

Abstract
Web-based user interfaces to scientific applications are important tools that allow researchers to utilize a broad range of software packages with just an Internet connection and a browser.1 One such interface, CHARMMing (CHARMM interface and graphics), facilitates access to the powerful and widely used molecular software package CHARMM. CHARMMing incorporates tasks such as molecular structure analysis, dynamics, multiscale modeling, and other techniques commonly used by computational life scientists. We have extended CHARMMing’s capabilities to include a fragment-based docking protocol that allows users to perform molecular docking and virtual screening calculations either directly via the CHARMMing Web server or on computing resources using the self-contained job scripts generated via the Web interface. The docking protocol was evaluated by performing a series of “re-dockings” with direct comparison to top commercial docking software. Results of this evaluation showed that CHARMMing’s docking implementation is comparable to many widely used software packages and validates the use of the new CHARMM generalized force field for docking and virtual screening.
Introduction
In the past, the cost and effort of developing a new drug has largely confined successes to large pharmaceutical companies or otherwise well-funded research institutions.2 Although development and use of computer-aided drug design (CADD) techniques has provided numerous benefits to the overall process, the expertise required to create powerful commercial software packages has resulted in high licensing costs,3,4 thus limiting access to academic groups. Fortunately, this trend has started to shift with the emergence of freely available software, such as Autodock5 and several other packages,4 largely developed by the academic computational chemistry community. However, for the most part, these software packages require familiarity with CADD methodologies and are better suited for computer savvy users that are at least comfortable if not familiar with the computational component of drug discovery.6 This has hampered the proliferation of CADD tools into less computationally minded drug discovery laboratories. The need for intuitive and easy to use CADD solutions has largely been met by the commercial software companies such as Accelrys, Schrödinger, and others that have incorporated full-featured graphical user interfaces (GUI) into their programs.7−9 However, as alluded to above, the cost of these packages is typically prohibitive to academic groups and/or institutions. Further, it has proven increasingly difficult to strike a balance between software that is user-friendly yet incorporates a wide range of advanced functionality and customizability. Another aspect of concern is portability. For example stand-alone software that requires local installation on every computer may find less use in today’s world where researchers expect both the application and the data to be accessible from any machine on any platform from any location.10
Another hurdle, faced by the nonexpert, to incorporating computational modeling into drug discovery efforts is the difficulty of obtaining reliable small molecule parameters.11−13 Most widely used and well-tested force fields have been developed with proteins and nucleic acids rather than small molecules in mind.14 Until recently this has meant that drug-like molecule parameters have been less reliable, with assignment often arbitrary. Lately, however, there has been a significant amount of effort devoted to improving the reliability of small molecule parameters and developing efficient protocols to generate them for a much greater and more diverse chemical space.11,12,14,15
The CHARMM interface and graphics (CHARMMing)16 is a Web interface to the popular macromolecular modeling package CHARMM.17,18 The goal of the CHARMMing project is to provide a platform-independent Web-based front-end that allows its users to set up and perform a wide variety of molecular modeling tasks. CHARMMing’s users range from small academic laboratories that benefit from the portal’s functionality to educators that include molecular modeling in their curricula and use the portal to facilitate their teaching.19−21 Moreover, the open source nature of the project allows outside developers to utilize the framework and build on the existing infrastructure, further expanding the range of features it includes. The framework can be easily installed on a private network or adopted into a new Web-based interface; this approach was utilized when developing a virtual target screening (VTS)22 server. Herein, we describe a similar effort using the CHARMMing infrastructure (i.e., built on a Python-based23 Django24 framework with a MySQL25 database); the implementation of a new drug design module that incorporates a fragment-based docking protocol includes a diverse set of drug-like compounds and facilitates creation of CHARMM friendly protein–small molecule systems for further modeling studies. We also assess the performance of the newly implemented docking protocol coupled to CHARMM’s new generalized force field (CGenFF) by reproducing a series of co-crystallized protein–ligand complexes and comparing the results against a leading commercially available docking package.
Implementation Details
Target Preparation
Target proteins begin their preparation via CHARMMing’s structure submission section. Here, tasks such as the addition of hydrogens, identification of any nonprotein moieties, and assignment of final parameters are carried out (using the latest CHARMM36 protein force field).26,27 Co-crystallized small molecules (i.e., ligands) are automatically parametrized using the CGenFF.12 Specifically, ligand atom-typing and parametrization is performed by sequentially attempting several automated parametrization tools. The default order is (1) ParamChem,12,28,29 (2) MATCH,30 (3) Antechamber,31 and (4) GENRTF.32 As an alternative to the default order, a user can specify the exact build procedure to use for parametrization.
Compound Library and Ligand Upload
CHARMMing docking module provides a preloaded library of drug-like compounds for virtual screening experiments. The library consists of approximately 8000 molecules from the Maybridge Hitfinder set (www.maybridge.com). All of the provided molecules have been atom typed according to CGenFF convention to comply with CHARMM requirements and confirmed to decompose into at least three sufficiently sized fragments to meet the fragment-based docking criteria. CHARMMing also allows users to upload ligands by providing a coordinate file in mol2 format. Upon uploading, the ligand undergoes atom-typing and parametrization as previously described. The ligand and corresponding parameter, topology, and structure files are then saved on a disk as well as cataloged in the database. Unlike the preloaded compound library, any user-uploaded ligands are restricted to their account only and are not visible to other users. The user is also given the ability to create custom sets of molecules based on any preloaded or user-uploaded compounds. This can be done via the “Ligand Sets” section (Figure 1) of the docking module. Any custom or preloaded set can be docked in its entirety or by selecting individual molecules on the docking submission page (Figure 2).
Figure 1.

“Ligand Set Details” page allows the user to manage custom ligand sets. The user can define and describe a custom ligand set as well as add ligands to it from any of the other sets including the preloaded public library.
Figure 2.

“Submit Docking Job” page presents the user with the ability to select the target coordinates for docking, define the binding pocket (vide infra), and select ligands to dock from the list of available small molecules. Native ligands and ligands available for docking can be visualized in 3D using the embedded visualization application.
Binding Site Definition
To provide maximum flexibility with respect to job setup, two different ways of specifying the binding region of interest are implemented. The first approach identifies the binding pocket using the position of a co-crystallized ligand that may be present. In this case, when launching a docking job, a user is presented with a list of all co-crystallized small molecules along with their 2D structural representations. Once the desired small molecule is chosen, the binding site is defined via proximity to the aforementioned small molecule. In cases where no co-crystallized ligand is present, or if a user simply wishes to investigate alternative binding sites, we have implemented an interactive and graphical binding site definition tool (Figure 3). To use this tool, two residues should be selected that roughly correspond to the edges of the desired binding region. The midpoint between these residues is then determined and defined as the approximate center of the binding site. On the basis of a user-defined radius, a list of all residues within this distance is compiled and both visually highlighted and presented as a list. The user can then add or remove residues to/from this list by either modifying the text of the residue list, changing the specified search radius, or modifying it via graphical selection (i.e., clicking). Ultimately, all user-defined binding sites are saved and presented as options with any existing co-crystallized ligands at the docking job setup page.
Figure 3.

“Binding Site Selection” page provides the user multiple ways to select a custom binding site. This can be done either by manually typing in the residue numbers, graphically selecting residues, or defining the centroid and specifying the radius in Å.
Docking Protocol
Docking algorithms used in this protocol are based on the popular grid-based paradigm used by most current docking programs.33−38 In this approach, the solvent accessible surface area of the target and the ligand as well as the target’s binding site are discretized onto a 3D lattice. The lattice then either stores information about the atoms enclosed by a cubic unit of the grid or contains the potential contributions projected onto the grid’s vertices. Precomputed grids allow for efficient calculation of both van der Waals and electrostatic contributions to the scoring function, facilitating rapid evaluation of ligand placements within the binding site.
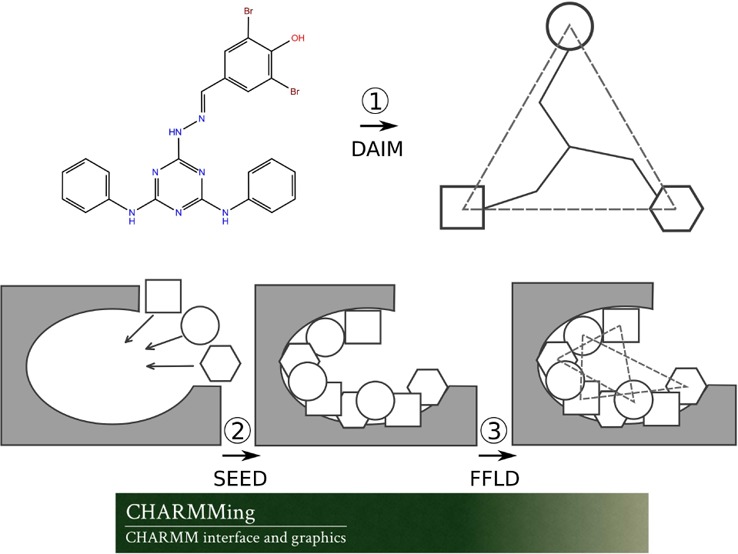
The docking procedure consists of several steps where different programs perform distinct tasks. To streamline the communication between the programs and ensure compatibility of input and output data, a series of scripts were written in Python, Perl, and Linux shell scripting languages. The OpenBabel39 file conversion utility was used to interconvert between different representations of the protein and compound structures. The program MATCH30 was used to generate CGenFF compatible topologies and parameters. The fragment-based docking protocol implemented in CHARMMing is outlined in Figure 4 and described as follows:
Figure 4.
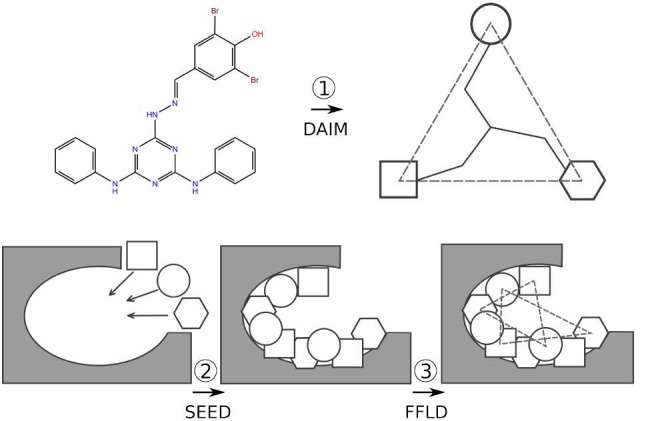
Schematic of the fragment-based docking protocol implemented into the CHARMMing Web user interface. Depicted are the three main stages of the docking: decomposition by DAIM, fragment docking by SEED, and ligand placement by FFLD.
(1) Each compound to be docked is first broken down into fragments. A fingerprint describing chemical richness is generated for each fragment and its parent compound. The three most chemically rich, but not necessarily different, fragments are identified to serve as anchors for docking. These steps are carried out by the program DAIM (Decomposition and Identification of Molecules).40
(2) The user then identifies the binding site to be used in the docking job. All nonprotein nonsolvent compounds present in the submitted target structure are displayed on the “Submit Docking Job” page (Figure 2). On the basis of the user selected compound, the proximal residues are identified and the binding site defined.
(3) The previously identified anchor fragments (step 1) are then docked into the binding site using the program SEED (Solvation Energy for Exhaustive Docking).41 The placement of fragments within the binding site is determined by matching either the direction of polar vectors between ligand and receptor atoms to form a hydrogen bond or the apolar vectors on the solvent accessible surface area of the receptor. The SEED score, used in fragment placement, accounts for the solvent effects by including terms for both receptor and fragment desolvation as well as a solvent-screened receptor-fragment electrostatic interaction term.
(4) The docked fragments are reconnected into the original ligand while undergoing refinement using the FFLD (Fragment-based Flexible Ligand Docking) program.42 FFLD uses a genetic algorithm that generates and evaluates populations of conformations and positions them within the binding site, as guided by fragment anchor locations. The fitness of a placed conformation is evaluated using a scoring function that is aimed at approximating the steric effects as well as hydrogen bonding contributions of the protein–ligand interactions. This function includes intraligand and protein–ligand van der Waals interaction terms as well as polar contributions based on the number of hydrogen bonds and unfavorable donor–donor and acceptor–acceptor interactions.
(5) Poses generated by FFLD that are within a user-defined energy cutoff (10 kcal/mol by default) are then clustered using a leader clustering algorithm implemented in the program FLEA (FFLD Leader Clustering).43
(6) Following the clustering, the protein–ligand complex is converted to native CHARMM format and saved. Using these files, in addition to the CHARMM protein and generalized force fields (i.e., CHARMM36 and CGenFF), protein structure (psf) and coordinate (crd) files are generated. Each ligand then undergoes 1000 steps of minimization using the adopted Newton–Rhapson (ABNR) algorithm while keeping protein atoms fixed. The “minimized” protein–ligand complexes are then scored using SEED and FFLD in their “evaluation only” mode, producing their own estimation of electrostatic, van der Waals, and total energy contributions for each pose. The final ranking of the docked poses is performed using a consensus approach. For this, energies (i.e., interaction energy from CHARMM and total energies from SEED and FFLD) are used to create three lists in which individual poses are sorted and ranked. The final rank of each pose is then set to the median of the three ranks as assigned in the individual lists. The consensus approach to scoring or ranking compounds when performing molecular docking or virtual screening studies has been shown to be more accurate than single scoring methods.44−49
Job Submission and Monitoring
When a docking job is launched, the PBS50 (Portable Batch System)-based queuing system TORQUE51 accepts the job as a wrapper shell script that controls the entire docking procedure. Using the interface, a job can be monitored in real time as it progresses and generates final poses for each docked compound. Basic job statistics such as submission time and job status can be monitored along with the output file reflecting the job progression (Figure 5). In addition, important files associated with job progress and results (e.g., final docked ligand poses, job output, etc.) can be downloaded to a local disk. Protein, ligands, compounds in the library, and final docked poses can all be visualized directly in CHARMMing. The 3D structure of each of the above elements can be rendered with the JSmol52 or GLmol53 visualization tools. Structures can be visualized using a variety of representations to highlight important structural features or interactions of the molecules and their complexes.
Figure 5.

“Job Details” page provides general job information as well as the list of docked poses and their respective scores. The docked poses can be visualized in 3D within the binding pocket of the protein using the embedded visualization application. An archive of the job directory can also be downloaded from this page for execution on local resources.
A walk-through outlining the entire process of performing a redock on a sample system is included in the tutorial covering basic CHARMM and CHARMMing functionality at www.charmmtutorial.org. Additionally, a docking lesson that guides a user through the redocking procedure has been added to the lessons section of the CHARMMing Web site.
Performance and Local Execution
Currently, all docking jobs executed via the Web interface are carried out sequentially. However, after initial setup of the docking job, all necessary files are available for download and execution on local computational resources. To improve performance of this procedure, we have developed a protocol that can be carried out in parallel as outlined in Figure 6. This is achieved by spawning a new execution branch for each of the most time-consuming steps in the protocol via a user-modifiable job queuing command. For example, each fragment of each molecule is docked (step 3, vide supra) as a separate submitted job. Once all of a molecule’s anchor fragments are docked, the placement of a ligand within the binding site by FFLD is also spawned as a series of separate jobs. Furthermore, to increase sampling by FFLD and improve performance, the protocol performs multiple docking iterations per ligand, again each as a separate job. Thus, instead of one docking job that attempts to sequentially sample a large conformational space per ligand, multiple shorter iterations with different random seeds are run in parallel, taking less real time and still sufficiently sampling ligand conformational space. The number of iterations per ligand as well as the amount of energy evaluations per iteration are all user modifiable parameters.
Figure 6.

Parallelization of the docking protocol is achieved by spawning new job execution threads at both the fragment docking (i.e., one per fragment) and ligand placement (i.e., one per iteration per ligand) steps. Clustering and scoring threads are also spawned for each docked ligand.
In order to execute a job on local resources, the following programs need to be downloaded and installed: VMD,54 DAIM, SEED, FFLD, FLEA, MATCH, and CHARMM. Except for CHARMM, all of these programs are free for academic use. VMD can be downloaded from the University of Illinois at Urbana–Champaign’s Theoretical and Computational Biophysics group (www.ks.uiuc.edu/Research/vmd). DAIM, SEED, FFLD, and FLEA can be obtained from the University of Zurich’s Computational Structural Biology lab (www.biochem-caflisch.uzh.ch/download). Further, a more general description of the installation process is included as part of the CHARMM tutorial and can be found at the following Web address: www.charmmtutorial.org/index.php/Installation_of_CHARMMing.
Once the job directory is downloaded and the software is installed on local resources, the provided settings file should be used to specify the location of program executables. In addition, job details (e.g., protein file name, number of docking iterations, clustering energy cutoff, etc.) can be modified via the settings file. This file is also where PBS/TORQUE commands can be modified for local resources. Because there is no limit to the number of possible parallel processes spawned, the protocol checks for available resources and will wait for current processes to complete if the queue is full. The protocol will automatically take advantage of all available resources to speed up job completion while at the same time adhering to the local queuing system policies.
Results and Discussion
To assess the performance of the docking protocol, a diversity set was constructed from the publicly available CCDC/Astex test set55 containing high-resolution X-ray complexes and an augmented version of that set, which has been used to compare the performance of a number of docking programs.56 Our final set contained 24 protein–ligand complexes with X-ray resolutions ranging from 1.50–2.30 Å. In particular, we selected complexes where the ligand could be decomposed into three fragments (i.e., at least three rotatable bonds) using the default settings of DAIM, as the ultimate goal was to evaluate the implementation of the decomposition-based approach.
Redock validation involved removing the co-crystallized ligand from the complex, redocking it via the fragment-based protocol, and comparing the docked pose to that of the original crystal structure. Each complex was processed using CHARMMing’s “Submit Structure” section that downloads the structure based on the PDB code, adds hydrogen atoms, and prepares the structure for modeling using CHARMM. Further, each system containing the protein, solvent, and ligand molecules was briefly minimized for 100 steps using the Steepest Descent method followed by 1000 steps of ABNR using CHARMMing’s “Calculations” module. Using the “Ligand Upload” section of CHARMMing’s docking module, the previously downloaded ligand was processed. The docking calculation for each minimized system was set up by selecting a native ligand to define a binding pocket and user-uploaded ligand for docking, all from the “Submit Docking Job” page of the docking module. The progress of each job was monitored using the job monitoring section of the docking module. To assess the performance of the dockings, root-mean-square deviation (RMSD) between the heavy atoms of the docked poses and the crystal structures was calculated using VMD.
To compare the docking protocol’s performance, a commercially available docking package was also used. Redockings were performed using Schrödinger’s Glide34−36,57 Standard Precision (SP) docking protocol. Glide’s SP protocol attempts to dock multiple conformations of a ligand into a receptor grid, subsequently calculating the effective ligand–receptor interactions using a proprietary scoring function. Conformational sampling of the ligand is achieved via varying torsion angles around rotatable bonds. Prior to docking, each target was prepared using Maestro’s7 Protein Preparation Wizard.58−62 The preparation included removal of solvent molecules, addition of hydrogens, and brief minimization. As Glide is also a grid-based docking protocol, the grids, similar to CHARMMing’s procedure, were built using the co-crystal ligand to define the binding region. The native ligand was removed and redocked using default parameters of the SP docking protocol. The poses with the best docking scores were used to calculate their respective RMSD from the crystal structure using VMD.
Table 1 reports the RMSD of poses generated by CHARMMing’s fragment-based docking protocol and Glide SP docking (w.r.t. crystal structure). Results reported from CHARMMing’s fragment-based docking protocol correspond to the pose closest to the crystal structure. This set yields a 71% success rate using RMSD < 2.0 Å as the metric; this criteria is commonly employed for evaluating the performance of docking algorithms.56,63−65 This clearly shows that the protocol can successfully recover the crystal pose in the majority of the cases. We are currently optimizing a consensus scoring function based on this diversity set; results of that effort will be reported in a subsequent publication. Nevertheless, virtual screening is known to suffer from a high false-positive rate, which does not diminish its value in drug discovery as the unfit compounds are screened out during the experimental stages of the discovery campaigns.66 Regardless, we are encouraged by the success of fragment-based docking, which shows approximately the same performance as widely used docking programs, i.e., within the range of 40–90%.56,63−65
Table 1. RMSDs of Docking Poses Generated by CHARMMing’s Fragment-Based Docking Protocol and Glide SP and success Rates (defined by the percentage of the ligands whose reported RMSD is below 2.0 Å)a.
| PDB ID | resolution (Å) | best RMSD (Å) | Glide SP RMSD (Å) |
|---|---|---|---|
| 1A4Q | 1.90 | 2.61 | 3.30 |
| 1A6W | 2.00 | 1.01 | 6.72 |
| 1AOE | 1.60 | 3.13 | 1.80 |
| 1AQW | 1.80 | 1.88 | 0.96 |
| 1ATL | 1.80 | 1.68 | 1.09 |
| 1BMA | 1.80 | 2.76 | 1.55 |
| 1D3H | 1.80 | 0.99 | 0.81 |
| 1FCZ | 1.38 | 1.06 | 0.31 |
| 1GLQ | 1.80 | 4.71 | 1.01 |
| 1HFC | 1.50 | 2.63 | 2.36 |
| 1HVR | 1.80 | 3.84 | 0.75 |
| 1JAP | 1.82 | 1.41 | 0.92 |
| 1KE5 | 2.20 | 1.07 | 1.75 |
| 1MLD | 1.83 | 1.29 | 1.07 |
| 1MMQ | 1.90 | 0.50 | 0.30 |
| 1MTS | 1.90 | 1.96 | 0.54 |
| 1MVC | 1.90 | 0.29 | 0.94 |
| 1NHZ | 2.30 | 0.78 | 1.89 |
| 1NQ7 | 1.50 | 0.94 | 1.26 |
| 1QBR | 1.80 | 9.31 | 0.98 |
| 1SRJ | 1.80 | 1.33 | 0.51 |
| 1TXI | 1.90 | 1.66 | 1.64 |
| 3ERT | 1.90 | 0.71 | 1.59 |
| 4DFR | 1.70 | 1.66 | 10.48 |
| Success Rate: | 71% | 84% | |
“Best RMSD” refers to the pose closest to the crystal structure. Glide SP RMSD is of the top scoring pose of Glide’s standard precision docking.
The fragment-based approach that was implemented into CHARMMing yields a substantial amount of information about the characteristics of each docked pose. At each step, from decomposition to minimization of docked poses, users have the ability to closely analyze results. The binding modes of each individual fragment can be inspected, and a number of modifiable parameters, such as decomposition criteria, can be used to optimize the protocol. Moreover, information gained from docking a fragment library into a particular target can be used to mine large libraries for compounds containing those fragments that form the most favorable interactions with the target.67−69
There are potentially a number of improvements that can be made to improve the performance and usability of CHARMMing’s docking protocol. The most obvious limitation is the current requirement of three fragments to be used as anchors. As shown by the number of ligands eliminated from the original benchmarking set, this limits the applicability of this protocol in its current form to medium- to large-sized molecules with a sufficient number of rotatable bonds. Although partially this problem can be alleviated by decreasing the fragment richness threshold at the decomposition step, this will only increase the “eligibility” rate of molecules by a small margin. Alternatively, when docking these small and/or rigid molecules is desired, the decomposition step could be omitted, at which point the molecules would undergo docking only by SEED. This however will require prior conformation sampling step as SEED currently does not sample the internal conformation of docked fragments. The conformational sampling of the fragments is an obvious improvement to the docking protocol even in its current state. This addition will help ensure that larger fragments sample their orientations within the binding site while varying their internal geometry, thus ensuring greater enrichment of anchor positions for the final ligand placement. Efforts to incorporate these functionality improvements are currently underway.
Conclusions
We have implemented a fragment-based docking protocol into the CHARMMing Web interface. The protocol allows users to perform docking and virtual screening calculations online as well as generates self-contained scripts to execute these in parallel on local HPC resources. The performance of the docking protocol was evaluated by carrying out a series of redockings and comparing the results against a top commercial docking package. The fragment-based docking protocol yielded results comparable to both the commercial package used herein and a wide variety of additional docking software. Specifically, the rate of recovering the correct X-ray pose with CHARMMing’s protocol was 71%, well within the 40–90% range that numerous benchmarking studies have reported.
While the scoring function can still be improved, the tool lays substantial groundwork for allowing academic laboratories to set up and perform molecular docking and virtual screening studies. It is important to note that the protocol is able to create CHARMM-formatted protein–ligand systems giving users the ability to access the wide range of functionality that exists in CHARMM. For example, docked poses can easily be refined with MD simulations, and predocked proteins can be coupled with simulations or normal-mode analysis to proceed via an ensemble docking approach. These, in addition to other improvements are currently being developed.
Acknowledgments
H.L.W. would acknowledges the NIH (1K22HL088341-01A1), DOE (DE-SC0011297TDD), NSF (CHE1156853) and the University of South Florida (start-up) for funding. Computations were performed at the USF Research Computing Center. The authors thank Dr. Sandra Rennebaum for her assistance with troubleshooting docking programs used in the protocols, Dr. Peter Kolb for help with DAIM, Prof. Alex MacKerell and Dr. Kenno Vanommeslaeghe for assistance with small molecule atom-typing and access to the Maybridge library. Finally, we especially thank Mr. Tim Miller, Dr. Bernard Brooks, and the National Heart, Lung, and Blood Institute of the NIH for support and collaboration.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Bello M.; Martínez-Archundia M.; Correa-Basurto J. Automated docking for novel drug discovery. Expert Opin. Drug Discovery 2013, 8, 821–834. [DOI] [PubMed] [Google Scholar]
- Moors E. H. M.; Cohen A. F.; Schellekens H. Towards a sustainable system of drug development. Drug Discovery Today 2014, 1–10. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. The case for open-source software in drug discovery. Drug Discovery Today 2005, 10, 213–217. [DOI] [PubMed] [Google Scholar]
- Geldenhuys W. J.; Gaasch K. E.; Watson M.; Allen D. D.; Van der Schyf C. J. Optimizing the use of open-source software applications in drug discovery. Drug Discovery Today 2006, 11, 127–132. [DOI] [PubMed] [Google Scholar]
- Goodsell D. S.; Olson A. J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [DOI] [PubMed] [Google Scholar]
- Lill M. A.; Danielson M. L. Computer-aided drug design platform using PyMOL. J. Comput.-Aided. Mol. Des. 2011, 25, 13–19. [DOI] [PubMed] [Google Scholar]
- Maestro; Schrödinger, LLC: New York, 2013. [Google Scholar]
- Discovery Studio Modeling Environment; Accelrys Software, Inc.: San Diego, CA, 2013.
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group, Inc.: Montreal, Quebec, Canada, 2013.
- Ebejer J.-P.; Fulle S.; Morris G. M.; Finn P. W. The emerging role of cloud computing in molecular modelling. J. Mol. Graph. Model. 2013, 44, 177–187. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. L.; Brooks C. L. Validating CHARMM parameters and exploring charge distribution rules in structure-based drug design. J. Chem. Theory Comput. 2009, 5, 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Zoete V.; Cuendet M. A.; Grosdidier A.; Michielin O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [DOI] [PubMed] [Google Scholar]
- Miller B. T.; Singh R. P.; Klauda J. B.; Hodoscek M.; Brooks B. R.; Woodcock H. L. CHARMMing: A new, flexible web portal for CHARMM. J. Chem. Inf. Model. 2008, 48, 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar]
- Brooks B. R.; Brooks C. L.; Mackerell A. D.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. T.; Singh R. P.; Schalk V.; Pevzner Y.; Sun J.; Miller C. S.; Boresch S.; Ichiye T.; Brooks B. R.; Woodcock H. L. III Web-based computational chemistry education with CHARMMing I: Lessons and tutorial. PLoS Comput. Biol. 2014, 10, e1003719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard F. C. IV; Miller B. T.; Schalk V.; Lerner M. G.; Woodcock H. L. III; Brooks B. R. Web-based computational chemistry education with CHARMMing II: Coarse-grained protein folding. PLoS Comput. Biol. 2014, 10, e1003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin B. S. Jr; Miller B. T.; Schalk V.; Woodcock H. L.; Brooks B. R.; Ichiye T. Web-based computational chemistry education with CHARMMing III: Reduction potentials of electron transfer proteins. PLoS Comput. Biol. 2014, 10, e1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner Y.; Santiago D. N.; Fries J. S.; Metcalf R. S.; Daniel K. G.; Calcul L.; Woodcock H. L.; Baker B. J.; G W. C.; Brooks W. H. Virtual target screening to rapidly identify potential protein targets of natural products in drug discovery. AIMS Mol. Sci. 2014, 1, 49–66. [Google Scholar]
- Python, Python Software Foundation. www.python.org (accessed August 27, 2014).
- Django, Django Software Foundation. http://www.djangoproject.com (accessed August 27, 2014).
- MySQL; Oracle Corporation: Redwood Shores, CA, 2014.
- MacKerell A. D.; Bashford D.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Mackerell A. D.; Feig M.; Brooks C. L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. Automation of the CHARMM general force field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Automation of the CHARMM general force field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesselman J. D.; Price D. J.; Knight J. L.; Brooks C. L. MATCH: An atom-typing toolset for molecular mechanics force fields. J. Comput. Chem. 2012, 33, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
- Hodoscek M.GENRTF, 2008. http://code.google.com/p/genrtf/ (accessed August 27, 2014).
- Meng E. C.; Shoichet B. K.; Kuntz I. D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 2149–2159. [DOI] [PubMed] [Google Scholar]
- Wu G.; Robertson D. H.; Brooks C. L.; Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [DOI] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Caflisch A. Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking. J. Med. Chem. 2006, 49, 7384–7392. [DOI] [PubMed] [Google Scholar]
- Majeux N.; Scarsi M.; Caflisch A. Efficient electrostatic solvation model for protein-fragment docking. Proteins 2001, 42, 256–268. [DOI] [PubMed] [Google Scholar]
- Budin N.; Majeux N.; Caflisch A. Fragment-based flexible ligand docking by evolutionary optimization. Biol. Chem. 2001, 382, 1365–1372. [DOI] [PubMed] [Google Scholar]
- Dey F.; Caflisch A.. FLEA – FFLD Leader Clustering. http://www.biochem-caflisch.uzh.ch/download/ (accessed August 27, 2014).
- Charifson P. S.; Corkery J. J.; Murcko M. A.; Walters W. P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [DOI] [PubMed] [Google Scholar]
- Ece A.; Sevin F. The discovery of potential cyclin A/CDK2 inhibitors: A combination of 3D QSAR pharmacophore modeling, virtual screening, and molecular docking studies. Med. Chem. Res. 2013, 22, 5832–5843. [Google Scholar]
- Ji X.; Zheng Y.; Wang W.; Sheng J.; Hao J.; Sun M. Virtual screening of novel reversible inhibitors for marine alkaline protease MP. J. Mol. Graph. Model. 2013, 46, 125–131. [DOI] [PubMed] [Google Scholar]
- Houston D. R.; Walkinshaw M. D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [DOI] [PubMed] [Google Scholar]
- Friedman R.; Caflisch A. Discovery of plasmepsin inhibitors by fragment-based docking and consensus scoring. ChemMedChem. 2009, 4, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Klon A. E.; Glick M.; Davies J. W. Combination of a naive Bayes classifier with consensus scoring improves enrichment of high-throughput docking results. J. Med. Chem. 2004, 47, 4356–4359. [DOI] [PubMed] [Google Scholar]
- OpenPBS, PBS Works. www.openpbs.org (accessed August 27, 2014).
- TORQUE, Cluster Resources, Inc. (Adaptive Computing, Inc.).http://www.clusterresources.com (accessed August 27, 2014).
- Hanson R. M.; Prilusky J.; Renjian Z.; Nakane T.; Sussman J. L. JSmol and the next-generation Web-based representation of 3D molecular structure as applied to proteopedia. Isr. J. Chem. 2013, 53, 207–216. [Google Scholar]
- Nakane T. GLmol – Molecular Viewer on WebGL/Javascript, version 0.47. http://webglmol.sourceforge.jp/index-en.html (accessed August 27, 2014).
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Nissink J. W. M.; Murray C.; Hartshorn M.; Verdonk M. L.; Cole J. C.; Taylor R. A new test set for validating predictions of protein–ligand interaction. Proteins 2002, 49, 457–471. [DOI] [PubMed] [Google Scholar]
- Cross J. B.; Thompson D. C.; Rai B. K.; Baber J. C.; Fan K. Y.; Hu Y.; Humblet C. Comparison of several molecular docking programs: Pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [DOI] [PubMed] [Google Scholar]
- Small-Molecule Drug Discovery Suite 2014–1: Glide; Schrödinger, LLC: New York, 2014. [Google Scholar]
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided. Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Epik; Schrödinger, LLC: New York, 2013.
- Impact; Schrödinger, LLC: New York, 2013.
- Prime; Schrödinger, LLC: New York, 2014.
- Protein Preparation Wizard, Schrödinger Suite 2013; Schrödinger, LLC: New York, 2013.
- Perola E.; Walters W. P.; Charifson P. S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. [DOI] [PubMed] [Google Scholar]
- Srivastava H. K.; Chourasia M.; Kumar D.; Sastry G. N. Comparison of computational methods to model DNA minor groove binders. J. Chem. Inf. Model. 2011, 51, 558–571. [DOI] [PubMed] [Google Scholar]
- Neale D. S.; Thompson P. E.; White P. J.; Chalmers D. K.; Yuriev E.; Manallack D. T. Binding mode prediction of PDE4 inhibitors: A comparison of modelling methods. Aust. J. Chem. 2010, 63, 396–404. [Google Scholar]
- Shoichet B. K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Kipouros C. B.; Huang D.; Caflisch A. Structure-based tailoring of compound libraries for high-throughput screening: discovery of novel EphB4 kinase inhibitors. Proteins 2008, 73, 11–18. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Dong J.; Lafleur K.; Nevado C.; Caflisch A. Discovery of a novel chemotype of tyrosine kinase inhibitors by fragment-based docking and molecular dynamics. ACS Med. Chem. Lett. 2012, 3, 834–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Gartenmann L.; Dong J.; Spiliotopoulos D.; Caflisch A. Discovery of BRD4 bromodomain inhibitors by fragment-based high-throughput docking. Bioorg. Med. Chem. Lett. 2014, 24, 2493–2496. [DOI] [PubMed] [Google Scholar]