurons against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced dopaminergic neurotoxicity. Synapse. 2003;50:7–13. doi: 10.1002/syn.10239. [DOI] [PubMed] [Google Scholar]
- 48.Ebadi M, Sharma S, Shavali S, Sangchot P, Brekke L. The multiple actions of selegiline. Proc West Pharmacol Soc. 2002;45:39–41. [PubMed] [Google Scholar]
- 49.Jyoti A, Sethi P, Sharma D. Bacopa monniera prevents from aluminium neurotoxicity in the cerebral cortex of rat brain. J Ethnopharmacol. 2007;111:56–62. doi: 10.1016/j.jep.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 50.Coyle JT, Puttfarken P. Oxidative stress, glutamate and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 51.Pieczenik SR, Neustadt J. Mitochondrial dysfunction and molecular pathways of disease. Exp Mol Pathol. 2007;83:84–92. doi: 10.1016/j.yexmp.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Kaizer RR, Correa MC, Spanevello RM, Morsch VM, Mazzanti CM, Gonçalves JF. Acetylcholinesterase activation and enhanced lipid peroxidation after long-term exposure to low levels of aluminum on different mouse brain regions. J Inorg Biochem. 2005;99:1865–70. doi: 10.1016/j.jinorgbio.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Hargreaves IP, Lane A, Sleiman PM. The coenzyme Q10 status of the brain regions of Parkinson's disease patients. Neurosci Lett. 2008;447:17–9. doi: 10.1016/j.neulet.2008.09.069. [DOI] [PubMed] [Google Scholar]
- 54.Satoh E, Okada M, Takadera T, Ohyashiki T. Glutathione depletion promotes aluminum- mediated cell death of PC12 cells. Biol Pharm Bull. 2005;28:941–6. doi: 10.1248/bpb.28.941. [DOI] [PubMed] [Google Scholar]
- 55.Gutteridge JM. Free radicals in disease processes: A compilation of cause and consequence. Free Radic Res Commun. 1993;19:141–58. doi: 10.3109/10715769309111598. [DOI] [PubMed] [Google Scholar]
- 56.Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 57.Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 58.McCay PB. Vitamin E: Interactions with free radicals and ascorbate. Annu Rev Nutr. 1985;5:323–40. doi: 10.1146/annurev.nu.05.070185.001543. [DOI] [PubMed] [Google Scholar]
- 59.Kaikkonen J, Nyyssönen K, Tomasi A, Iannone A, Tuomainen TP, Porkkala-Sarataho E, et al. Antioxidative efficacy of parallel and combined supplementation with coenzyme Q10 and d-α-tocopherol in mildly hypercholesterolemic subjects: A randomized placebo-controlled clinical study. Free Radic Res. 2000;33:329–40. doi: 10.1080/10715760000301501. [DOI] [PubMed] [Google Scholar]
- 60.Feher J, Nemeth E, Nagy V, Gabriella L. The preventive role of coenzyme Q10 and other antioxidants in injuries caused by oxidative stress. Arch Med Sci. 2007;3:305–14. [Google Scholar]