

Abstract
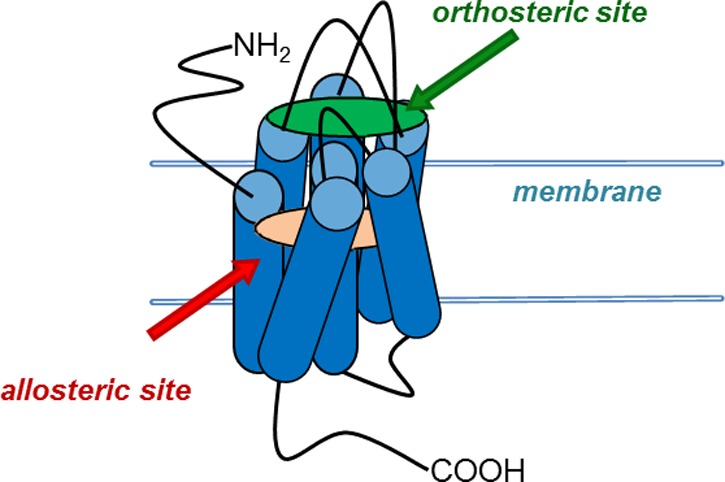
The identification of sites on receptors topographically distinct from the orthosteric sites, so-called allosteric sites, has heralded novel approaches and modes of pharmacology for target modulation. Over the past 20 years, our understanding of allosteric modulation has grown significantly, and numerous advantages, as well as caveats (e.g., flat structure–activity relationships, species differences, “molecular switches”), have been identified. For multiple receptors and proteins, numerous examples have been described where unprecedented levels of selectivity are achieved along with improved physiochemical properties. While not a panacea, these novel approaches represent exciting opportunities for tool compound development to probe the pharmacology and therapeutic potential of discrete molecular targets, as well as new medicines. In this Perspective, in commemoration of the 2013 Philip S. Portoghese Medicinal Chemistry Lectureship (Lindsley C. W.Adventures in allosteric drug discovery. Presented at the 246th National Meeting of the American Chemical Society, Indianapolis, IN, September 10, 2013; The 2013 Portoghese Lectureship), several vignettes of drug discovery campaigns targeting novel allosteric mechanisms will be recounted, along with lessons learned and guidelines that have emerged for successful lead optimization.
I. Introduction: Background on Allosteric Modulation. Novel Approaches for Therapeutics
While the first concepts regarding allosterism were put forth in the 1960s, only in the past decade, with advances in molecular pharmacology and functional screening technology, has the impact of this alternative approach for target modulation been realized.2−12 Indeed, the discovery of topologically distinct allosteric (from the Greek as “other site”) binding sites for a diverse range of receptor and protein families (GPCRs, ion channels, caspases, kinases, and phospholipases) has provided unparalleled opportunities to obtain druggable small molecules with exquisite selectivity and unique pharmacological profiles.2−12 Here, an allosteric ligand binds the target at a topographically distinct allosteric site and either potentiates or inhibits the binding and/or signaling of an orthosteric ligand by taking advantage of conformational flexibility of the receptor and/or protein.2−12 The clinical success and safety of benzodiazepines (BZDs) 1–3 (Figure 1), the first allosteric modulator drugs, which potentiate the effect of γ-aminobutyric acid (GABA) at the ionotropic GABAA receptor are in direct opposition to the adverse and potentially lethal effects of orthosteric GABAA agonists.4,11,13 Further exploration within the BZD class elucidated multiple modes of allosteric pharmacology: positive allosteric modulators (PAMs), which potentiate GABAA receptor response, negative allosteric modulators (NAMs), which decrease channel activity, and silent allosteric modulators (SAMs, or no affect ligands, NALs) that bind to the allosteric site and block both PAM and NAM activity without any effect on receptor signaling alone.4,11,13 These data fueled the concept of allosteric modulation in modern drug discovery leading to the identification of allosteric modulators for other ion channels, kinases, phospholipases, and G-protein-coupled receptors (GPCRs).11,13,14 Moreover, multiple allosteric modulators are now in various stages of clinical development11,13,14 as well as marketed therapeutics (cinacalcet, 4, a calcium sensing receptor PAM, and maraviroc, 5, a CCR5 NAM).15,16
Figure 1.

The first allosteric modulators with clinical success were benzodiazepines (BZDs), GABAA PAMs. The generic BZD core 1 and important medications 2 (Valium) and the tricylic analog 3 (Xanax) are shown. Also shown are structures of the two marketed GPCR allosteric modulators: cinacalcet (4), a calcium sensing receptor PAM, and maraviroc (5), a CCR5 NAM.
Over the past 13 years, our laboratories at Merck and within the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD)17 have pioneered allosteric modulation as a pharmacological approach to modulate kinases, GPCRs, ion channels, and phospholipases,11,13,14 and we have introduced a plethora of important small molecule tools for use by the biomedical research community (via the VCNDD and the Molecular Libraries Probe Center Network, or MLPCN).17,18 Clearly, allosteric ligands afford unprecedented selectivity (by targeting evolutionary less conserved binding sites), enhanced chemical tractability, and improved physiochemical properties.2−12 In the course of our research programs, we have encountered numerous caveats surrounding allosteric ligand pharmacology and chemical optimization (ligand bias, species differences, “molecular switches”, flat SAR, the “fluorine walk”) for which we have developed guidelines and strategies to enhance the odds of a successful lead optimization campaign.2−12,14 These general concepts have all been extensively reviewed elsewhere;11,12,14 thus, this Perspective will focus on the defining allosteric modulator programs that gave rise to these principles along with programs that transitioned from conceptual preclinical postulates into human clinical trials.
II. Allosteric Modulation of Kinases: The Case of Akt
Protein phosphorylation is a ubiquitous cellular signaling process mediated by the action of kinases, and dysfunction within this system gives rise to numerous human diseases from diabetes to cancer.19−21 Despite the presence of >500 kinases in humans and a highly conserved orthosteric ATP (6) binding site, numerous small molecule drugs have received FDA approval; however, selectivity versus the kinome remains a major challenge, especially for kinase family with multiple isoforms/isoenzymes.22,23 Today, we recognize four major types of kinase inhibitors: type I (classical ATP mimetics), type II, (ATP-site binders that extend into an adjacent allosteric site), type III (bind exclusively at an allosteric site, near the ATP-site), and type IV (bind exclusively at an allosteric site, distinct and remote from the ATP-site).24 Type IV inhibitors pharmacologically can induce structural reorganization, stabilize inactive conformations, prevent substrate recognition, induce degradation, and/or occupy autoinhibitory sites by targeting a limited number of inactive kinase conformations, such as disruption of a conserved αC-helix.25−28 To date, allosteric kinase inhibitors have been developed for Akt, Abl, JNK1, mTOR, CDK2, CHK1, IGF-1R, and PDK1, which possess chemotypes distinct from classical ATP mimetics and excellent selectivity versus the kinome.29 However, with respect to a nascent Akt kinase inhibitor program at Merck in early 2001, this was virgin territory and to pursue a novel, nontraditional (not ATP-competitive ligand) approach, was viewed with skepticism.
In 2001, the modulation of discrete signaling targets in PTEN/PI3K pathway was a major focus for oncology drug discovery.30 At this time, Akt (PKB), a serine/threonine kinase in the AGC family of kinases (e.g., PKA, PKC, SGK), was known to be an oncogene, and an attractive kinase target though small molecule ligands were classical ATP-competitive, typified by the pan-kinase inhibitor staurosporine 7 or lipid analogs such as 8 (Figure 2).30−32 In mammals, there are three isoforms/isoenyzmes of Akt (Ak1, Akt2, and Akt3) with different physiological roles that share greater than 85% homology (>95% at the ATP pocket). Structurally, each Akt isoform contains an N-terminal pleckstrin homology (PH) domain, a kinase domain, and a C-terminal regulatory domain. Akt was known to be comformationally flexible existing in the cytoplasm in a closed, inactive PH-in conformation, where the PH domain shields the ATP binding pocket and blocks phosphorylation of Ser473/T308.30−37 Upon PIP3 recruitment to the plasma membrane, Akt then adopts a PH-out conformation that exposes Ser473/T308 for phosphorylation by PDK1 and mTORC2 (in 2001, it was believed to be a putative PDK2).30−37 Could this unique conformational flexibility of Akt engender allosteric sites that could effectively “lock” Akt into an inactive (PH-in) conformation and thus inhibit both the activity and the activation of Akt (Figure 3A)? Beyond this critical question, the objectives for this nascent project involved the development of small molecule tools to assess the apoptotic response of selective inhibition of Akt1, Akt2, and dual Akt1/2 inhibition, and once clear, the development of an oral Akt clinical candidate for the treatment of cancer. Notwithstanding the issues surrounding kinome selectivity, also developing Akt inhibitors with isoform selective inhibition by targeting the ATP-binding site seemed improbable and unlikely, as no selective Akt kinase inhibitors were known, and no one had yet attempted to achieve isoform selective inhibition of Akt. Thus, we chose a different and unprecedented, for that time, approach.
Figure 2.

Structures of adenosine triphosphate (ATP) 6, the prototypical ATP-competitive, pan-kinase inhibitor staurosporine 7, and a PIP lipid analog 8. In 2001, 6 and 7 were examples of known Akt inhibitors, but neither were selective versus the kinome or other PH-domain containing proteins.
Figure 3.

Akt biology and allosteric inhibitors. (A) Loss of function mutations or deletions in PTEN leads to high levels of PIP3 which recruits Akt from the cytoplasm, where it exists in an inactive, PH-in conformation to the plasma membrane, where the PH domains binds to PIP3 leading to a PH-out conformation exposing T308 and Ser473 for phosphorylation by PDK1 and mTORC2, and active Akt. Inset: a model by which an allosteric inhibitor could stabilize the PH-in conformation, thus blocking both the activity and activation of Akt. Detailed mutagenesis, biochemical, and later X-ray studies confirmed that allosteric Akt inhibitors 9–13 adhere to this model of inhibition. (B) HTS lead 9 was found to be the first allosteric inhibitor of Akt. An iterative parallel synthesis and MAOS approach (TES) delivered key tool compounds with unprecedented selectivity for Akt1 (10), Akt2 (11), or both Akt1 and Akt2 (12). Subsequent intense medicinal chemistry provided MK-2206 (13) an orally bioavailable pan-allosteric Akt inhibitor that advanced into clinical trials and displayed efficacy against solid tumors in phase II.
A high-throughput screen (HTS) was performed, and as expected, the “usual suspects” (ATP mimetics) resulted. An exceptional biochemist on the program, Stanley Barnett, then performed ATP kinetics with the HTS hits and found a lone compound 9, a functionalized quinoxaline, that was noncompetitive with ATP and was dissimilar from classical ATP-competitive ligands (Figure 3B).38 Moreover, 9 was active at Akt1 and Akt2 but inactive (IC50 values of >250 μM) at Akt3 or against Akt 1 and Akt2 mutants lacking the PH domain as well as PKA, PKC, and SGK. Moreover, 9 inhibited both the activity and the activation of Akt. These data strongly suggested that the binding site for 9 resided within the PH domain or required the PH domain, thus suggesting an allosteric mode of inhibition.38 Extensive mutagenesis work and biochemical studies supported a model wherein 9 was a two-site, allosteric binder, with a high affinity site in the PH domain, inducing a conformational change to a second site in the catalytic domain, locking Akt into a closed, PH-in conformation.38 As this was a nascent program, chemistry support was limited to a single medicinal chemist, but significant optimization of 8 was required to meet the program objectives. Here, we developed the technology enabled synthesis (TES) approach of iterative parallel synthesis and fragment libraries, coupled with microwave-assisted organic synthesis and mass-directed preparative LCMS, to leverage technology for accelerating lead optimization with limited human resources.39 In short order, we had identified a key piperidine benzimidazole moiety that significantly enhanced Akt activity along with cores that afforded the required Akt1-selective 10, Akt2-selective 11, and dual Akt1/2 selective inhibitors 12.36 Like 9, 10–12 were PH-domain dependent inhibitors, noncompetitive with ATP, and allosteric. Early proof-of-concept studies demonstrated that inhibition of both Akt1 and Akt2 was required for maximal apoptotic effect and that 12 could inhibit both basal and IGF-stimulated Akt1 and Akt2 phosphorylation in mouse lung.36 Effort then focused on development of a clinical candidate32 and addition of a larger medicinal chemistry team that ultimately delivered MK-2206 (13), the first allosteric, oral Akt kinase inhibitor that enhanced antitumor efficacy by standard chemotherapeutics and was efficacious in patients with solid tumors in phase II (Figure 3B).40 Years later, the proposed mechanism of Akt inhibition was confirmed both by X-ray crystallography, whereby a cocrystal of Akt and 12 illustrated that 12 binds in a hydrophobic pocket formed by residues within the PH and kinase domains, and by fluorescence resonance energy transfer (FRET) data showing that 12 locked Akt into a PH-in conformation, preventing phosphorylation of S473 and T308.14,41 Serendipitously, the allosteric approach with 13 also enabled our project team to side-step a major issue only recently disclosed. Upon binding, ATP-competitive Akt inhibitors induce hyperphosphorylation engendering regulatory phosphorylation; in contrast, 13 inhibits drug-induced hyperphosphorylation,42 thus increasing the therapeutic window noted for 13 and newer back-up compounds from Merck. Overall, this first venture into allosteric drug space was fruitful and demonstrated the value of a commitment to basic science and novel approaches for target modulation to advance into the clinic and paved the way for a career focused on allosteric mechanisms of target modulation.
III. Allosteric Modulation of GPCRs
G-protein-coupled receptors, also referred to as seven-transmembrane receptors (7TMRs), account for >50% of all known drugs, and with the exception of 4(15) and 5,16 the remaining FDA-approved drugs bind at the orthosteric site and modulate receptor function by blocking the action of the native agonist (competitive antagonism), inhibition of constitutive activity (inverse agonism), or direct activation (agonism).2−12,14 The historical reason for this trend lies in the assays employed in their discovery: radioligand displacement assays that targeted the orthosteric site.2−12,14 Despite this success, many GPCRs are still without ligands and selectivity remains an issue, with desensitization resulting from prolonged activation, and based on the nature of the orthosteric ligands (neurotransmitters (family A), amino acids (family C), and large peptides (family B)), ligands often possess poor physiochemical properties (especially for CNS targets). With the advent of high throughout functional assays, it became possible to identify ligands that modulate GPCR function without regard to the binding site, which heralded the dramatic growth of allosteric modulators and a new frontier in pharmacology.2−12,14 Like the BZDs,13 GPCRs allosteric modulators can act as PAMs, NAMs, NALs (SAMs), ago-PAMs, partial antagonists, and even allosteric agonists (activating the GPCR in the absence of native agonist). It has been demonstrated time and again the advantages of allosteric modulation: (1) both subtype and overall selectivity, (2) maintenance of activity dependence (state dependence), (3) temporal and spatial aspects of endogenous physiological signaling, (4) less densensitization, and (5) fewer side effects. However, there are also challenges: (1) steep shallow SAR, (2) species differences (due to less evolutionary conservation of allosteric sites), (3) signal bias, (4) “molecular switches”, (5) allosteric ligands that can act at multiple distinct, overlapping, and nonoverlapping sites on the same receptor, and (6) the impact of homo- versus heterodimer pharmacology.2−12,14 This field has been extensively reviewed in a Journal of Medicinal Chemistry Perspective11 as well as in multiple other venues;2−10,12,14 therefore, this section will serve to highlight the GPCR allosteric modulator programs from our laboratories that helped establish this field and defined guiding principles (Chart 1) for allosteric ligand optimization.
Chart 1.

Once again, we return to Merck in 2001, where Jeff Conn was the Director of Neuroscience and my medicinal chemistry group was supporting his nascent programs targeting metabotropic glutamate receptors (mGlus) where the orthosteric agonist is glutamate (14).43 At this time, mGlu5 NAMs, such as MPEP (15), were well-known (Figure 4),44 and Dr. Conn postulated that mGlu5 PAMs should then be pharmacologically possible. If so, PAMs could avoid the epileptiform activity of mGlu5 agonists43 and represent a potential novel therapeutic mechanism, via the NMDA hypofunction hypothesis,45 to improve cognition and treat schizophrenia.
Figure 4.
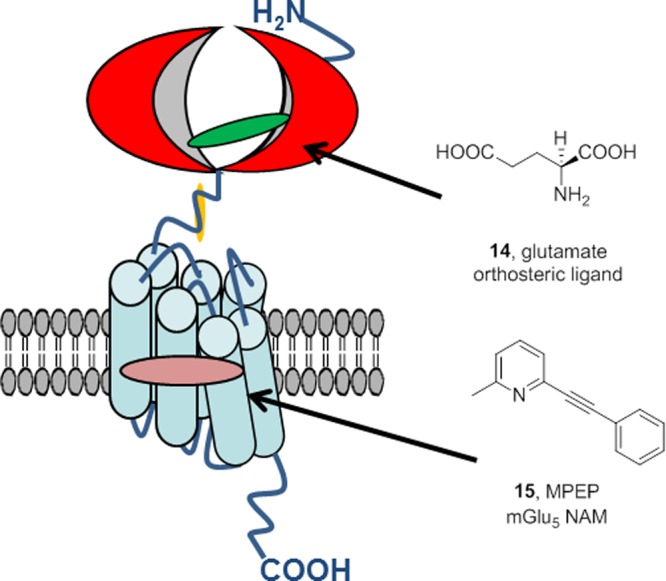
Cartoon of the metabotropic glutamate receptor subtype 5 (mGlu5) showing the structure and binding site of the endogenous agonist glutamate (14) in the extracellular Venus fly trap domain and the prototypical allosteric ligand, MPEP (15) an mGlu5 NAM, that binds within the transmembrane domain.
However, there were no reports of mGlu5 PAMs, and management was not supportive of an HTS campaign. Therefore, to initiate a PAM effort, we examined the historical mGlu5 NAM HTS data and looked for compounds that potentiated the glutamate EC80 (as opposed to the classical PAM EC20 screening paradigm). Despite “noisy” data, we identified two compounds worthy of follow-up. The first was a benzaldazine series represented by DFB (16), the first mGlu5 PAM (EC50 = 2.6 μM), with the anticipated PAM pharmacology and was shown to bind at the well-characterized MPEP NAM site (Figure 5).46 SAR around 16 was surprising in that subtle changes led to an equipotent mGlu5 NAM (17) and 18, the first mGlu5 SAM (now termed a NAL).46 At this point, we had yet to see this as a general phenomenon which we later (in 2008) coined “molecular switches”,47−49 but rather it was viewed as an anomaly. Interestingly, the other hit 19 also behaved as a PAM and potentiated NMDA receptor currents, but pharmacological characterization indicated that it, as well as an advanced mGlu5 PAM in this series CPPHA (20),50,51 did not bind at the MPEP site, thus providing evidence that there are two allosteric sites on mGlu5 that can enable positive allosteric modulation of the receptor. In parallel to the iterative parallel synthesis that delivered 20, we also performed fragment libraries around 20 that led to CDPPB (21), the first centrally active mGlu5 PAM that validated this novel mechanism in preclinical models of schizophrenia and cognition.52,53 Surprisingly, 21 was shown to bind at the MPEP site, and many other laboratories utilized 21 to demonstrate antipsychotic-like effects in multiple animal models as well as enhancement of synaptic plasticity and cognitive function. However, Merck elected to stop the mGlu5 PAM program in favor of other schizophrenia programs.
Figure 5.

The first mGlu5 PAMs 16 and 20 and the subsequent optimization that led to the discovery of two distinct allosteric sites on mGlu5 that can potentiate receptor function and the key in vivo proof of concept mGlu5 PAM 21 that validated the mechanism and pharmacological approach for treatment of multiple symptom clusters of schizophrenia.
After these initial reports, multiple companies and academic laboratories initiated mGlu5 PAM programs, and now there are over 40 distinct chemotypes reported as mGlu5 PAMs, both for the MPEP and for the non-MPEP binding sites, with increasing examples of therapeutic efficacy.54 Upon moving to Vanderbilt to once again partner with Jeff Conn and establish the VCNDD,17 we initiated a new mGlu5 PAM project with a full “triple add” HTS campaign,2−12,14 which afforded a plethora of fundamentally new mGlu5 PAM and NAM leads. Key to our new Center was to have a dedicated, NIH-funded basic science effort to parallel the drug discovery effort that generated in vivo tool compounds that enabled a “deep dive” into target biology to inform the drug discovery teams. Shortly after developing patented mGlu5 PAMs, we partnered the program with Janssen and quickly developed a broad intellectual property suite en route to a new medical entity (NME), while our basic science team provided keen insights into the biology of mGlu5 potentiation. For example, we identified mGlu5 PAMs that closely resembled MPEP, yet did not bind in a competitive manner with the MPEP binding site, leading us to realize that PET tracers for the program had to be generated from within the exact same scaffold as the candidate to ensure translational utility (Chart 1).54 Furthermore, a critical observation with MPEP site ligands, as an extension to the early observations with 16–18,46 was a general phenomenon we coined “molecular switches”.47−49 Here, we noted that the mode of pharmacology of an mGlu5 partial antagonist 22 could be switched to a PAM 23 by the addition of a methyl group in the 4-position of the distal phenyl ring (Figure 6). The addition of a 2-aminomethyl group to the 2-position of the pyrimidine provided 24, a potent mGlu5 PAM that displayed in vivo efficacy in antipsychotic models.48 All other structural modifications to 22 engendered full mGlu5 NAM activity. Other series proved impervious to “molecular switches”, and these series were more favorable as leads toward an NME. As we will discuss later, the phenomenon of “molecular switches” is not only limited to mGlu5 or class C GPCRs but is also prevalent with allosteric ligands for class A and class B GPCRs as well.12,14,49
Figure 6.

“Molecular switches” within MPEP site mGlu5 allosteric ligands. Subtle structural modifications modulate the mode of pharmacology. 24 proved to be a potent PAM with in vivo activity in antipsychotic models, demonstrating the mode of pharmacology “switch” in vitro was mirrored in vivo.
The observation that incorporation of a small, polar moiety could engender a “switch” in the mode of pharmacology caused serious concern regarding the potential for CYP-mediated “molecular switches” from oxidative metabolism. Here, the academic effort with “tool compounds” informed the drug discovery team to judiciously characterize metabolites. About this time, Merck reinitiated an mGlu5 PAM program through a partnership with Addex Pharmaceuticals and had published a report on mechanism-based toxicity based on data within a series of mGlu5 PAMs, represented by 5PAM523 (25).55 In this study, fluorojade staining showed necrotic neurons in the auditory cortex and hippocampus (Figure 7); moreover, these findings, in part, led Merck to once again abandon their mGlu5 PAM program. We were aware of this, as well as the potential for neurotoxicity and seizure liabilities (known for group I mGlu agonists) with PAMs that possessed agonist activity, e.g., ago-PAMs. Once again, our academic, deep science effort realized the need for both high- and low-expressing mGlu5 cell lines as well as the need to have a native preparation, which was found in astrocytes (they natively express mGlu5). For PAMs such as VU0242465 (26) which displayed ago-PAM activity in both cell lines and also displayed mGlu5 agonism in astrocytes, we knew they potentiated long-term depression (LTD) alone and induced behavioral disturbances (seizures) when dosed in rats and were to be avoided. However, we were very surprised when VU0403602 (27), a pure PAM both in cell lines and in astrocytes, was found to induce significant seizure activity.56 Studies with MTEP demonstrated that the adverse seizure activity was mGlu5 mediated, with a full blockade of seizure activity with 10 mg/kg MTEP. On the basis of our concern of CYP-mediated “molecular switches” and the presence of a hydroxyl moiety in the amide of 26, we also pretreated rats with aminobenzotriazole (ABT), a pan-CYP inhibitor, prior to 27 and found that the seizure activity was completely suppressed and equivalent to 10 mg/kg MTEP. Thus, the seizures were mGlu5 mediated, most likely mGlu5 allosteric agonism, and the activity was due to a metabolite and not the parent 27. Metabolic identification studies then showed that the major metabolite M1 (28) was due to oxidation of the cyclobutyl moiety, and this secondary alcohol proved to be a potent mGlu5 allosteric agonist, a similar moiety to that found in 26. NOE studies and synthesis later demonstrated that there was also stereochemical bias in the “molecular switch” with the trans-isomer 29 possessing potent (EC50 = 400 nM) allosteric agonist activity, while the cis-isomer 30 remained a pure PAM (EC50 = 33 nM).56 Thus, this was the first reported example of an in vivo, CYP-mediated “molecular switch”, and subsequent allosteric modulator programs now closely monitor major metabolites and their pharmacology/selectivity.56
Figure 7.

mGlu5 PAMs and ago-PAMs leading to seizure liability and neurotoxicity. Pure PAM 27 also induced seizures, but studies found that a CYP-mediated “molecular switch” afforded a racemic secondary alcohol metabolite 28 that modulated the mode of pharmacology from pure PAM to allosteric agonist and gave rise to the adverse effect liability and not the parent.
Finally, the concept of stimulus/signal bias was well-known in the context of GPCR allosteric modulators, and in the case of mGlu5, potentiation of NMDA receptor currents represented the major adverse effect liability once allosteric agonism had been addressed. Under this principle, we evaluated several mGlu5 PAMs and found a compound that did not potentiate NMDA receptor currents directly, yet displayed robust efficacy, akin to mGlu5 PAMs that potentiated NMDA receptor currents, in preclinical rodent models of antipsychotic activity, potentiated hippocampal LTD and LTP and yet showed no acute adverse effects or short-term excito- or neurotoxicity (fluorojade staining) in rats, in contrast to 25;1 however, these results do not ensure an absence of toxicity in other species or upon higher and/or chronic dosing regimens.57 Nevertheless, the value of a strong basic science effort, operating in parallel to the drug discovery effort, afforded unique insights that enabled the delivery of an approved mGlu5 PAM NME in less than 3 years from the initiation of the collaboration.
It is important to note that the “molecular switch” phenomenon can also be beneficial.14,49 We and others have reported numerous examples where a small structural modification to a core has engendered unique, desired pharmacology or afforded access, through a change in subtype selectivity, to ligands for a receptor subtype that were previously unavailable.58−63 Figure 8 shows three recent examples of the latter. Taking advantage of the promiscuity of MPEP site mGlu5 PAM ligands, we noted that mGlu5 PAM 30 (mGlu5 EC50 = 270 nM) had weak activity as an mGlu3 NAM (mGlu3 IC50 > 10 μM) and felt this could be exploited to access the first selective mGlu3 NAMs.64 A diversity-oriented synthesis effort identified a “molecular switch”, again in the 4-position of the distal phenyl ring, in the form of a methoxy moiety. SAR was “flat” as anticipated, yet we were able to access VU0463597 (31, ML289) as the first highly selective mGlu3 NAM (mGlu3 IC50 = 650 nM, mGlu2 IC50 > 10 μM), which was devoid of mGlu5 activity (both PAM and NAM).64 Further lead optimization and utilization of the “fluorine walk” provided the more selective mGlu3 NAM probe VU06024017 (32, ML337); however, the key moiety that engender activity at mGlu3, the 4-methoxy group, was also the lone metabolic liability, undergoing a rapid oxidative dealkylation to an inactive congener.65 Steric and electronic modification to both the ether and the pendent aromatic ring led to a significant diminution in potency. Ultimately, this led to the introduction of another valuable tool in the allosteric modulator tool box: deuterium incorporation and reliance on the stronger C–D bond. Replacement of the OCH3 with a OCD3 (33) maintained the pharmacological profile, yet reduced in vitro and in vivo clearance by ∼50%, enabling 33 to serve as a valuable in vivo tool to study selective mGlu3 inhibition.65
Figure 8.

Beneficial “molecular switches”. (A) Delivery of the first highly selective mGlu3 NAM 31 by virtue of a 4-OMe “molecular switch” on an mGlu5 PAM scaffold. Subsequent optimization and “fluorine walk” provided 32, which was further optimized for DMPK properties by deuterium incorporation, as in 33, to overcome “flat” SAR. (B) The allosteric sites on mGlu4 and mGlu1 historically cross-talk, and taking advantage of a “double molecular switch”, an mGlu1 PAM with properties suitable for in vivo studies resulted. (C) An M1 HTS identified 37 as an M1, M3, M5 PAM. A “molecular switch” in the form of a 5-OCF3 group engendered selectivity for M5, while addition of small heterocycles to replace the 4-Br moiety afforded selective M1 PAMs.
Likewise, many mGlu4 PAMs and mGlu1 NAMs are known in the literature, but there are few mGlu1 PAMs and none with properties suitable for in vivo studies.66 It is well established since the first report of (−)-PHCCC as an mGlu4 PAM (and weak mGlu1 NAM) that there was cross-talk between the mGlu4 and mGlu1 allosteric sites.67 However, optimization efforts were able to separate these activities delivering highly selective mGlu4 PAMs, such as 34.68 Further chemical optimization and modification to the imide moiety provided 35, an equally potent and highly selective mGlu1 NAM via a “molecular switch”.69 Replacement of the imide in 35 with a phthalimide, as in 36, induced another “molecular switch”, in essence a “double molecular switch” from 34, leading to a highly selective mGlu1 PAM, with properties enabling dissection of an emerging role of GRM1 mutation in schizophrenia as well as in vivo studies (Figure 8B).70
Finally, this is not restricted to class C GPCRs or mGluRs. The phenomenon of “molecular switches” is prevalent across all families of GPCRs, where it has been instrumental in the development of highly subtype selective muscarinic acetylcholine (mAChR) ligands (Figure 8C). For instance, an HTS to identify M1 PAMs generated a unique hit: a pan-Gq coupled, M1, M3, M5-triple PAM 37 with equivalent low micromolar activity across the three subtypes.71,72 A matrix library (9 × 12), surveying nine functionalized isatins and 12 different benzyl moieties, uncovered a 5-OCF3 moiety as “molecular switch” that provided the first highly selective M5 PAM, 38 (ML129).71 Further optimization with iterative libraries and the “fluorine walk” afforded an N-Me pyrrole as a “molecular switch” leading to a highly selective M1 PAM, 39 (ML137).72 Both of these findings led to further optimization programs and high quality in vivo tools to probe M1 and M5 function in the CNS.
Before leaving GPCRs, it is important to discuss a related pharmacological approach that is often confused with PAMs: allosteric agonism. While PAMs require the orthosteric agonist and potentiate its activity, an allosteric agonist binds at a site distinct from the orthosteric site and is capable of activating the receptor in the absence of the orthosteric ligand.2−12 This approach has been largely isolated to the M1 mACh receptor, based on the clinical proof of concept studies with xanomeline (40), an M1/M4 preferring agonist, in patients with either Alzheimer’s disease or schizophrenia (Figure 9).73 However, like all mAChR M1 agonists developed in the 1990s, the true lack of subtype selectivity led to activation of peripheral M2/M3 receptors and adverse events precluded further development, despite efficacy noted in PhII trials.2−12 These data led to the quest for selective M1 and/or M4 allosteric agonists. Many weak partial and functionally selective M1 allosteric agonists were disclosed, and a prototypical example is TBPB (41);74 yet 41 and related congeners were found to be bitopic, meaning the ligand bound to an allosteric site on M1 that led to functionally selective M1 activation, but when screened in antagonist mode, they were full antagonists of the M2–M5, and in competition binding studies, as the concentration was increased, the orthosteric radioligand, [3H]NMS was fully displaced.60,74−76 Therefore, the “allosteric agonists” bind with high affinity at an allosteric site, but all also bind at the orthosteric site. This was further supported by modulating the basicity of the central piperidine ring of 41 with the β-F congener 42 and disrupting a key hydrogen bind at the allosteric site that led to pan-mAChR (M1-M5) antagonism.76 Finally, we demonstrated that two additional partial M1 allosteric agonists 43 and 44(75) displayed receptor reserve-dependent pharmacology in native brain tissue preparations that translated into in vivo behavioral outcomes.77 Thus, although there was early optimism with the approach, it became clear that allosteric agonists were actually a “second coming” of the M1 agonists of the 1990s, laden with unpredictable activity across brain regions and susceptibility to peripheral M2/M3 issues due to high receptor reserve and bitopic binding/pharmacology.77 Therefore, after years of effort and dedicated basic science, it became clear that M1 (e.g., 45 and 46)78,79 and M4 PAMs (e.g., 47 and 48)80,81 would be the desired pharmacological approach to achieve true mAChR subtype selectivity and advance into the clinic to validate the clinical data with 40. Preclinically, it is clear that M4 plays a larger role as an antipsychotic agent with modest cognition enhancing activity, whereas the reverse is true for M1.78−81 A program we initiated at Merck provided the first reported M1 PAM, BQCA (45), where all the principles discussed (iterative parallel synthesis, matrix libraries, and the “fluorine walk”) were critical in lead optimization and ultimately led to the development of MK-7622 (structure not officially disclosed) which is currently in phase II clinical trials, and efficacy data are eagerly awaited.
Figure 9.

In pursuit of a better xanomeline. Shown are structures of xanomeline (40) and subsequent M1 allosteric agonists 41–44, which demonstrated that all were bitopic with receptor reserve-dependent pharmacology (e.g., brain region-specific activity) and thus not tractable as a therapeutic approach. PAMs of either M1 (45 and 46) or M4 (47 or 48) have emerged as important in vivo tools to demonstrate that this mode of pharmacology is superior to allosteric agonism with true mAChR selectivity.
Finally, our group has contributed important small molecule allosteric modulators for other GPCRS including mGlu1 (PAMs and NAMs), mGlu3 (NAMs), mGlu4 (PAMs and NAMs), mGlu5 (PAMs and NAMs), pan group III mGluRs (PAMs), M1 (PAMs and allosteric agonists), M4 (PAMs), M5 (PAMs and NAMs), and even class B GPCRs such as GLP-1 (PAMs).2−12,14 These programs gave rise to the principles outlined in Chart 1 and referred to throughout this Perspective. While targeting allosteric sites has provided highly selective druglike compounds with good CNS exposure to understand the physiological underpinnings and therapeutic potential of many discrete GPCRs, it is not a panacea. Judicious evaluation of species differences must be adhered to along with the “triple add” primary screen to capture molecular switches, which carries forward into DMPK to understand the pharmacology of metabolites. Furthermore, stimulus bias can be beneficial as well as harmful; therefore, pharmacological characterization across a broader panel of assays to assess stimulus bias must be performed on HTS lead series and prior to focusing on a given scaffold. Although insights from mutagenesis work as to the origin of “molecular switches” have been helpful, the surge of new X-ray crystal structures of relevant GPCRS, some with allosteric ligands bound,82 is very exciting and will hopefully pave the way for a greater understanding of allosteric SAR.
IV. Allosteric Modulation of Phopsphodiesterases: The Case of Phospholipase D (PLD)
Phospholipase D (PLD) is a phospholipase that catalyzes the production of choline and phosphatidic acid (PA), an important lipid second messenger involved in a myriad of critical signaling and metabolic pathways by the hydrolysis of phosphatidylcholine.83 Mammals possess two isoforms of PLD, termed PLD1 and PLD2 (sharing 53% homology), both of which are differentially regulated and possess independent physiological roles. Structurally, PLD, like Akt, is a highly flexible enzyme that consists of a highly conserved active site (composed of two histidine-lysine-aspartate amino acid domains) and both N-terminal phox homology (PX) and PH regulatory domains.83 Extensive biochemical data and genetic studies have implicated dysfunction in PLD signaling and/or PLD overexpression in cancer, viral infection, and CNS disorders; however, the therapeutic potential of modulating PLD function remained elusive for over 20 years since its discovery because of a lack of selective and PLD isoform specific small molecules. Indeed, the field has been driven by the use of n-butanol (49), which competes for water in a transphosphatidylation reaction with water (Figure 10).83
Figure 10.

Development of allosteric, isoform selective PLD inhibitors 51–54, utilizing DOS, iterative parallel synthesis, the “fluorine walk” and “molecular switches”, all of which were borrowed from the allosteric GPCR field.
A 2007 short report from Novartis described halopemide (50), an atypical antipsychotic agent, and a collection of 12 related congers as PLD inhibitors.84 Upon recognition of clinical trial data with 50, wherein both isoforms of PLD were inhibited in man with normal biochemistry and without adverse events generated great interest in a field lacking small molecule tools.85 Because of the presence of a PH domain in PLD and the piperdine benzimidazolone moiety in 50, we believed 50 may be inhibiting the enzyme through an allosteric mechanism, akin to Akt, and that the dual PLD1/2 inhibitor 50 could represent an attractive lead from which to access PLD1 and PLD2 selective compounds. Thus, our laboratory in collaboration with Alex Brown and his laboratory initiated a diversity-oriented synthesis campaign around 50, followed by iterative parallel synthesis and the “fluorine walk” (Figure 10), to develop the first direct, isoform selective PLD inhibitors 51 (1700-fold PLD1 selective) and 52 (75-fold PLD2 selective).86−88 Mutants lacking the N-terminus (PX and PH domains) lost PLD activity (again similar to Akt), and when combined with other biochemical and enzyme kinetic studies, it became clear that these compounds were indeed allosteric. Despite value in aiding to more clearly define the individual contributions of PLD1 and PLD2 in various systems and diseases, poor DMPK and physiochemical properties precluded robust in vivo studies.86−88 Further chemical optimization led to the discovery of the potent dual PLD1/2 inhibitor ML299 (53) and a second generation PLD2 selective probe ML298 (54).89 Both showed improved ancillary pharmacology but only marginally improved DMPK profiles and physiochemical properties. Here, we noted a “molecular switch” in the form of a “magic methyl” that enhanced PLD1 activity ∼50-fold in the piperidine benzimidazolone series represented by 51 but could reverse PLD2 selectivity over 250-fold in the triazaspirone series, represented by 53.89 While further optimization to improve DMPK continues, the tools in hand elucidated a wealth of information regarding the therapeutic potential of PLD inhibition, especially PLD2. For example, 52 was critical in studies that identified PLD as a novel regulator of Akt in glioblastoma multiforme (GBM).9052 enabled studies that showed PA as an essential component for the membrane recruitment and activation of Akt, as well as a direct protein–protein interaction between PLD2 and Akt. Inhibition of PLD2 by 52 decreases activation of Akt leading to cell death through inhibition of autophagic flux and a back door by which to inhibit Akt.90 Furthermore, we demonstrated that infection by influenza virus stimulates phospholipase D (PLD) activity and PLD colocalizes with influenza during infection.91 Inhibition of PLD2 with 52 delayed viral entry and reduced viral titers in vitro and in vivo, as well as enhancing survival against a broad panel of influenza strains (H1, H3, H5, and H7).91 Once again, development of highly isoform selective PLD inhibitors, by targeting an allosteric mechanism, advanced our understanding of the deeper signaling biology and uncovered therapeutic potential in oncology and virology. Moreover, principles, strategies, and issues for optimization of GPCR allosteric ligands carried over into phospholipases.
V. Conclusion
Allosteric modulation has fundamentally altered our ability to prosecute “tough” targets and successfully develop ligands. Here, in commemoration of the 2013 Philip S. Portoghese Medicinal Chemistry Lectureship, several vignettes of drug discovery campaigns targeting novel allosteric mechanisms for kinases, phospholipases, and G-protein-coupled receptors were described that gave rise to general principles for successful optimization. On the basis of the broad applicability and success of allosteric modulation, we wish to move beyond classical drug repurposing to “receptor repurposing” and re-engage targets that failed because of the ligands/chemotypes with new functional HTS campaigns and subsequent development of allosteric ligands and exploit fundamentally new chemotypes and biased signaling profiles. Phase II clinical data are eagerly awaited for multiple allosteric modulators (Akt, M1 PAMs, etc.) to further validate the approach to improve human health and impact unmet medical needs.
Acknowledgments
First and foremost, medicinal chemists must acknowledge Professor Philip S. Portoghese, Editor-in-Chief of the Journal of Medicinal Chemistry for almost 40 years, who helped shape the practice of medicinal chemistry into the discipline it is today. Second, I must thank Jeff Conn, my colleague and partner in our adventures through allosteric drug discovery; there is no better molecular pharmacologist or drug hunter. Funding from NIH for support of our research in allosteric modulators of GPCRS and in particular NIMH (Grant MH082867), NIDA (Grant DA023947), and the MLPCN (Grant U54MH084659), as well as Janssen, AstraZeneca, Bristol-Myers Squibb, and Seaside Therapeutics, is acknowledged. Dr. Lindsley also thanks the Warren family and Foundation for establishing the William K. Warren Jr. Chair in Medicine. Finally, thanks are given to Merck (2001–2006) for establishing a culture enabling advances in fundamental science in pursuit of novel drug discovery approaches.
Glossary
Abbreviations Used
- CNS
central nervous system
- GPCR
G-protein-coupled receptor
- ATP
adenosine triphosphate
- CNS
central nervous system
- PAM
positive allosteric modulator
- NAM
negative allosteric modulator
- SAM
silent allosteric modulator
- NAL
no affect ligand
- BZD
benzodiazepine
- GABAA
γ-aminobutyric acid
- mAChR
muscarinic acetylcholine receptor
- ACh
acetylcholine
- mGluR
metabotropic glutamate receptor
- NMDA
N-methyl-d-aspartate
- DMPK
drug metabolism/pharmacokinetic
- SAR
structure–activity relationship
- GLP1
glucagon-like peptide 1
- PET
positron emission tomography
- CRC
concentration–response curve
- FRET
fluorescence resonance energy transfer
- HTS
high-throughput screen
- DFB
3,3′-difluorobenzaldazine
- CDPPB
3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- MPEP
2-methyl-6-(phenylethynyl)pyridine
Biography
Craig W. Lindsley is Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery in Nashville, TN, and holds the William K. Warren, Jr. Chair in Medicine. He received his doctorate in 1996 from the University of California, Santa Barbara, and pursued postdoctoral studies at Harvard University, MA. In 2001, he moved to Merck and developed a streamlined approach for lead optimizaton that resulted in the delivery of six preclinical candidates, including the first isoenzyme-selective allosteric AKT-kinase inhibitors, as well as the first positive allosteric modulators of the mGlu5 and M1. In 2006, he accepted Associate Professor appointments in Pharmacology and Chemistry at Vanderbilt University, and in 2009 he was promoted to full Professor and is the founding Editor-in-Chief for ACS Chemical Neuroscience.
The authors declare the following competing financial interest(s): Funding was received from BMS and AZ for allosteric modulators of mGlu4 and M4, respectively.
Funding Statement
National Institutes of Health, United States
Footnotes
This Award Address was presented at the 246th National Meeting of the American Chemical Society in Indianapolis, IN, September 13, 2013.
References
- Lindsley C. W.Adventures in allosteric drug discovery. Presented at the 246th National Meeting of the American Chemical Society, Indianapolis, IN, September 10, 2013; The 2013 Portoghese Lectureship.
- Kenakin T.; Miller L. J. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 2010, 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton A. W. Allostery: an illustrated definition for the “second secret of life”. Trends Biochem. Sci. 2008, 33, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Christopolous A.; Lindsley C. W. Allosteric modulators of GPCRs as a novel approach to treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges T. M.; Lindsley C. W. G-protein coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem. Biol. 2008, 3, 530–542. [DOI] [PubMed] [Google Scholar]
- Christopoulos A.; Kenakin T. G protein-coupled receptors allosterism and complexing. Pharmacol. Rev. 2002, 54, 323–374. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. Allosteric binding sites on cell surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discovery 2002, 1, 198–210. [Google Scholar]
- Kenakin T. P. 7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol. Sci. 2009, 30, 460–469. [DOI] [PubMed] [Google Scholar]
- Lane R. J.; Abdul-Ridha A.; Canals M. Regulation of G protein-coupled receptors by allosteric ligands. ACS Chem. Neurosci. 2013, 4, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J.-P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1956, 88–118. [DOI] [PubMed] [Google Scholar]
- Melancon B. J.; Hopkins C. R.; Wood M. R.; Emmitte K. A.; Niswender C. M.; Christopoulos A.; Conn P. J.; Lindsley C. W. Allosteric modulation of 7 transmembrane spanning receptors: theory, practice and opportunities for CNS drug discovery. J. Med. Chem. 2012, 55, 1445–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Meiler J.; Niswender C. M. Opportunities and challenges in discovery of allosteric modulators of GPCRs for treatment of CNS disorders. Nat. Rev. Drug Discovery 2014, 13, 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler H. F.; Fritschy J. M.; Rudolph U. A new benzodiazepine pharmacology. J. Pharmacol. Exp. Ther. 2002, 300, 2–8. [DOI] [PubMed] [Google Scholar]
- Wenthur C. J.; Gentry P. R.; Matthews T. P.; Lindsley C. W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E. F.; Heaton W. H.; Miller M.; Fox J.; Balandrin M. F.; Van Wagenen B. C.; Colloton M.; Karbon W.; Scherrer J.; Shatzen E.; Rishton G.; Scully S.; Qi M.; Harris R.; Lacey D.; Martin D. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J. Pharmacol. Exp. Ther. 2004, 308, 627–635. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A.; Kadow J. F. Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr. Opin. Invest. Drugs 2007, 8, 669–681. [PubMed] [Google Scholar]
- For information on the VCNDD, see the following: http://www.vcndd.com/ (accessed August 1, 2014).
- For information on the MLPCN and available probe compounds, see the following: https://mli.nih.gov/mli/mlpcn/ (accessed August 1, 2014).
- Johnson L. N.; Lewis R. J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- Pawson T.; Scott J. D. Protein phosphorylation in signaling. Trends Biochem. Sci. 2005, 30, 286–290. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases—the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Eyck L.; Karlsson R.; Xuong N.; Taylor S. S.; Sowadski J. M. Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry 1993, 32, 2154–2161. [DOI] [PubMed] [Google Scholar]
- Hantschel O. Structure, regulation, signaling, and targeting of Abl kinases in cancer. Genes Cancer 2012, 3, 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri L.; Giulio R. αC Helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discovery Today 2012, 1–8. [DOI] [PubMed] [Google Scholar]
- Betzi S.; Riazul A.; Mathew M.; Lubbers D. J.; Han H.; Jakkaraj S. R.; Georg G. I.; Schönbrunn E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busschots K.; Lopez-Garcia L. A.; Lammi C.; Stroba A.; Zeuzem S.; Piiper A.; Alzari P. M.; Neimanis S.; Arencibia J. M.; Engel M.; Schulze J. O.; Biondi R. M. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem. Biol. 2012, 19, 1152–1163. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia L.; Schulze J. O.; Fröhner W.; Zhang H.; Süss E.; Weber N.; Navratil J.; Amon S.; Hindie V.; Zeuzem S.; Jorgensen T. J. D.; Alzari P. M.; Engel M.; Biondi R. M. Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem. Biol. 2011, 18, 1463–1473. [DOI] [PubMed] [Google Scholar]
- Fischmann T. O.; Smith C. K.; Mayhood T. W.; Myers J. E.; Reichert P.; Mannarino A.; Carr D.; Zhu H.; Wong J.; Yang R.-S.; Le H. V.; Madison V. S. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry 2009, 48, 2661–2674. [DOI] [PubMed] [Google Scholar]
- Fang Z.; Christian G.; Daniel R. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS Chem. Biol. 2013, 8, 58–70. [DOI] [PubMed] [Google Scholar]
- Hennessy B. T.; Smith D. L.; Ram P. T.; Lu Y.; Mills G. B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discovery 2005, 4, 988–1004. [DOI] [PubMed] [Google Scholar]
- Barnett S. F.; Bilodeau M. T.; Lindsley C. W. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation. Curr. Top. Med. Chem. 2005, 5, 109–125. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation: a 2009 update. Curr. Top. Med. Chem. 2010, 10, 458–477. [DOI] [PubMed] [Google Scholar]
- Kumar C.; Madison V. AKT crystal structure and AKT-specific inhibitors. Oncogene 2005, 24, 7493–7501. [DOI] [PubMed] [Google Scholar]
- Cheng J. Q.; Lindsley C. W.; Cheng G. Z.; Yang H.; Nicosia S. V. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [DOI] [PubMed] [Google Scholar]
- Kim D.; Cheng G. Z.; Lindsley C. W.; Yang H.; Cheng J. Q. Targeting phosphatidylinositol-3 kinase/Akt pathway for cancer therapy. Curr. Opin. Invest. Drugs 2005, 6, 1250–1258. [PubMed] [Google Scholar]
- Lindsley C. W.; Zhao Z.; Duggan M. E.; Barnett S. F.; Defeo-Jones D.; Huber H. E.; Huff J. R.; Hartman G. D.; Leister W.; Kral A.; Fu S.; Hancock P. J.; Haskell K. A.; Jones R. E.; Robinson R. Allosteric Akt (PKB) kinase inhibitors. Discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 761–764. [DOI] [PubMed] [Google Scholar]
- Defeo-Jones D.; Barnett S. F.; Fu S.; Hancock P. J.; Haskell K. M.; Leander K. R.; McAvoy E.; Lindsley C. W.; Duggan M. E.; Hartman G. D.; Huber H. E. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol. Cancer Ther. 2005, 4, 271–279. [PubMed] [Google Scholar]
- Barnett S. F.; Defeo-Jones D.; Fu S.; Hancock P. J.; Haskell K. M.; Jones R. E.; Kahana J. A.; Kral A. M.; Leander K.; Lee L. L.; Malinowski J.; McAvoy E. M.; Nahas D. D.; Robinson R. G.; Huber H. E. Identification and characterization of pleckstin-homology-domain-dependent and isoenzyme-specfic Akt inhibitors. Biochem. J. 2005, 385, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy J. P.; Williams L.; Bridges T. M.; Daniels R. N.; Weaver C. D.; Lindsley C. W. Application of combinatorial chemistry science on modern drug discovery. J. Comb. Chem. 2008, 10, 345–354. [DOI] [PubMed] [Google Scholar]
- Hirai H.; Sootome H.; Nakatsuru Y.; Miyama K.; Taguchi S.; Tsukioka K.; Ueno Y.; Hatch H.; Majumder P. K.; Pan B.-S.; Kotani H. MK-2206, an allosteric Akt inhibitor, enhances tumor efficacy by standard chemotherapetuic agents or molecular targetd drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Calleja V.; Laguerre M.; Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009, 7, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuzumi T.; Fielder D.; Zhang C.; Gray D. C.; Aizenstein B.; Hoffman R.; Shokat K. M. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009, 5, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmittee K. A. Recent advances in the design and development of novel negative allosteric modulators of mGlu5. ACS Chem. Neurosci. 2011, 2, 411–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley C. W.; Shipe W. D.; Wolkenberg S. E.; Theberge C. R.; Williams D. L. Jr.; Sur C.; Kinney G. G. Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr. Top. Med. Chem. 2006, 6, 771–785. [DOI] [PubMed] [Google Scholar]
- O’Brien J. A.; Lemaire W.; Chen T. B.; Chang R. S. L.; Jacobson M. A.; Ha S. N.; Lindsley C. W.; Schaffhauser H. J.; Sur C.; Pettibone D. J.; Conn P. J.; Williams D. L. Jr. A family of highly selectiveallosteric modulators of the metabotropic glutamate receptor subtype 5. Mol. Pharmacol. 2003, 64, 731–740. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Rodriguez A. L.; Conn P. J.; Lindsley C. W. Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett. 2008, 18, 4098–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Kedrowski J.; Rodriguez A. L.; Niswender C. M.; Jones C. K.; Conn P. J.; Lindsley C. W. Discovery of molecular switches that modulate modes of mGlu5 pharmacology in vitro and in vivo within a series of functionalized, regioisomeric 2- and 5-(phenylethynyl)pyrimidines. J. Med. Chem. 2009, 52, 4103–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. R.; Hopkins C. R.; Brogan J. T.; Conn P. J.; Lindsley C. W. “Molecular Switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry 2011, 50, 2403–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J. A.; Lemaire W.; Wittmann M.; Jacobson M. A.; Ha S. N.; Wisnoski D. D.; Lindsley C. W.; Schaffhauser H. J.; Rowe B.; Sur C.; Williams D. L. Jr.; Conn P. J. A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J. Pharmacol. Exp. Ther. 2004, 309, 568–577. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wisnoski D. D.; Lemaire W.; O’Brien J. A.; Williams D. L. Jr.; Jacobson M. A.; Lindsley C. W. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg. Med. Chem. Lett. 2007, 17, 1386–1391. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Wisnoski D. D.; Duggan M. E.; Leister W. H.; Hartman G. D.; Huff J. R.; Williams D. L. Jr.; O’Brien J. A.; Lemaire W.; Sur C.; Kinney G. G.; Conn P. J.; Duggan M. E. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 (mGluR5) that potentiate receptor function in vivo. J. Med. Chem. 2004, 47, 5825–5828. [DOI] [PubMed] [Google Scholar]
- Kinney G. G.; O’Brien J. A.; Lemaire W.; Burno M.; Bickel D. J.; Clements M. K.; Chen T. B.; Wisnoski D. D.; Lindsley C. W.; Tiller P. R.; Conn P. J.; Williams D. L. Jr. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J. Pharmacol. Exp. Ther. 2005, 313, 199–206. [DOI] [PubMed] [Google Scholar]
- Stauffer S. R. Progress toward positive allosteric modulators of the metabotropic glutamate receptor subtype 5 (mGlu5). ACS Chem. Neurosci. 2011, 2, 450–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier-Batteur S.; Hutson P. H.; Menzel K.; Uslaner J. M.; Mattson B. A.; O’Brien J. A.; Magliaro B. C.; Forest T.; Stump C. A.; Tynebor R. M.; Anthony N. J.; Tucker T. J.; Zhang X. F.; Gomez R.; Huszar S. L.; Lambeng N.; Fauré H.; Le Poul E.; Poli S.; Rosahl T. W.; Rocher J. P.; Hargreaves R.; Williams T. M. Mechanism based neurotoxicity of mGlu5 positive allosteric modulators: development challenges for a promising novel antipsychotic target. Neuropharmacology 2014, 82, 161–173. [DOI] [PubMed] [Google Scholar]
- Bridges T. M.; Rook J. R.; Noetzel M. J.; Morrison R. D.; Zhou Y.; Gogliotti R. D.; Vinson P. N.; Xiang Z.; Jones C. K.; Niswender C. M.; Lindsley C. W.; Stauffer S. R.; Conn P. J.; Daniels S. J. Biotransformation of a novel positive allosteric modulator of metabotropic glutamate receptor subtype 5 contributes to seizure-like adverse events in rats involving a receptor agonism-dependent mechanism. Drug Metab. Dispos. 2013, 41, 1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J. M.; Noetzel M. J.; Pouliot W. A.; Bridges T. M.; Vinson P. N.; Cho H. P.; Zhou Y.; Stauffer S. R.; Niswender C. M.; Daniels S. J.; Lindsley C. W.; Conn P. J. Unique signaling profiles of positive allosteric modulators of metabotropic mlutamate receptor subtype 5 determine differences in in vivo activity. Biol. Psychiatry 2013, 73, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schann S.; Mayer S.; Franchet C.; Frauli M.; Steinberg E.; Thomas M.; Baron L.; Neuville P. Chemical switch of a metabotropic glutamate receptor 2 silent sllosteric modulator into dual metabotropic glutamate receptor 2/3 negative/positive allosteric modulators. J. Med. Chem. 2010, 53, 8775–8779. [DOI] [PubMed] [Google Scholar]
- Utley T.; Haddenham D.; Salovich J. M.; Zamorano R.; Vinson P. N.; Lindsley C. W.; Hopkins C. R.; Niswender C. M. Synthesis and SAR of a novel metabotropic glutamate receptor 4 (mGlu4) antagonist: unexpected “molecular switch” from a closely related mGlu4 positive allosteric modulator. Bioorg. Med. Chem. Lett. 2011, 21, 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby G. J.; Utley T. J.; Lamsal A.; Sevel C.; Sheffler D. J.; Lebois E. P.; Bridges T. M.; Lindsley C. W.; Conn P. J. Chemical modification of the M(1) agonist VU0364572 reveals molecular switches in pharmacology and a bitopic binding mode. ACS Chem. Neurosci. 2012, 3, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luker T.; Bonnert R.; Schmidt R.; Sargent C.; Paine S. W.; Thom S.; Pairaudeau G.; Patel A.; Mohammed R.; Akam E.; Dougall I.; Davis A. M.; Abbott S.; Millichip I.; McInally T. Switching between agonists and antagonists at CRTh2 in a series of highly potent and selective biaryl phenoxyacetic acids. Bioorg. Med. Chem. Lett. 2011, 21, 3616–3621. [DOI] [PubMed] [Google Scholar]
- Cheung Y. Y.; Haibo Yu.; Kaiping Xu.; Beiyan Zou; Lindsley C. W.; Li M.; Hopkins C. R. Discovery of a series of 2-phenyl-N-(2-(pyrrolidin-1-yl) phenyl) acetamides as novel molecular switches that modulate modes of Kv7. 2 (KCNQ2) channel. J. Med. Chem. 2012, 55, 6975–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory K. J.; Noetzel M. J.; Rook J. M.; Vinson P. N.; Stauffer S. R.; Rodriguez A. L.; Emmitte K. A.; Niswender C. M.; Lindsley C. W.; Conn P. J. Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure–function studies and structure–activity relationships. Mol. Pharmacol. 2012, 82, 860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler D. J.; Wenthur C. R.; Bruner J. A.; Carrington S. J. S.; Vinson P. N.; Gogi K. K.; Blobaum A. L.; Morrison R. D.; Vamos M.; Cosford N. D. P.; Stauffer S. R.; Daniels S. J.; Niswender C. M.; Conn P. J.; Lindsley C. W. Development of a novel, CNS-penetrant, metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) derived from a closely related mGlu5 PAM. Bioorg. Med. Chem. Lett. 2012, 22, 3921–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthur C. J.; Morrison R.; Felts A. S.; Smith K. A.; Engers J. L.; Byers F. W.; Daniels J. S.; Emmitte K. A.; Conn P. J.; Lindsley C. W. Discovery of (R)-(2-fluoro-4-((-4-methoxyphenyl)ethynyl)phenyl) (3-hydroxypiperidin-1-yl)methanone (ML337), an mGlu3 selective and CNS penetrant negative allosteric modulator (NAM). J. Med. Chem. 2013, 56, 5308–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notartomaso S.; Zappulla C.; Biagioni F.; Cannella M.; Bucci D.; Mascio G.; Scarselli P.; Fazio F.; Weisz F.; Lionetto L.; Simmaco M.; Gradini R.; Battaglia G.; Signore M.; Puliti A.; Nicoletti F. Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1. Mol. Brain 2013, 19, 1650–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.; Zhou Y.; Niswender C. M.; Luo Q.; Conn P. J.; Lindsley C. W.; Hopkins C. R. Re-exploration of the PHCCC scaffold: discovery of improved positive allosteric modulators of mGlu4. ACS Chem. Neurosci. 2010, 1, 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. K.; Engers D. W.; Thompson A. D.; Field J. R.; Blobaum A. L.; Lindsley S. R.; Zhou Y.; Gogliotti R. D.; Jadhav S.; Zamorano R.; Smith Y.; Morrsion R.; Daniels S. J.; Weaver C. D.; Conn P. J.; Lindsley C. W.; Niswender C. M.; Hopkins C. R. Discovery, synthesis, and structure–activity relationship development of a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): characterization of a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4) with oral efficacy in an antiparkinsonian animal model. J. Med. Chem. 2011, 54, 7639–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. P.; Engers D. W.; Venable D. F.; Niswender C. M.; Lindsley C. W.; Conn P. J.; Emmitte K. A.; Rodriguez A. L. A novel class of succinimide-derived negative allosteric modulators of metabotropic glutamate receptor subtype 1 provides insight into a disconnect in activity between the rat and human receptors. ACS Chem. Neurosci. 2014, 5, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. P.; Garcia-Barrantes P. M.; Brogan J. T.; Hopkins C. R.; Niswender C. M.; Rodriguez A. L.; Venable D.; Morriosn R. D.; Bubser M.; Daniels S. J.; Jones C. K.; Conn P. J.; Lindsley C. W. Chemical modulation of mutant mGlu1receptors derived from deleterious GRM1 mutations found in schizophrenics: development of novel mGlu1 PAMs via a “double molecular switch” of an mGlu4 PAM chemotype. ACS Chem. Biol. 2014, 10.1021/cb500560h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges T. M.; Marlo J. E.; Niswender C. M.; Jones C. K.; Jahhav S. B.; Gentry P. R.; Cho H. P.; Weaver C. D.; Conn P. J.; Lindsley C. W. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J. Med. Chem. 2009, 52, 3445–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges T. M.; Kennedy J. P.; Noetzel M. J.; Breininger M. L.; Gentry P. R.; Conn P. J.; Lindsley C. W. Chemical lead optimization of a Ppan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) lead. Part II: Development of a potent and highly selective M1 PAM. Bioorg. Med. Chem. Lett. 2010, 20, 1972–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza N. R.; Peters D.; Sparks R. G. Xanomeline and the antipsychotic potential of muscarinic receptor subtype selective agonists. CNS Drug Rev. 2003, 9, 159–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. K.; Brady A. E.; Davis A. A.; Xiang Z.; Bubser M.; Tawawy M. N.; Kane A. S.; Bridges T. M.; Kennedy J. P.; Bradley S. R.; Peterson T. E.; Ansari M. S.; Baldwin R. M.; Kessler R. M.; Deutch A. Y.; Lah J. L.; Levey A. I.; Lindsley C. W.; Conn P. J. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J. Neurosci. 2008, 28, 10422–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebois E. P.; Bridges T. M.; Lewis M. L.; Dawson E. S.; Kane A. S.; Xiang Z.; Jadhav S. B.; Yin H.; Kennedy J. P.; Meiler J.; Niswender C. M.; Jones C. K.; Conn P. J.; Weaver C. D.; Lindsley C. W. Discovery and characterization of novel subtype-selective allosteric agonists for the investigation of M1 receptor function in the central nervous system. ACS Chem. Neurosci. 2010, 1, 104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler D. J.; Sevel C.; Le U.; Lovell K. M.; Tarr J. C.; Carrington S. J. S.; Cho H. P.; Digby G. J.; Nisewnder C. M.; Conn P. J.; Hopkins C. R.; Wood M. R.; Lindsley C. W. Further exploration of M1 allosteric agonists: subtle structural changes abolish M1 allosteric agonism and result in pan-mAChR orthosteric antagonism. Bioorg. Med. Chem. Lett. 2013, 23, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby G. J.; Noetzel M. J.; Bubser M.; Utley T. J.; Walker A. G.; Byun N. E.; Lebois E. P.; Xiang Z.; Sheffler D. J.; Cho H. P.; Davis A. A.; Nemirovsky N. E.; Mennenga S. E.; Camp B. W.; Bimonte-Nelson H. A.; Bode J.; Italiano K.; Morrsion R.; Daniels J. S.; Niswender C. M.; Olive M. F.; Lindsley C. W.; Jones C. K.; Conn P. J. Novel allosteric agonists of M1 muscarinic acetylcholine receptors induce brain region-specific responses that correspond with behavioral effects in animal models. J. Neurosci. 2012, 32, 8532–8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Seager M. A.; Wittmann M.; Jacobson M.; Bickel D.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; Pearson M.; McCampbell A.; Gaspar R.; Shughrue P.; Danziger A.; Regan C.; Flick R.; Pascarella D.; Garson S.; Doran S.; Kreatsoulas C.; Veng L.; Lindsley C. W.; Shipe W.; Kuduk S.; Sur C.; Kinney G.; Seabrook G. R.; Ray W. J. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr J. C.; Turlington M. L.; Reid P. R.; Utley T. J.; Sheffler D. J.; Cho H. P.; Klar R.; Pancani T.; Klein M. T.; Bridges T. M.; Morrison R. D.; Blobaum A. L.; Xiang Z.; Daniels J. S.; Niswender C. M.; Conn P. J.; Wood M. R.; Lindsley C. W. Targeting selective activation of M1 for the treatment of Alzheimer’s disease: further chemical optimization and pharmacological characterization of the M1 positive allosteric modulator ML169. ACS Chem. Neurosci. 2012, 3, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N. E.; Grannan M.; Bubser M.; Barry R. L.; Thompson A.; Rosanelli J.; Gowrishankar R.; Kelm N. D.; Damon S.; Bridges T. M.; Melancon B. J.; Tarr J. C.; Brogan J. T.; Avison M. J.; Deutch A. Y.; Wess J.; Wood M. R.; Lindsley C. W.; Gore J. C.; Conn P. J.; Jones C. K. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100. Neuropsychopharmacology 2014, 39, 1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubser M.; Bridges T. M.; Dencker D.; Gould R. W.; Grannan M. D.; Wes J.; Duggan M. E.; Dunlop J.; Wood M. R.; Lindsley C. W.; Conn P. J.; Jones C. K. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem. Neurosci. 2014, 10.1021/cn500128b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore A. S.; Okrasa K.; Patel J. C.; Serrano-Vega M.; Bennett K.; Cooke R. M.; Errey J. C.; Jazayeri A.; Khan S.; Tehan B.; Weir M.; Wiggin G. R.; Marshall F. H. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [DOI] [PubMed] [Google Scholar]
- Selvy P. E.; Lavieri R.; Lindsley C. W.; Brown H. A. Phospholipase D: enzymology, signaling, and chemical modulation. Chem. Rev. 2011, 111, 6064–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monovich L.; Mugrage B.; Quadros E.; Toscano K.; Tommasi R.; LaVoie S.; Liu E.; Du Z. M.; LaSala D.; Boyar W.; Steed P. Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 2007, 17, 2310–2311. [DOI] [PubMed] [Google Scholar]
- De Cuyper H.; Verstraeten D. The clinical significance of halpemide, a dopamine-blocker related to butyrphenones. Neuropsychobiology 1984, 12, 211–216. [DOI] [PubMed] [Google Scholar]
- Scott S. A.; Selvy P. E.; Buck J.; Cho H. P.; Criswell T. L.; Thomas A. L.; Armstrong M. D.; Arteaga C.; Lindsley C. W.; Brown H. A. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 2009, 5, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. A.; Scott S. A.; Lavieri R.; Buck J. R.; Selvy P. E.; Stoops S. L.; Armstrong M. D.; Brown H. A.; Lindsley C. W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: Impact of alternative halogenated privileged structures on PLD1 specificity. Bioorg. Med. Chem. Lett. 2009, 19, 1916–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri R.; Scott S. A.; Selvy P. E.; Brown H. A.; Lindsley C. W. Design, synthesis and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazasprio[4.5]decan-8-yl)ethylbenzamides: discovery of an isoform selective small-molecule phospholipase D2 (PLD2) inhibitor. J. Med. Chem. 2010, 53, 6706–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M. C.; Scott S. A.; Brown K. A.; Oquin T. H. III; Thomas P. G.; Daniels J. S.; Morrison R.; Brown H. A.; Lindsley C. W. Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-triazaspiro[4.5]decane core; discovery of ML298 and ML299 that decrease invasive migration in U87-MG glioblastoma cells. J. Med. Chem. 2013, 56, 2695–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunz R.; Taylor H. E.; Lindsley C. W.; Brown H. A. Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. J. Biol. Chem. 2014, 289, 600–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguin T.; Sharma S.; Stuart A. D.; Jones C. K.; Daniels J. S.; Lindsley C. W.; Thomas P. G.; Brown H. A. Phospholipase D facilitates efficient entry of influenza virus allowing escape from innate mmune inhibition. J. Biol. Chem. 2014, 10.1074/jbc.M114.558817. [DOI] [PMC free article] [PubMed] [Google Scholar]