

Abstract
The Fragile X syndrome (FXS) is the most frequent form of inherited mental disability and is considered a monogenic cause of autism spectrum disorder. FXS is caused by a triplet expansion that inhibits the expression of the FMR1 gene. The gene product, the Fragile X Mental Retardation Protein (FMRP), regulates mRNA metabolism in brain and nonneuronal cells. During brain development, FMRP controls the expression of key molecules involved in receptor signaling, cytoskeleton remodeling, protein synthesis and, ultimately, spine morphology. Symptoms associated with FXS include neurodevelopmental delay, cognitive impairment, anxiety, hyperactivity, and autistic-like behavior. Twenty years ago the first Fmr1 KO mouse to study FXS was generated, and several years later other key models including the mutant Drosophila melanogaster, dFmr1, have further helped the understanding of the cellular and molecular causes behind this complex syndrome. Here, we review to which extent these biological models are affected by the absence of FMRP, pointing out the similarities with the observed human dysfunction. Additionally, we discuss several potential treatments under study in animal models that are able to partially revert some of the FXS abnormalities.
The Fragile X syndrome: clinical features
Complex cognitive brain functions are affected in patients with learning and intellectual disabilities (IDs), adult onset dementias such as Alzheimer disease, aging-dependent dementias and the various amnesias. A large body of evidence suggests that local protein synthesis regulates memory formation and cognition, two processes that rely on activity-dependent synaptic plasticity (Kang and Schuman 1996; Antion et al. 2008; Kelleher and Bear 2008; Wang et al. 2009; Ho et al. 2011; Redondo and Morris 2011; Jung et al. 2012). Dysregulation of such mechanisms contribute to spine dysmorphogenesis and a variety of other neuropathological conditions (Bagni and Greenough 2005; Sala and Segal 2014). Synaptic inputs dictate the time, place, and amount of protein synthesis necessary for individual synapses: events often impaired in individuals with IDs.
Fragile X syndrome (FXS) is the most frequent monogenic cause of inheritable mental disability (Bagni and Greenough 2005). The syndrome's name refers to a cytogenetic marker on the X chromosome at Xq27.3, a “fragile site” in which the chromatin fails to condense properly during mitosis (Hecht and Sutherland 1985). The brittle point, denoted as FRAXA (FRAgile site, X chromosome, A site), is evident in ∼50% of the metaphase chromosomes of virtually all clinically affected humans (Fu et al. 1991; Bagni et al. 2012).
FXS has a greater incidence in males (one in 4000 males and one in 6000 females; Mandel and Biancalana 2004; Bagni et al. 2012), because the X chromosome with the fragile X site is more often inactivated compared with the nonaffected X. FXS leads to a spectrum of physical, intellectual, emotional, and behavioral characteristics ranging from mild to severe. Symptoms include, among others, developmental and behavioral deficits, attention deficits, autistic behaviors, aggression, anxiety and hyperactivity, sleep disorders, epileptic seizures, hypersensitivity to sensory stimulation including noise and touch, and deficits in social-personal skills. Physical features displayed by affected individuals include flat feet, flexible joints, and low muscle tone, large forehead or ears with a prominent jaw, long face, soft skin, strabismus, and enlarged testes (Berry-Kravis 2014; Jacquemont et al. 2014). Boys with FXS are usually diagnosed within the first 3 yr of life, when they experience delays in speech development and social interactions. Hypotonia, hand flapping, poor eye contact, and autistic-like behavior can also be observed at this age (Cordeiro et al. 2011; Bagni et al. 2012). As girls have fewer symptoms, they tend to be diagnosed much later, often in their early teens, when a high percentage exhibit ovarian failure (Sherman 2000). The ID which is the hallmark of this condition is milder or absent in females.
Recently exome sequencing of samples from patients diagnosed with schizophrenia (SCZ) (Fromer et al. 2014; Purcell et al. 2014) or autism (AD) (Iossifov et al. 2012; Waltes et al. 2014) identified several mutations in genes targeted by FMRP (for review, see Fernandez et al. 2013). In addition, proteins interacting with FMRP such as the cytoplasmic interactor CYFIP1 were shown to be associated with AD (Waltes et al. 2014) and SCZ (Fromer et al. 2014; Purcell et al. 2014). Furthermore, our laboratory has recently identified (De Rubeis et al. 2013) several proteins encoded by genes involved in SCZ, ASD, and ID that are associated with CYFIP1 in the mouse model. This strongly suggests that FMRP expression needs to be tightly controlled, because any alteration of the FMRP pathway might lead to multiple dysregulated processes that probably underlie these conditions and syndromes.
From genetic causes to model systems for FXS
In most cases FXS is the result of loss or functional silencing of the FMR1 gene due to methylation of the CGG triplet expansion in the 5′ untranslated region of the gene (Pieretti et al. 1991; Verheij et al. 1993). Additional, but less frequent, cases have been reported with point mutations in the FMR1 gene (De Boulle et al. 1993; Lugenbeel et al. 1995; Wang et al. 1997) leading to classic features of FXS. The gene encodes for the Fragile Mental Retardation protein (FMRP), an RNA-binding protein with four RNA-binding domains affecting different aspects of mRNA metabolism including transport, stability, and translation (Bagni et al. 2012; Doyle and Kiebler 2012; Wang et al. 2012; Darnell and Klann 2013; Hornberg and Holt 2013; Rajan 2014). The absence of FMRP leads to a deregulated protein translation resulting in excessive accumulation of certain proteins and reduction of others (for review, see Bagni et al. 2012). Furthermore, FMRP appears to be involved in developmental decisions at the level of neurite extension, guidance, and branching (for review, see Doll and Broadie 2014). Golgi studies of human postmortem brain from patients with FXS, as well as from the Fmr1 KO mouse brains, revealed that the absence of FMRP leads to longer and thinner dendritic spines compared with normal. These “filopodia-like shape” resemble immature spines (for review, see Bagni and Greenough 2005). Notably, dysgenesis of dendritic spines is a feature shared by several ID disorders (for review, see Fiala et al. 2002; Penzes et al. 2011; De Rubeis et al. 2012).
In the Fmr1 KO mice, the absence of FMRP leads to an increased long-term depression (LTD; Huber et al. 2002), dependent on the metabotropic glutamate receptor 5 (mGluR5), which is protein synthesis independent (Nosyreva and Huber 2006). Based on these findings, Bear et al. proposed the “mGluR theory of FXS” (Bear et al. 2004), postulating that alterations of the mGluR pathway-dependent plasticity contribute to FXS pathophysiology. Accordingly, a broad range of affected phenotypes observed in animal models to study FXS have been rescued by pharmacological and genetic reduction of mGluR5 activity as well as by targeting downstream signaling pathways (discussed below and for review, see Krueger and Bear 2011; Bagni et al. 2012). However, in addition to the mGluR5 signaling, other receptors and key molecules have been implicated in the development and manifestation of this disability, such as the ionotropic glutamate receptors (NMDA and AMPA), GABAergic receptors (GABA A and B), and muscarinic acetylcholine receptors (for review, see Sethna et al. 2013). The majority of the drugs developed to correct the behavioral phenotypes observed in patients with FXS target these receptors or downstream effectors as shown in Figure 1.
Figure 1.
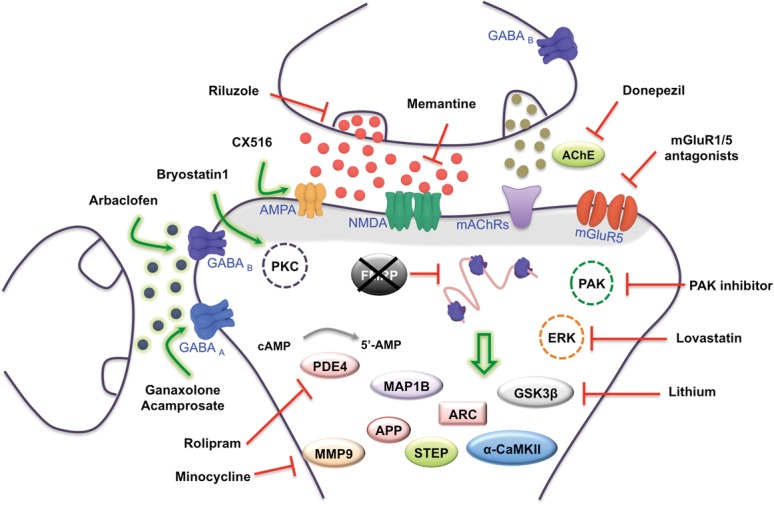
Molecular signaling altered in Fragile X syndrome. In the absence of FMRP protein translation is enhanced and several proteins encoded by FMRP targets such as APP, STEP, MAP1B, ARC, CaMKIIα, and others are up-regulated. In addition, many receptors are deregulated in FXS. Therefore, to normalize the observed behavioral phenotypes, several drugs have been used—in fly, animal models, and affected patients—to correct or revert some of the abnormalities. Among them: mGluR5 antagonists (MPEP, Fenobam, AFQ056, STX107, Acamprosate, RO491752, CTEP, LY341495, MPPG, and MTPG), agonists of the GABAergic pathway (Asbaclofen, Ganaxolene, and Acamprosate) and AMPAR signaling (CX516), antagonist of the cholinergic pathway (Donepezil), among others. Studies using fly and animal models have shown that targeting specific molecules/pathways corrected some defects and this information may be used to develop nontoxic drugs (denoted by the dashed line), such as ERK, PAK, and PKC inhibitors.
The ideal animal model to study FXS, or any other human disease, should re-create most of the complex deficits observed in the human condition, including cellular, physiological, and behavioral alterations. Intraspecies characteristics and differences in genetic background contribute to genetic disease variability and therefore make the generation of a biological model a difficult task; nevertheless, following the discovery of FMR1 gene in 1991 (Pieretti et al. 1991; Verkerk et al. 1991) the first animal model to study FXS was generated. In this case, a Neo cassette was inserted into exon 5 of the Fmr1 coding region of the mouse ortholog (Bakker 1994). This biological model can partially mimic human FXS pathology inasmuch as it leads to several phenotypes similar to the ones observed in patients. The use of this mouse model has enormously advanced the FXS field. The subsequent capitalizing of the power and advanced genetics of Drosophila melanogaster, a fruit fly model for FXS, further contributed to the understanding of FXS (Zhang et al. 2001; Dockendorff et al. 2002; Inoue et al. 2002; Morales et al. 2002; Lee et al. 2003; Xu et al. 2004).
In this review, we discuss some of the most relevant studies that enhanced the understanding of the behavioral deficits observed in patients with FXS, using the Fmr1 KO mouse and the dFmr1 mutant fruit fly as a model system. Ultimately, we briefly introduce some strategies aiming at rescuing the FXS phenotypes that hopefully will open new routes into more direct disease-targeted therapies.
The Mouse model for FXS
Fmr1 KO mice show anxiety-like behavioral phenotype
Because FXS patients are often described as hyperactive and under hyperarousal (Berry-Kravis 2014), the Fmr1 KO mice have been evaluated in anxiety-like and exploratory tasks. In the “light–dark compartment” assay, shown to be sensitive to anxiolytic drugs (Rogoz and Skuza 2011), the choice between the interest to explore novel environments and the aversion to brightly lit compartments is investigated. In this test, the Fmr1 KO mice were shown to present lower anxiety-like behaviors (Peier et al. 2000; Veeraragavan et al. 2012), as observed by a decreased transition between the light/dark compartments. The “open-field” test is commonly used to observe general exploratory activity. In this case, the time spent in the center, the numbers of crossings, and velocity are all used to monitor activity, which was found to be increased in Fmr1 KO mice compared with control animals. Because Fmr1 KO mice are hyperactive (Peier et al. 2000; Chen and Toth 2001; Yan et al. 2004; Restivo et al. 2005; Olmos-Serrano et al. 2011), they are usually reported as less anxious in the open-field arena. We should keep in mind that anxiety measured in this assay is highly influenced by locomotor activity (Peier et al. 2000; Spencer et al. 2005). However, an increase in anxiety-like behaviors (Restivo et al. 2005) in the open-field has also been reported for the Fmr1 KO mice. The “elevated-plus maze (EPM)” is usually the preferred test to investigate anxiety-like phenotypes comparing the time spent in open or closed arms. Similarly to the above-mentioned discrepancies, the Fmr1 KO showed both a decrease (Yuskaitis et al. 2010) and an increase (Bilousova et al. 2009) in anxiety during the EPM task when compared with control animals. Considering that FXS patients show anxiety when faced with certain social situations, it is crucial to dissociate social and nonsocial anxiety in the mouse model of FXS (Liu and Smith 2009). Further support towards increased anxiety of the Fmr1 KO mice came from the “mirrored test chamber” where a reflection of the mouse image is interpreted by the test mouse as another animal. In this context, the Fmr1 KO mice exhibited increased anxiety-like behaviors, measured by their greater avoidance of the central mirrored chamber (Spencer et al. 2005). Importantly, the reintroduction of human FMRP in the Fmr1 KO background alleviates deficits in anxiety and exploration (Peier et al. 2000), highlighting the role of FMRP for normal behavior.
Although the Fmr1 KO mouse model was fundamental for the identification of the cellular and molecular pathways affected in FXS, we believe that the study of the anxiety-like behaviors resulted in more difficult and sometimes with opposite outcomes possibly because in rodents anxiety is difficult to interpret due to multiple factors that interfere with the experimental settings.
Fmr1 KO mice show cognitive impairments
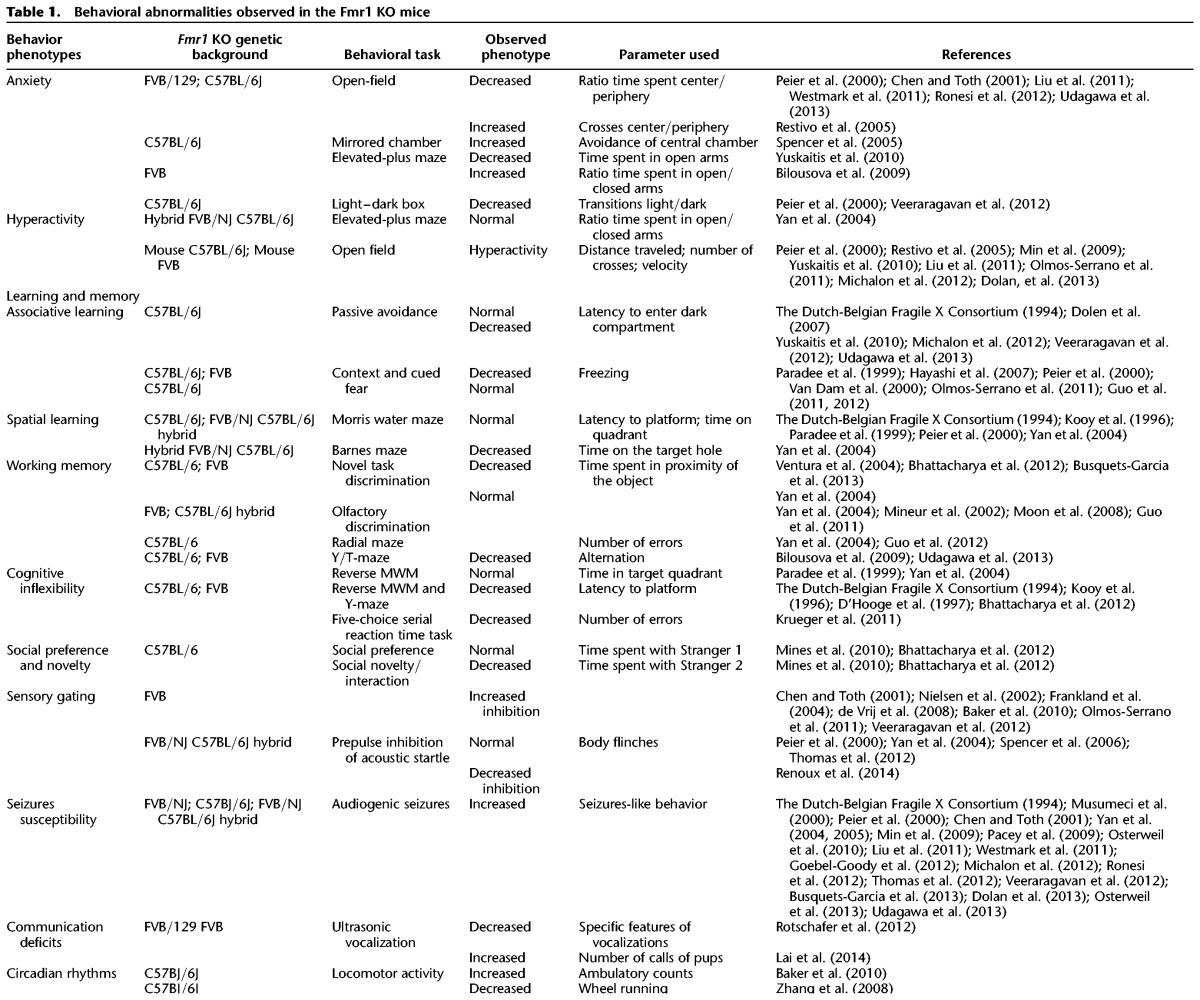
In addition to the anxiety aspect, the Fmr1 KO mouse has been used to model the intellectual disability of patients with FXS (Berry-Kravis 2014) with variable results, possibly due to differences in protocols, environment, and genetic background (see Table 1).
Table 1.
Behavioral abnormalities observed in the Fmr1 KO mice
Spatial learning
The Morris water maze (MWM) task is commonly used to evaluate hippocampal-dependent spatial learning in rodents. The latency to find an escape platform from a pool of opaque water decreases with the number of training trials. This test, initially developed by Richard Morris (Morris et al. 1982), is used as a readout for learning. Fmr1 KO mice do not show differences in latency compared with controls (Bakker 1994; Kooy et al. 1996; Paradee et al. 1999; Peier et al. 2000; Yan et al. 2004), suggesting that spatial learning is not affected by the absence of FMRP. Memory consolidation and retrieval are not affected either; both Fmr1 KO and WT mice spent more time in the target quadrant (Kooy et al. 1996; Paradee et al. 1999; Yan et al. 2004), indicating that both groups learned and remembered the initial position of the escape platform. To further understand the role of FMRP in spatial learning and memory, the performance of Fmr1 KO mice was also analyzed in other mazes. In the Barnes maze (BM), rodents are trained to find an escape hole based on distal cues, and contrary to what has been found in the MWM, in the BM the Fmr1 KO mice show significant differences in retrieval and memory consolidation compared with controls (Yan et al. 2004). In addition, recent evidence (Guo et al. 2012) indicated that Fmr1 KO mice, when studied in the radial arm maze apparatus, perform worse compared with wild-type littermates, as measured by the decreased ability of Fmr1 KO mice to preserve spatial information after food reward.
Associative learning
In the “passive avoidance” task, animals learn to avoid a location (dark compartment) in which they had previously received an unpleasant stimulus (for example as a footshock). While control mice take more time or even refuse to enter the dark compartment, because they associate it with the shock of their paws, the Fmr1 KO mice show a range of behavioral responses. It has been reported that the Fmr1 KO mice showed a deficit in associative learning (Yuskaitis et al. 2010; Michalon et al. 2012), but also normal performance (Bakker 1994; Dolen et al. 2007; Veeraragavan et al. 2012; Udagawa et al. 2013). However, memory extinction was exaggerated in the Fmr1 KO mice (Dolen et al. 2007; Michalon et al. 2012), as observed by shorter latencies to enter the dark compartment. In the “five-choice serial reaction time” task, an assay to measure visuospatial attention and impulsivity, mice are required to detect a random light in one of five holes and to respond with a nose-poke in the correct spatial location to receive a food reward. Fmr1 KO mice needed significantly more time to complete the task, as observed by the increased number of errors per trial during the training period (Krueger et al. 2011); however, they were able to complete the task. Additionally, mutant mice performed normally in the “olfactory discrimination task” (Mineur et al. 2002; Yan et al. 2004; Moon et al. 2008; Guo et al. 2011), suggesting that working memory (which correlates with IQ in humans) is not affected.
Associative learning dependent on hippocampus and amygdala (Phillips and LeDoux 1992) is usually monitored via “cued and contextual fear conditioning.” In this task the conditioned and nonaversive stimulus occurs in association with a harmful unconditioned stimulus. As a result of this pairing, the conditioned stimulus acquires the aversive properties of the unconditioned stimulus, inducing freezing as a readout of a defensive behavior (Curzon et al. 2009). When Fmr1 KO mice were tested in both cued (tone) and contextual (environment) fear conditioning, they were found to freeze less than the control littermates indicating memory deficits (Paradee et al. 1999; Hayashi et al. 2007; Guo et al. 2011, 2012; Olmos-Serrano et al. 2011). However, other groups using this test did not report memory deficits in the Fmr1 KO compared with control (Peier et al. 2000; Van Dam et al. 2000). The diverse outcome could stem from differences in the protocols used and/or to the influence of the strain used. Interestingly, one group reported a decreased freezing in cued fear conditioning, when the same tone is presented in an altered environment (different context) from the initial chamber where the training took place (Olmos-Serrano et al. 2011), suggesting that the amygdala-dependent learning is mostly impaired. These findings are consistent with an intact hippocampal function/s in the KO mouse, consistent with the electrophysiological results indicating normal long-term potentiation (LTP) of hippocampal Schaffer Collateral fibers after tetanic (Paradee et al. 1999) or high frequency stimulation (Godfraind et al. 1996).
Finally, the Fmr1 KO mice were studied to explore a possible behavioral inflexibility, a feature often associated to patients with ASD (Geurts et al. 2009). The test allows the monitoring of a performance of “reverse learning” after a task has been acquired. This behavior requires intact cortical function (Dalley et al. 2004). The Fmr1 KO mice, tested using the reverse MWM or reverse T/Y-maze, needed more time to learn the new position of the platform (Bakker 1994; Kooy et al. 1996; D'Hooge et al. 1997; Bhattacharya et al. 2012), suggesting that acquisition of the new spatial information is impaired in the absence of FMRP (Krueger et al. 2011). Other studies did not reach a similar conclusion (Paradee et al. 1999; Yan et al. 2004). Of note, cellular and molecular findings support a role of FMRP in the prefrontal cortex whose intact function/s is/are required for cognitive flexibility. Indeed, the expression of synaptic proteins, such as glutamate receptors, PSD95 and Arc is reduced in the prefrontal cortex of adult Fmr1 KO (Krueger et al. 2011) in accordance with the increased density of longer (and possibly immature) spines (Liu et al. 2011). Finally, the Fmr1 KO mice have a decreased neuronal activity in the prefrontal cortex, as observed by a reduction of c-fos positive neurons (Krueger et al. 2011).
Fmr1 KO mice show impaired social interaction and communication
Patients with FXS do not show major deficits in social interactions, although they show high levels of anxiety (for review, see Gross et al. 2012). However, problems with social communication are observed in patients with FXS and autistic features (Berry-Kravis 2014). In rodents, a test that mimics social preference and novelty in humans is the “three-chambered apparatus,” in which the target mouse is given the choice between exploring a cage containing a stranger mouse or an empty one. In the second phase, the test mouse can choose between the familiar mouse and a new (i.e., a second) stranger mouse. The numbers of approaches and the time spent in proximity with each mouse are scored (Moy et al. 2004). While the Fmr1 KO mice do not show any deficit in social preference (Mines et al. 2010; Bhattacharya et al. 2012), they are impaired in social novelty discrimination, because they do not show any preference for the unfamiliar stranger mouse (Mines et al. 2010; Bhattacharya et al. 2012). In addition, Fmr1 KO males display reduced social interaction with novel females (Mineur et al. 2006) as well as impaired social dominance with unfamiliar mice (Spencer et al. 2005). Such deficits may be explained by the enhanced anxiety aspect, as seen by increased rearing and digging behavior the Fmr1 KO mice exhibited in the presence of another mouse (McNaughton et al. 2008; Mines et al. 2010; Liu et al. 2011). Impaired preference for unfamiliar mice may indicate lack of interest in novelty and/or impairment to discriminate between familiar and novel mice. Indeed, when tested in the “novel object recognition” task, the Fmr1 KO mice fail to recognize the novel object (Ventura et al. 2004; Bhattacharya et al. 2012; Busquets-Garcia et al. 2013). These findings suggest that discrimination deficits are not only observed in social tasks.
Because problems in communication and speech are often reported in patients with FXS (for review, see Bagni et al. 2012; Hagerman et al. 2012), communication in the Fmr1 KO mice was also investigated by evaluating the pattern of “ultrasonic vocalizations (USV).” Pairing Fmr1 KO male with a female showed deficits in communication as observed by the reduced number of calls per second (Rotschafer et al. 2012), although no apparent difference in mating behavior was observed. After maternal separation, the type and duration of USV emitted by the Fmr1 KO pups (8 d old pups) were different from the control littermates with no apparent changes in the total number of calls (Roy et al. 2012). Recently, Lai et al. reported an increased number of USVs in younger (P7) Fmr1 KO pups (Lai et al. 2014) suggesting that the FMRP function on communication is age-dependent.
Fmr1 KO mice show deficits in sensory gating and audiogenic seizures
Abnormal sensory inhibition may reflect a deficit in processing and prioritizing incoming information, a feature of schizophrenic patients (Braff et al. 1978; Siegel et al. 1984; Brockhaus-Dumke et al. 2008; Hammer et al. 2013; Rihs et al. 2013). Treatments with antipsychotic drugs improve those deficits in rats and humans (Curzon et al. 1994; Sanchez-Morla et al. 2009; Suryavanshi et al. 2014). It is known that patients with FXS are hyperarousal in situations of excessive stimulation and habituate poorly to sensory stimuli (Berry-Kravis 2014). “Prepulse inhibition (PPI)” is used to evaluate the ability of human and rodents to filter irrelevant information in their surroundings. Therefore, Fmr1 KO mice and control littermates were tested in PPI in an acoustic startle task to investigate possible sensory gating deficits. In this task a weak stimulus, such as a tone, inhibits the subsequent response to a stronger, louder stimulus, if presented within 100 msec (Nielsen et al. 2002). Presently, how the Fmr1 KO mouse processes incoming information is still unclear, because either no differences (Peier et al. 2000; Yan et al. 2004; Spencer et al. 2006; Thomas et al. 2012) or enhanced startle responses were shown (Chen and Toth 2001; Nielsen et al. 2002; Frankland et al. 2004; de Vrij et al. 2008; Baker et al. 2010; Olmos-Serrano et al. 2011; Veeraragavan et al. 2012) compared with control animals. However, in one study (Frankland et al. 2004), Fmr1 KO mice showed an excessive reaction to auditory stimuli, similar to humans (Renoux et al. 2014). Despite the reported discrepancies, it is clear that FMRP plays a role in sensorimotor function/s underlying the altered sensitivity to sensory stimulation observed in both patients and mouse models.
Susceptibility to audiogenic seizures is one of the most robust features of FXS and indeed ∼20% of patients with FXS experience epileptic episodes (Musumeci et al. 1999; Berry-Kravis 2002). In accordance with the human data, Fmr1 KO mice display enhanced susceptibility to audiogenic seizures (Bakker 1994; Musumeci et al. 2000; Peier et al. 2000; Chen and Toth 2001; Yan et al. 2004, 2005; Min et al. 2009; Liu et al. 2011; Goebel-Goody et al. 2012; Thomas et al. 2012; Veeraragavan et al. 2012; Osterweil et al. 2013; Udagawa et al. 2013). Although the severity of the phenotype may be affected by the genetic background (Table 1; Yan et al. 2004), susceptibility to audiogenic seizures is the most consistent behavioral phenotype and is usually implemented to screen for potential drugs to ameliorate FXS.
Sleep problems are a common feature of patients with FXS (Berry-Kravis 2014), and in the mouse model FMRP has been suggested to regulate circadian rhythmicity as measured by locomotor analysis. In complete darkness, FMRP was found to regulate the length of circadian period (Zhang et al. 2008) since shorter activity periods of wheel running were observed in the Fmr1 KO mice compared with controls. Interestingly, FMRP affects circadian rhythmicity differently in females and males since ambulatory activity during the light phase was enhanced only in the Fmr1 KO females (Baker et al. 2010) and no changes were reported in males.
The fly model for FXS
In contrast to the two FMR1-related genes found in the mammalian genome (i.e., FXR1P and FXR2P), in Drosophila there is only one FMR1 homolog referred to as dFmr1, or more rarely as dfxr. Sequence comparison of the human and fly genes shows a high level of similarity between the functional regions of the proteins, with a total 56% similarity and 35% identity (Zhang et al. 2001; Gao 2002). The protein encoded by the dFmr1 (dFMRP) presents an analogous structure and shows similar RNA-binding properties to mammalian FMRP (Wan et al. 2000). Immunohistochemical analysis showed that dFMRP is continuously expressed during early embryogenesis, with strong expression in the mesoderm, the lobes of the brain and the abdominal (ventral) ganglia in larval and adult stages (Wan et al. 2000; Zhang et al. 2001; Dockendorff et al. 2002). Additional tissues where dFMRP is detected are the embryonic wing discs, testes, and ovaries (Zhang et al. 2001; Zarnescu et al. 2005). Like the homologous protein in mammals, dFMRP is highly expressed in the cytoplasm of neurons; low levels have been detected in glia cells (Wan et al. 2000; Zhang et al. 2001; Morales et al. 2002). To characterize the physiological functions of dFMRP, several loss-of-functions mutations have been generated in the dFmr1 gene (Zhang et al. 2001; Dockendorff et al. 2002; Inoue et al. 2002; Morales et al. 2002; Lee et al. 2003).
Lack of a functional dFmr1 gene results in many phenotypes reminiscent of those observed in the human condition, thus Drosophila is an appropriate and simple genetic model to study molecular, cellular, and behavioral features associated with the syndrome. At the cellular level, loss of dFmr1 results in neuron overgrowth, over-branching, and abnormalities in synapse formation, both at the peripheral nervous system (PNS) and the central nervous system (CNS), including the relatively mild defect of aberrant midline crossing of the Mushroom bodies (MB) β lobes (Morales et al. 2002; Michel et al. 2004; Pan et al. 2004; McBride et al. 2005).
dFmr1 mutant flies have abnormal circadian rhythms
As mentioned above, patients with FXS have sleep problems reflecting abnormal circadian behavior (Hagerman et al. 1996; Gould et al. 2000; Berry-Kravis 2014). “Circadian rhythm” studies in dFmr1 mutant flies, using the Trikinetics system (http://www.trikinetics.com/), revealed normal circadian rhythms but interrupted sleep and arrhythmic locomotor activity under constant darkness (Dockendorff et al. 2002; Inoue et al. 2002; Morales et al. 2002; Sekine et al. 2008). These behavioral defects are not due to motor dysfunctions because dFmr1 mutant flies did not show differences in the total activity in constant darkness when compared with the control (Dockendorff et al. 2002). These results are in agreement with the clinical observations that patients with FXS have attention deficit hyperactivity disorder (ADHD) (Hagerman 1997; Torrioli et al. 2008). Analysis of the circadian molecular clock revealed that cycling of the two key elements of the circadian clock PER and TIM proteins and their respective mRNA levels are not affected in dFmr1 mutants (Dockendorff et al. 2002; Inoue et al. 2002; Morales et al. 2002; Helfrich-Forster 2005; Chang 2006; Sehgal et al. 2007). However, the eclosion rhythm, which is controlled by the circadian system, is affected in dFmr1 mutant flies and the circadian oscillation of cAMP response element binding protein (CREB), a known output of the circadian clock in Drosophila (Belvin et al. 1999), is dramatically affected in dFmr1 mutant flies (Dockendorff et al. 2002; Morales et al. 2002). These data suggest that dFmr1 mutants present an abnormal circadian output pathway. It is well established that a molecular clock mechanism in the small ventro-lateral neurons (sLNs) of the brain controls the rest–activity rhythms (Helfrich-Forster 2005; Chang 2006; Nitabach et al. 2006). Notably, several laboratories have observed overextended and overgrowth axons in the sLNs (Dockendorff et al. 2002; Morales et al. 2002; Reeve et al. 2005; Gatto and Broadie 2008; Sekine et al. 2008; Gatto and Broadie 2009). These findings suggest that the axonal defects in sLNs might underlie the observed deficits in circadian behaviors. Interestingly, Gatto and Broadie found that in the sLNs dFMRP has a very restricted function in late brain development at complete synaptogenesis. Genetic expression of dFMRP in the dFmr1 mutant background restored the synaptic defects observed in sLNs only when expressed at a late brain developmental stage and failed to do so at early developmental stages or in the adult (Gatto and Broadie 2009). Additional evidence of dFMRP function in the regulation of circadian rhythm derives from its interaction with the RNA-binding protein, LARK. LARK is a known clock output factor, and when overexpressed causes arrhythmic locomotor activity. Accordingly, the double heterozygous mutant for dFmr1 and Lark revealed improvements in these defects (Sofola et al. 2008). FMRP and its cytoplasmic interactor CYFIP1, also known as specific RAC-1 activated protein (SRA-1), (Kobayashi et al. 1998; Schenck et al. 2003) control in an activity-dependent manner actin remodeling and local protein synthesis (Napoli et al. 2008; Galy et al. 2011; De Rubeis et al. 2013; Zhao et al. 2013). These findings suggest that the disorganization of the FMRP-CYFIP complex may result in the abnormal synaptic arborization and the defective sLNs observed in dFrm1 mutant flies. In addition to the arrhythmic activity, a prolonged “sleep phase” has been observed in dFmr1 mutant flies. The MBs, functionally analogous to the mammalian hippocampus, are essential for different types of behavior, such as sleep, associative learning, and memory (Davis 1993; Roman and Davis 2001; Joiner et al. 2006). The MBs contain the neuronal population required to regulate normal sleep activity; overexpression of dFmr1 in the MBs reduced the prolonged sleep episodes of mutant flies (Bushey et al. 2009). In another recent study, dFmr1 mutants showed a deeper night-like sleep phenotype during the day, which was regulated by two molecules, dFMRP and cAMP (van Alphen et al. 2013).
dFmr1 mutant flies show impairment in social interaction
The majority of neuropsychiatric and neurodevelopmental disorders entail defects in social behavior. The “courtship paradigm” in the fruit fly is used to probe potential social impairments upon dFmr1 loss in the fly. Courtship in Drosophila is based on visual, auditory and pheromonal cues and is used to assess behavioral interactions between male and female flies (Hall 1994; Greenspan and Ferveur 2000; Dockendorff et al. 2002; Yamamoto and Koganezawa 2013). The courtship repertoire is composed of four basic phases starting with orientation of a male towards a female and culminating with the final step of copulation. dFmr1 mutant males spend less time in active courtship in comparison with control flies and do not proceed to more advanced stages of courtship such as copulation. Therefore, dFmr1 mutant flies present shorter courtship duration in comparison with control flies. This behavior is consistent with lack or loss of interest by the mutant flies, a common characteristic of patients with FXS and ADHD (Dockendorff et al. 2002).
FXS patients present an increased rate of cognitive impairments with age (Galvez and Greenough 2005; Larson et al. 2005). Therefore, social interactions such as naïve courtship were studied in aged dFmr1 mutant flies, but no further differences were reported in comparison to young flies (Choi et al. 2010). Additional studies using dFmr1 transgenic flies expressing only proteins with mutations in the KH domains (I244N or I307N) revealed reduced naïve courtship activity, suggesting that this effect is likely due to the lack of dFMRP's ability to bind a specific subset of mRNAs (Banerjee et al. 2007). Furthermore, it was shown that dFmr1 mutant flies exhibit decreased locomotor activity and exploratory performance and reduced conspecific interactions compared with control flies (Bolduc et al. 2010), presenting additional evidence of defects in social interactions. These findings further support the use of dFmr1 mutant flies to address questions about the affected neuronal circuitry and molecular pathways controlling these behaviors.
Learning and memory in dFmr1 mutant flies
Two main behavioral paradigms have been described to investigate learning and memory in the Drosophila model of FXS, courtship, and olfactory conditioning (McBride et al. 2005; Bolduc et al. 2008; Coffee et al. 2010, 2012; Kanellopoulos et al. 2012; Gatto et al. 2014). In 2005, using the “courtship-conditioning paradigm,” McBride et al. investigated learning in dFmr1 mutant flies for the first time. In courtship conditioning a naïve male alters his courtship efforts towards any female after experiencing rejection by an unreceptive female: this constitutes a learning paradigm in flies (Hall 1994). dFmr1 mutant flies were found to have normal learning during the training phase of conditioned courtship but impairments in the immediate recall and short-term memory phase (McBride et al. 2005). Learning and memory were further assessed through the “olfactory classical conditioning paradigm,” which couples aversive olfactory stimuli (conditioned stimulus) with electric shock (unconditioned stimulus) (Tully 1984; Tully and Quinn 1985). Using this paradigm, it has been shown that dFmr1 mutant flies exhibit a robust associative learning deficit in comparison with wild-type flies (McBride et al. 2005; Bolduc et al. 2008; Coffee et al. 2010, 2012; Gatto et al. 2014). Similarly to the mouse model, dFmr1 homozygous mutants also have deficits in long-term memory. It has been proposed that protein synthesis through the miRNA pathway involving Staufen and AGO1 may be affected in those flies (Bolduc et al. 2008). These findings might explain the long-term memory deficits. In addition, heterozygous loss of function of both dFmr1 and cheerio, an ortholog of filamin A, revealed long-term memory deficits (Bolduc et al. 2010). These data suggest that microRNA patways acting on filamin A mRNA and/or other processes might be responsible for these behavioral phenotypes. Finally, a recent study by Kanellopoulos et al. showed that dFmr1 mutant heterozygotes exhibited a robust learning deficit under sensitive conditions of limited US/CS stimulus pairings (Moressis et al. 2009) and verified that under the typically used extended training conditions (Bolduc et al. 2008) these flies appear normal. In this study, using genetic and pharmacological tools, it was found that the effects of increased mGluR levels upon dFMRP reduction are linked to reduced cAMP, which in turn likely results in the associative learning and memory deficits (Kanellopoulos et al. 2012).
The Drosophila MBs are the main part of the brain involved in the two learning and memory paradigms, such as conditioned courtship and classical conditioning (Davis 1993; McBride et al. 1999; Pascual and Preat 2001). Several studies revealed that dFmr1 mutant flies have MBs developmental defects. Specifically, the β-lobe axons of the MBs do not terminate in the midline, leading to the formation of fused lobes (Michel et al. 2004; Pan et al. 2004; McBride et al. 2005; Bolduc et al. 2008; Chang et al. 2008; Coffee et al. 2010, 2012; Gatto et al. 2014). Absence of dFMRP causes over-elaboration and overgrowth of axons of the MB neurons (Pan et al. 2004; Tessier and Broadie 2008) suggesting that the structural abnormalities of the MBs may explain the learning and memory deficits of the dFmr1 mutant flies.
Pharmacological and genetic rescue of behavioral deficits observed in the mouse and fly models for FXS
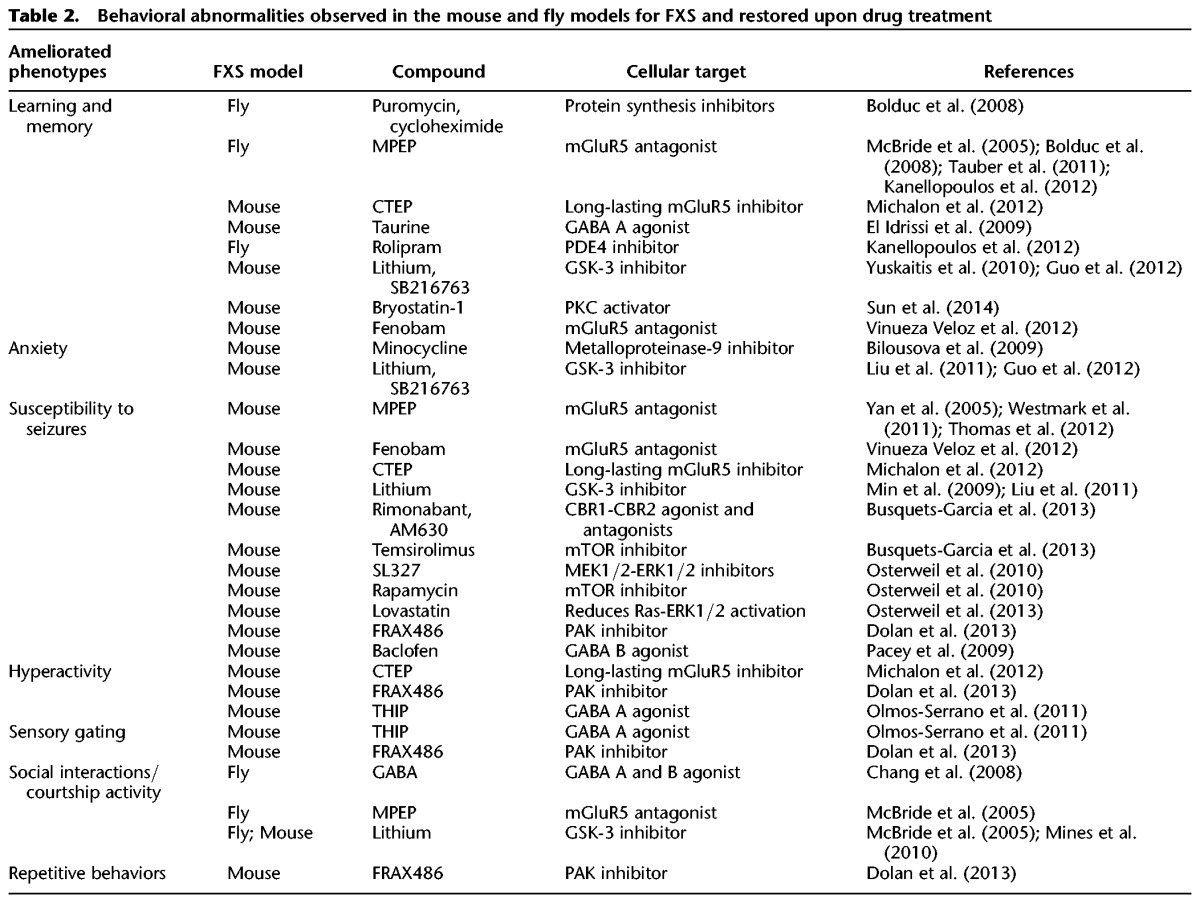
In this section, we review some of the strategies used with the Fmr1 KO mouse and the fly mutant model to ameliorate the FXS phenotypes (Table 2). Due to the initial discovery, i.e., increased mGluRs signaling in FXS (Huber et al. 2002) many of the therapeutic strategies are based on targeting of key dysregulated molecules of the mGluR pathway and downstream players regulating protein translation (www.clinicaltrial.org).
Table 2.
Behavioral abnormalities observed in the mouse and fly models for FXS and restored upon drug treatment
Over the past 10 years, additional receptor pathways have been shown to be affected in FXS, including the GABAergic, NMDA, TrkB, and cannabinoid systems (for review, see Bagni et al. 2012), and specific molecules have been used to ameliorate those pathways. Furthermore, since FMRP plays a central role as translational repressor (for review, see Bagni et al. 2012; Darnell and Klann 2013), a few other approaches have been developed to tackle the increased protein synthesis.
A hallmark of FXS is the increased susceptibility to seizures, and administration of several drugs targeting the mGluR pathway such as MPEP (Yan et al. 2005; Westmark et al. 2011; Thomas et al. 2012) and Fenobam (Westmark et al. 2011) were able to ameliorate these deficits. Similar effects were observed by targeting specific molecules which are dysregulated in the Fmr1 KO mouse (for review, see Bagni et al. 2012) with in vivo administration of compounds such as lithium (Min et al. 2009; Liu et al. 2011), GSK-3β inhibitors (Min et al. 2009), mTOR inhibitors (Busquets-Garcia et al. 2013), and MEK1/2-ERK1/2 inhibitors (Osterweil et al. 2010). The long-lasting mGluR5 inhibitor, CTEP, was also shown to correct elevated protein synthesis, hypersensitivity to sensory stimuli and hyperactivity in the Fmr1 KO mice (Michalon et al. 2012). In addition, the mGluR5 inhibitors showed an effect on learning and memory deficits (Michalon et al. 2012; Vinueza Veloz et al. 2012).
Genetic and pharmacological manipulations reducing the mGluRs activity were able to restore many phenotypes observed in dFmr1 mutant flies. Using the naïve courtship and memory recall assays, McBride et al. (2005) showed that dFMRP mutant flies fed with mGlu5R antagonists (MPEP, MPPG, MTPG, and LY341495) exhibit reversed social and learning behaviors. Some of these affected behaviors could only be reestablished if the drug was administered during development (McBride et al. 2005). In addition, administration of MPEP could restore the following characteristics of the dFmr1 mutant flies: the structural defects observed in the MBs (McBride et al. 2005; Pan et al. 2008), the impaired axon arborization (Pan et al. 2008), the increased embryonic lethality on glutamate enriched food (Chang et al. 2008), the social learning deficits (Tauber et al. 2011), and short- and long-term olfactory memory deficits (Bolduc et al. 2008; Kanellopoulos et al. 2012). In contrast, MPEP failed to restore the circadian deficits of the dFmr1 mutant flies (McBride et al. 2005). Kanellopoulos et al. demonstrated that the administration of MPEP and/or the PDE inhibitor Rolipram, which increases cAMP levels, restores the olfactory associative learning and memory deficits of dFmr1 heterozygous mutants. In addition, genetic abrogation of mGluRs in dFmr1 mutant background or the double-mutant dFmr1 and dnc (PDE4) could also restore cAMP levels and the corresponding behavioral phenotypes.
These improvements using the fly model for FXS are similar to those observed in the mouse model indicating that the behavioral plasticity deficits could be attributed to a large degree to exaggerated responses to glutamate.
Because these findings pointed out the central role of the mGluR and downstream signaling pathway/s, a few compounds targeting the mGluR activity have been used in clinical trials. RO4917523 (Roche Pharmaceuticals) and STX107 (Seaside Therapeutics) are in Phase 2 trials. The outcome of a clinical trial with Fenobam showed an improvement of 20% in the PPI (Berry-Kravis et al. 2009), although the mGluR5 antagonist AFQ056 (Novartis) was very recently discontinued due to the lack of positive outcome measurements (Busquets-Garcia et al. 2014; Gomez-Mancilla et al. 2014).
As previously mentioned, animal and fly models for FXS express certain subunits of the GABA receptors at lower level compared with WT (for review, see D'Hulst and Kooy 2007). This defect suggests an enhancement of excitatory signaling, consistent with the high susceptibility to seizures observed in patients with the syndrome (Hagerman et al. 2012) confirmed in the mouse model. In accordance with these findings, treatment with GABA agonists normalized hyperactivity and sensory gating defects (Pacey et al. 2009; Olmos-Serrano et al. 2011) and significantly improved conditioned learning (El Idrissi et al. 2009) in Fmr1 KO mice. Similarly, treatment with GABA also restored the courtship defects of mutant flies (Chang et al. 2008). Taking advantage of the fly model, a large screening was performed using 2000 chemical compounds (Chang et al. 2008). This study has led to the identification of a few molecules, implicated in the GABAergic pathway, able to restore the courtship phenotype (Chang et al. 2008). In a more recent study, GABAergic modulation after feeding dFmr1 mutant flies with GABA or Nipecotic Acid (a GABA reuptake inhibitor) failed to restore the associative learning deficits, suggesting that the GABAergic signaling alterations in the dFmr1 mutant flies do not appear to underlie their associative learning deficits (Gatto et al. 2014). The alterations in the GABAergic system led to the development of clinical trials with compounds able to restore the GABA receptor signaling. For example, patients with FXS given Arbaclofen (STX209), an agonist of GABA B receptor, showed an improvement in social behavior (Berry-Kravis et al. 2012). Ganaxolone and Acamprosate, agonists of GABA A receptor, are also in clinical trials (for review, see Hagerman et al. 2014).
In addition to targeting the receptors, other strategies have envisioned the use of compounds able to target the dysregulated proteins in the absence of FMRP. For example, GSK-3β activity is increased in Fmr1 KO mice (Min et al. 2009; Guo et al. 2012), and because lithium acts as a nonspecific inhibitor of GSK-3β (Min et al. 2009; Guo et al. 2012), it has been used in both mouse and fly models for FXS. Lithium administration improved performance in several deficient behavioral tasks in the Fmr1 KO mouse such as open-field (Min et al. 2009; Yuskaitis et al. 2010; Liu et al. 2011), passive avoidance memory (Yuskaitis et al. 2010), and social preference (Mines et al. 2010). Lithium had also a positive effect on courtship and MBs structural abnormalities in the fly model (McBride et al. 2005). However, some symptoms reappeared when the treatment was discontinued (King et al. 2013). Finally, a specific inhibitor of GSK-3β was shown to normalize hippocampus-dependent learning deficits in Fmr1 KO mice (Guo et al. 2012), highlighting a role for GSK-3β dysfunction in FXS pathology. While lithium is a common mood stabilizer that seems to improve the anxiety and hyperactivity features of patients with FXS, no additional clinical trials have been envisioned so far (Hagerman et al. 2014).
The intra-signaling cascade regulating protein synthesis, spine shaping, and synaptic plasticity, such as mTOR (Busquets-Garcia et al. 2013), ERK (Osterweil et al. 2010, 2013), and PKC (Weiler et al. 2004), are also impaired in FXS. Consistent with this finding, a potent PKC activator (bryostatin-1) was reported to restore cellular, molecular, and behavioral phenotypes in the Fmr1 KO mice. In addition, the phosphorylation levels of GSK-3β, affected in FXS, were also restored (Sun et al. 2014).
Finally FMRP links local protein synthesis to actin remodeling (Schenck et al. 2003; De Rubeis et al. 2013) via its cytoplasmic interactor CYFIP1 (Napoli et al. 2008). Additional contacts with the cytoskeleton occur via the adenomatous polyposis coli (APC) tumor suppressor (Mili et al. 2008) and p21-activated kinase (PAK1) (Hayashi et al. 2007). PAK1 has been shown to antagonize the role of FMRP by reducing both spine number and the proportion of longer and thinner dendrites (Hayashi et al. 2004). Consistently, genetic manipulation or drug treatment leading to PAK1 activity reduction (Hayashi et al. 2007; Dolan et al. 2013) correct hyperactivity (Hayashi et al. 2007; Dolan et al. 2013) and repetitive behaviors as well as reduce the frequency of seizures (Dolan et al. 2013) of mutant mice.
En masse, in the absence of FMRP, many molecules that are members of second messenger pathways including mTOR, PKC, ERK1/2, GSK-3β, cAMP, and PAK1 are dysregulated. Because these pathways converge on the translational initiation factor eIF4E, changes in their activity might explain the increased protein synthesis observed in FXS. In support of these findings, the use of protein synthesis inhibitors appears to ameliorate some of the cellular and molecular defects. For example, cycloheximide and puromycin, two inhibitors of general protein synthesis, restored long-term memory deficits of dFmr1 mutant flies (Bolduc et al. 2008). Additionally, minocycline, thought to exert its antimicrobial effect by the inhibition of protein synthesis (Garrido-Mesa et al. 2013), restores the anxiety-like phenotype (Bilousova et al. 2009) in the Fmr1 KO mice leading to long-term improvements after drug withdrawal (Dansie et al. 2013). Intracellular minocycline has been shown to inhibit metalloproteinase-9 (MMP-9) involved in synaptic plasticity and dendritic structure (Michaluk et al. 2011; Janusz et al. 2013). Accordingly, MMP-9 activity is enhanced in hippocampi of Fmr1 KO mice and restored to control levels after minocycline treatment (Bilousova et al. 2009). Treatment of dFmr1 null animals with minocycline restored the abnormal synaptic morphology observed in three different neuronal areas: the neuromuscular junction (NMJ), the clock neurons, and the Kenyon cells of MBs (Siller and Broadie 2011). Finally, minocycline administration in patients with FXS was shown to improve attention deficits, language use, and communication skills (Paribello et al. 2010; Utari et al. 2010; Leigh et al. 2013).
Besides investigations of drug effects and potential treatments, several groups have explored genetic rescue approaches of FXS deficits. Expression of several key molecules implicated in FXS pathology has been modulated to reestablish some of the phenotypes characterizing FXS.
The increased receptor activity in FXS was first observed upon genetic reduction of mGluR5 expression (Dolen et al. 2007), or modulation of mGluR5-Homer interaction (Ronesi et al. 2012). In both genetic rescues the authors observe a normalization of the enhanced susceptibility to auditory stimuli displayed by Fmr1 KO mice. Recently, double-mutant Fmr1 KO mice with reduced expression of endocannabinoid receptor 1 (CBR1) were shown to revert to normalcy with respect to several phenotypic deficits, such as cognitive impairment and audiogenic seizure susceptibility (Busquets-Garcia et al. 2013). Interestingly, the authors of this study show that treatment with inhibitors of the endocannabinoid system is also effective, both acutely and chronically, in normalizing altered spine morphology and several behavioral phenotypes.
Three up-regulated molecules involved in the intracellular signaling and cytoskeleton remodeling, such as amyloid Precursor Protein (APP), striatal enriched protein tyrosine phosphatase (STEP), and microtubule associated Protein 1 (MAP1b), are encoded by known FMRP target mRNAs (for review, see Bagni et al. 2012). The genetic cross of the APP KO with the Fmr1 KO lead to amelioration of cellular and behavioral FXS effects (Westmark et al. 2011). Reduction of STEP (Goebel-Goody et al. 2012), a target of FMRP, corrects some behavioral phenotypes of Fmr1 KO mice. Furthermore, the doubly mutant dfmr1 and futsch (a Map1b homolog and target of FMRP) in flies could rescue the synaptic defects in the neuromuscular junction and in the eye, but failed to rescue the locomotor activity (Zhang et al. 2001).
Finally, the modulation of proteins involved in protein synthesis seems to counteract the absence of FMRP. For instance, one of the RNA-binding proteins colocalizing with FMRP in dendrites is the cytoplasmic polyadenylation element-binding protein 1 (CPEB1) (Ferrari et al. 2007), which regulates polyadenylation and mRNA metabolism. Double Fmr1 and CPEB1 KO restored the enhanced susceptibility to seizures of Fmr1 KO and working memory deficits (Udagawa et al. 2013). Furthermore, in agreement with increased protein translation in Fmr1 KO mice, genetic reduction of S6K1, a key translation initiation and elongation factor, corrected deficits in cortical-dependent tasks, such as novel object recognition and inflexibility, while reduction of S6K1 was not sufficient to prevent hyperactivity (Bhattacharya et al. 2012). These findings suggest that multiple key proteins may regulate brain function in a tissue and developmental-specific manner ultimately causing the complex FXS symptomatology. Further investigations are required to understand the molecular events underlying all these successful different genetic rescues.
Conclusions
Over the past few years, intense research on FXS has led to the identification of several hundreds of putative FMRP mRNA targets (for review, see Fernandez et al. 2013). This large number of FMRP mRNA targets might explain the extensive and heterogeneous behavioral deficits in FXS, suggesting that FMRP loss influences different circuitries and causes alterations in various receptor pathways. Although, it is very well established that FMRP loss alters the glutamatergic signaling (mGluR theory), many other molecular pathways such as BDNF, mTOR, ERK1/2, cAMP, and PKC cascades are affected. In addition, different neuronal circuits, such as the GABAergic, cholinergic, dopaminergic, and serotonergic systems, are modified by FMRP loss.
Although the genetic cause leading to FXS is the absence of a single gene, the FMR1, the wide spectrum of disabilities and heterogeneity of clinical and cognitive features among patients with FXS renders the cure of the disease difficult. The combination of pharmaceutical treatments targeting different molecules altered in FXS might be the key for the amelioration of FXS deficits. On the other hand, the use of fly and mouse models has been of utmost importance. Both models re-create the cellular and molecular alterations caused by the absence of a functional FMRP, suggesting that the pathophysiology of FXS is evolutionarily conserved between these species.
Furthermore, as discussed above, loss of FMRP leads to several behavioral deficits in both mice and flies. Taking advantage of such deficits, several drug therapies have been tested and developed using these models. While, due to the diversity of the phenotypes and genetic heterogeneity observed in FXS patients, the clinical impairments are only partially ameliorated, it is remarkable that the responses to treatment in flies and mice are so well conserved. These findings strongly suggest that how these treatments manifest behaviorally depends on the complexity of the nervous system in different species.
Because a single animal model cannot always fully re-create the FXS behavioral phenotypes, the search for new and targeted therapies should also be focused on the development of assays to properly evaluate the effect of a specific drug on more than one model and in the context of different genetic backgrounds.
Acknowledgments
We thank Efthimios M.C. Skoulakis (BSRC Alexander Fleming, Greece) for reading the manuscript and providing insightful comments and suggestions. We are extremely grateful to Myles W. Jackson (NYU) for his thoughtful proofreading of the text. This work was supported by grants from VIB, SAO, FWO-G.0705.11, and FWO-G.0667.09, Associazione Italiana Sindrome X Fragile, Queen Elisabeth Foundation (Belgium), CARIPLO, HEALTH-2009-2.1.2-1 EU-FP7 “SynSys.” Ana Rita Santos is recipient of a Marie Curie-COFUND VIB fellowship (omics@vib). We thank Eef Lemmens for her excellent administrative support.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.035956.114.
References
- Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, Schuman EM, Rosenblum K, Klann E 2008. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem 15: 29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Greenough WT 2005. From mRNP trafficking to spine dysmorphogenesis: the roots of Fragile X syndrome. Nat Rev Neurosci 6: 376–387 [DOI] [PubMed] [Google Scholar]
- Bagni C, Tassone F, Neri G, Hagerman R 2012. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest 122: 4314–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KB, Wray SP, Ritter R, Mason S, Lanthorn TH, Savelieva KV 2010. Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav 9: 562–574 [DOI] [PubMed] [Google Scholar]
- Banerjee P, Nayar S, Hebbar S, Fox CF, Jacobs MC, Park JH, Fernandes JJ, Dockendorff TC 2007. Substitution of critical isoleucines in the KH domains of Drosophila fragile X protein results in partial loss-of-function phenotypes. Genetics 175: 1241–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST 2004. The mGluR theory of fragile X mental retardation. Trends Neurosci 27: 370–377 [DOI] [PubMed] [Google Scholar]
- Belvin MP, Zhou H, Yin JC 1999. The Drosophila dCREB2 gene affects the circadian clock. Neuron 22: 777–787 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E 2002. Epilepsy in Fragile X syndrome. Dev Med Child Neurol 44: 724–728 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E 2014. Mechanism-based treatments in neurodevelopmental disorders: Fragile X syndrome. Pediatr Neurol 50: 297–302 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, et al. 2009. A pilot open label, single dose trial of fenobam in adults with Fragile X syndrome. J Med Genet 46: 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis EM, Hessl D, Rathmell B, Zarevics P, Cherubini M, Walton-Bowen K, Mu Y, Nguyen DV, Gonzalez-Heydrich J, Wang PP, et al. 2012. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med 4: 152ra127. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E 2012. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76: 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM 2009. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet 46: 94–102 [DOI] [PubMed] [Google Scholar]
- Bolduc FV, Bell K, Cox H, Broadie KS, Tully T 2008. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci 11: 1143–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc FV, Valente D, Mitra P, Tully T 2010. An assay for social interaction in Drosophila Fragile X mutants. Fly (Austin) 4: 216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L 1978. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15: 339–343 [DOI] [PubMed] [Google Scholar]
- Brockhaus-Dumke A, Schultze-Lutter F, Mueller R, Tendolkar I, Bechdolf A, Pukrop R, Klosterkoetter J, Ruhrmann S 2008. Sensory gating in schizophrenia: P50 and N100 gating in antipsychotic-free subjects at risk, first-episode, and chronic patients. Biol Psychiatry 64: 376–384 [DOI] [PubMed] [Google Scholar]
- Bushey D, Tononi G, Cirelli C 2009. The Drosophila fragile X mental retardation gene regulates sleep need. J Neurosci 29: 1948–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busquets-Garcia A, Gomis-Gonzalez M, Guegan T, Agustin-Pavon C, Pastor A, Mato S, Perez-Samartin A, Matute C, de la Torre R, Dierssen M, et al. 2013. Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19: 603–607 [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Maldonado R, Ozaita A 2014. New insights into the molecular pathophysiology of fragile X syndrome and therapeutic perspectives from the animal model. Int J Biochem Cell Biol 53C: 121–126 [DOI] [PubMed] [Google Scholar]
- Chang DC 2006. Neural circuits underlying circadian behavior in Drosophila melanogaster. Behav Processes 71: 211–225 [DOI] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST 2008. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol 4: 256–263 [DOI] [PubMed] [Google Scholar]
- Chen L, Toth M 2001. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103: 1043–1050 [DOI] [PubMed] [Google Scholar]
- Choi CH, McBride SM, Schoenfeld BP, Liebelt DA, Ferreiro D, Ferrick NJ, Hinchey P, Kollaros M, Rudominer RL, Terlizzi AM, et al. 2010. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 11: 347–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee RL Jr, Tessier CR, Woodruff EA III, Broadie K 2010. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Model Mech 3: 471–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee RL Jr, Williamson AJ, Adkins CM, Gray MC, Page TL, Broadie K 2012. In vivo neuronal function of the fragile X mental retardation protein is regulated by phosphorylation. Hum Mol Genet 21: 900–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro L, Ballinger E, Hagerman R, Hessl D 2011. Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J Neurodev Disord 3: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curzon P, Kim DJ, Decker MW 1994. Effect of nicotine, lobeline, and mecamylamine on sensory gating in the rat. Pharmacol Biochem Behav 49: 877–882 [DOI] [PubMed] [Google Scholar]
- Curzon P, Rustay NR, Browman KE 2009. Cued and contextual fear conditioning for rodents. In Methods of behavior analysis in neuroscience (ed. Buccafusco JJ), pp. 19–37, chapter 2 CRC Press, Boca Raton, FL: [PubMed] [Google Scholar]
- Dalley JW, Cardinal RN, Robbins TW 2004. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev 28: 771–784 [DOI] [PubMed] [Google Scholar]
- Dansie LE, Phommahaxay K, Okusanya AG, Uwadia J, Huang M, Rotschafer SE, Razak KA, Ethell DW, Ethell IM 2013. Long-lasting effects of minocycline on behavior in young but not adult Fragile X mice. Neuroscience 246: 186–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Klann E 2013. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16: 1530–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL 1993. Mushroom bodies and Drosophila learning. Neuron 11: 1–14 [DOI] [PubMed] [Google Scholar]
- De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA, Willems PJ 1993. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet 3: 31–35 [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Fernandez E, Buzzi A, Di Marino D, Bagni C 2012. Molecular and cellular aspects of mental retardation in the Fragile X syndrome: from gene mutation/s to spine dysmorphogenesis. Adv Exp Med Biol 970: 517–551 [DOI] [PubMed] [Google Scholar]
- De Rubeis S, Pasciuto E, Li KW, Fernandez E, Di Marino D, Buzzi A, Ostroff LE, Klann E, Zwartkruis FJ, Komiyama NH, et al. 2013. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron 79: 1169–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R 2008. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis 31: 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, Kooy RF, Oostra BA, Willems PJ, De Deyn PP 1997. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76: 367–376 [DOI] [PubMed] [Google Scholar]
- D'Hulst C, Kooy RF 2007. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci 30: 425–431 [DOI] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA 2002. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34: 973–984 [DOI] [PubMed] [Google Scholar]
- Dolan BM, Duron SG, Campbell DA, Vollrath B, Rao BS, Ko HY, Lin GG, Govindarajan A, Choi SY, Tonegawa S 2013. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci 110: 5671–5676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF 2007. Correction of fragile X syndrome in mice. Neuron 56: 955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll CA, Broadie K 2014. Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front Cell Neurosci 8: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle M, Kiebler MA 2012. A numbers game underpins cytoplasmic mRNA transport. Nat Cell Biol 14: 333–335 [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium. 1994. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell 78: 23–33 [PubMed] [Google Scholar]
- El Idrissi A, Boukarrou L, Dokin C, Brown WT 2009. Taurine improves congestive functions in a mouse model of fragile X syndrome. Adv Exp Med Biol 643: 191–198 [DOI] [PubMed] [Google Scholar]
- Fernandez E, Rajan N, Bagni C 2013. The FMRP regulon: from targets to disease convergence. Front Neurosci 7: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Mercaldo V, Piccoli G, Sala C, Cannata S, Achsel T, Bagni C 2007. The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol Cell Neurosci 34: 343–354 [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM 2002. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 39: 29–54 [DOI] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ 2004. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 9: 417–425 [DOI] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. 2014. De novo mutations in schizophrenia implicate synaptic networks. Nature 506: 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG Jr, Warren ST, et al. 1991. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67: 1047–1058 [DOI] [PubMed] [Google Scholar]
- Galvez R, Greenough WT 2005. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A 135: 155–160 [DOI] [PubMed] [Google Scholar]
- Galy A, Schenck A, Sahin HB, Qurashi A, Sahel JA, Diebold C, Giangrande A 2011. CYFIP dependent actin remodeling controls specific aspects of Drosophila eye morphogenesis. Dev Biol 359: 37–46 [DOI] [PubMed] [Google Scholar]
- Gao FB 2002. Understanding fragile X syndrome: in sights from retarded flies. Neuron 34: 859–862 [DOI] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, Galvez J 2013. What is behind the non-antibiotic properties of minocycline? Pharmacol Res 67: 18–30 [DOI] [PubMed] [Google Scholar]
- Gatto CL, Broadie K 2008. Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135: 2637–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K 2009. Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Pereira D, Broadie K 2014. GABAergic circuit dysfunction in the Drosophila Fragile X syndrome model. Neurobiol Dis 65: 142–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts HM, Corbett B, Solomon M 2009. The paradox of cognitive flexibility in autism. Trends Cogn Sci 13: 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind JM, Reyniers E, De Boulle K, D'Hooge R, De Deyn PP, Bakker CE, Oostra BA, Kooy RF, Willems PJ 1996. Long-term potentiation in the hippocampus of fragile X knockout mice. Am J Med Genet 64: 246–251 [DOI] [PubMed] [Google Scholar]
- Goebel-Goody SM, Wilson-Wallis ED, Royston S, Tagliatela SM, Naegele JR, Lombroso PJ 2012. Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model. Genes Brain Behav 11: 586–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Mancilla B, Berry-Kravis E, Hagerman R, von Raison F, Apostol G, Ufer M, Gasparini F, Jacquemont S 2014. Development of mavoglurant and its potential for the treatment of fragile X syndrome. Expert Opin Investig Drugs 23: 125–134 [DOI] [PubMed] [Google Scholar]
- Gould EL, Loesch DZ, Martin MJ, Hagerman RJ, Armstrong SM, Huggins RM 2000. Melatonin profiles and sleep characteristics in boys with fragile X syndrome: a preliminary study. Am J Med Genet 95: 307–315 [PubMed] [Google Scholar]
- Greenspan RJ, Ferveur JF 2000. Courtship in Drosophila. Annu Rev Genet 34: 205–232 [DOI] [PubMed] [Google Scholar]
- Gross C, Berry-Kravis EM, Bassell GJ 2012. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 37: 178–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, et al. 2011. Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17: 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Murthy AC, Zhang L, Johnson EB, Schaller EG, Allan AM, Zhao X 2012. Inhibition of GSK3beta improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet 21: 681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R 1997. Fragile X: treatment of hyperactivity. Pediatrics 99: 753. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Staley LW, O'Conner R, Lugenbeel K, Nelson D, McLean SD, Taylor A 1996. Learning-disabled males with a fragile X CGG expansion in the upper premutation size range. Pediatrics 97: 122–126 [PubMed] [Google Scholar]
- Hagerman R, Lauterborn J, Au J, Berry-Kravis E 2012. Fragile X syndrome and targeted treatment trials. Results Probl Cell Differ 54: 297–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Des-Portes V, Gasparini F, Jacquemont S, Gomez-Mancilla B 2014. Translating molecular advances in fragile X syndrome into therapy: a review. J Clin Psychiatry 75: e294–e307 [DOI] [PubMed] [Google Scholar]
- Hall JC 1994. The mating of a fly. Science 264: 1702–1714 [DOI] [PubMed] [Google Scholar]
- Hammer TB, Oranje B, Skimminge A, Aggernaes B, Ebdrup BH, Glenthoj B, Baare W 2013. Structural brain correlates of sensorimotor gating in antipsychotic-naive men with first-episode schizophrenia. J Psychiatry Neurosci 38: 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi ML, Choi SY, Rao BS, Jung HY, Lee HK, Zhang D, Chattarji S, Kirkwood A, Tonegawa S 2004. Altered cortical synaptic morphology and impaired memory consolidation in forebrain- specific dominant-negative PAK transgenic mice. Neuron 42: 773–787 [DOI] [PubMed] [Google Scholar]
- Hayashi ML, Rao BS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S 2007. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci 104: 11489–11494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht F, Sutherland GR 1985. Detection of fragile sites on human chromosomes. Clin Genet 28: 95–96 [DOI] [PubMed] [Google Scholar]
- Helfrich-Forster C 2005. Neurobiology of the fruit fly's circadian clock. Genes Brain Behav 4: 65–76 [DOI] [PubMed] [Google Scholar]
- Ho VM, Lee JA, Martin KC 2011. The cell biology of synaptic plasticity. Science 334: 623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberg H, Holt C 2013. RNA-binding proteins and translational regulation in axons and growth cones. Front Neurosci 7: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF 2002. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci 99: 7746–7750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Shimoda M, Nishinokubi I, Siomi MC, Okamura M, Nakamura A, Kobayashi S, Ishida N, Siomi H 2002. A role for the Drosophila fragile X-related gene in circadian output. Curr Biol 12: 1331–1335 [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. 2012. De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Berry-Kravis E, Hagerman R, von Raison F, Gasparini F, Apostol G, Ufer M, Des Portes V, Gomez-Mancilla B 2014. The challenges of clinical trials in fragile X syndrome. Psychopharmacology 231: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janusz A, Milek J, Perycz M, Pacini L, Bagni C, Kaczmarek L, Dziembowska M 2013. The Fragile X mental retardation protein regulates matrix metalloproteinase 9 mRNA at synapses. J Neurosci 33: 18234–18241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner WJ, Crocker A, White BH, Sehgal A 2006. Sleep in Drosophila is regulated by adult mushroom bodies. Nature 441: 757–760 [DOI] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulos AK, Semelidou O, Kotini AG, Anezaki M, Skoulakis EM 2012. Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci 32: 13111–13124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM 1996. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: 1402–1406 [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Bear MF 2008. The autistic neuron: troubled translation? Cell 135: 401–406 [DOI] [PubMed] [Google Scholar]
- King M, Bottomley C, Bellon-Saameno J, Torres-Gonzalez F, Svab I, Rotar D, Xavier M, Nazareth I 2013. Predicting onset of major depression in general practice attendees in Europe: extending the application of the predictD risk algorithm from 12 to 24 months. Psychol Med 43: 1929–1939 [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Kuroda S, Fukata M, Nakamura T, Nagase T, Nomura N, Matsuura Y, Yoshida-Kubomura N, Iwamatsu A, Kaibuchi K 1998. p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J Biol Chem 273: 291–295 [DOI] [PubMed] [Google Scholar]
- Kooy RF, D'Hooge R, Reyniers E, Bakker CE, Nagels G, De Boulle K, Storm K, Clincke G, De Deyn PP, Oostra BA, et al. 1996. Transgenic mouse model for the fragile X syndrome. Am J Med Genet 64: 241–245 [DOI] [PubMed] [Google Scholar]
- Krueger DD, Bear MF 2011. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu Rev Med 62: 411–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF 2011. Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci 108: 2587–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai JK, Sobala-Drozdowski M, Zhou L, Doering LC, Faure PA, Foster JA 2014. Temporal and spectral differences in the ultrasonic vocalizations of fragile X knock out mice during postnatal development. Behav Brain Res 259: 119–130 [DOI] [PubMed] [Google Scholar]
- Larson J, Jessen RE, Kim D, Fine AK, du Hoffmann J 2005. Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J Neurosci 25: 9460–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Li W, Xu K, Bogert BA, Su K, Gao FB 2003. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development 130: 5543–5552 [DOI] [PubMed] [Google Scholar]
- Leigh MJ, Nguyen DV, Mu Y, Winarni TI, Schneider A, Chechi T, Polussa J, Doucet P, Tassone F, Rivera SM, et al. 2013. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr 34: 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Smith CB 2009. Dissociation of social and nonsocial anxiety in a mouse model of fragile X syndrome. Neurosci Lett 454: 62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZH, Chuang DM, Smith CB 2011. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol 14: 618–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL 1995. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet 10: 483–485 [DOI] [PubMed] [Google Scholar]
- Mandel JL, Biancalana V 2004. Fragile X mental retardation syndrome: from pathogenesis to diagnostic issues. Growth Horm IGF Res 14 Suppl A: S158–S165 [DOI] [PubMed] [Google Scholar]
- McBride SM, Giuliani G, Choi C, Krause P, Correale D, Watson K, Baker G, Siwicki KK 1999. Mushroom body ablation impairs short-term memory and long-term memory of courtship conditioning in Drosophila melanogaster. Neuron 24: 967–977 [DOI] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, et al. 2005. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45: 753–764 [DOI] [PubMed] [Google Scholar]
- McNaughton CH, Moon J, Strawderman MS, Maclean KN, Evans J, Strupp BJ 2008. Evidence for social anxiety and impaired social cognition in a mouse model of fragile X syndrome. Behav Neurosci 122: 293–300 [DOI] [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L 2012. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74: 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, Wawrzyniak M, Alot P, Szczot M, Wyrembek P, Mercik K, Medvedev N, Wilczek E, De Roo M, Zuschratter W, et al. 2011. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J Cell Sci 124: 3369–3380 [DOI] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL 2004. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci 24: 5798–5809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, Moissoglu K, Macara IG 2008. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature 453: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, Bauchwitz RP 2009. Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56: 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS 2010. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One 5: e9706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Sluyter F, de Wit S, Oostra BA, Crusio WE 2002. Behavioral and neuroanatomical characterization of the Fmr1 knockout mouse. Hippocampus 12: 39–46 [DOI] [PubMed] [Google Scholar]
- Mineur YS, Huynh LX, Crusio WE 2006. Social behavior deficits in the Fmr1 mutant mouse. Behav Brain Res 168: 172–175 [DOI] [PubMed] [Google Scholar]
- Moon J, Ota KT, Driscoll LL, Levitsky DA, Strupp BJ 2008. A mouse model of fragile X syndrome exhibits heightened arousal and/or emotion following errors or reversal of contingencies. Dev Psychobiol 50: 473–485 [DOI] [PubMed] [Google Scholar]
- Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR, Nelson DL, Hassan BA 2002. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34: 961–972 [DOI] [PubMed] [Google Scholar]
- Moressis A, Friedrich AR, Pavlopoulos E, Davis RL, Skoulakis EM 2009. A dual role for the adaptor protein DRK in Drosophila olfactory learning and memory. J Neurosci 29: 2611–2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J 1982. Place navigation impaired in rats with hippocampal lesions. Nature 297: 681–683 [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN 2004. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav 3: 287–302 [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, De Sarro GB, Elia M 1999. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40: 1092–1099 [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA 2000. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41: 19–23 [DOI] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, et al. 2008. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134: 1042–1054 [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Derber WJ, McClellan DA, Crnic LS 2002. Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res 927: 8–17 [DOI] [PubMed] [Google Scholar]
- Nitabach MN, Wu Y, Sheeba V, Lemon WC, Strumbos J, Zelensky PK, White BH, Holmes TC 2006. Electrical hyperexcitation of lateral ventral pacemaker neurons desynchronizes downstream circadian oscillators in the fly circadian circuit and induces multiple behavioral periods. J Neurosci 26: 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM 2006. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol 95: 3291–3295 [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano JL, Corbin JG, Burns MP 2011. The GABA(A) receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci 33: 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF 2010. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30: 15616–15627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF 2013. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77: 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacey LK, Heximer SP, Hampson DR 2009. Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol Pharmacol 76: 18–24 [DOI] [PubMed] [Google Scholar]
- Pan L, Zhang YQ, Woodruff E, Broadie K 2004. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol 14: 1863–1870 [DOI] [PubMed] [Google Scholar]
- Pan L, Woodruff E III, Liang P, Broadie K 2008. Mechanistic relationships between Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A signaling. Mol Cell Neurosci 37: 747–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST 1999. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94: 185–192 [DOI] [PubMed] [Google Scholar]
- Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, Ethell DW 2010. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol 10: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Preat T 2001. Localization of long-term memory within the Drosophila mushroom body. Science 294: 1115–1117 [DOI] [PubMed] [Google Scholar]
- Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL 2000. (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet 9: 1145–1159 [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM 2011. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14: 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE 1992. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci 106: 274–285 [DOI] [PubMed] [Google Scholar]
- Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL 1991. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66: 817–822 [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. 2014. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506: 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan NM 2014. Speaking human. Tenn Med 107: 33. [PubMed] [Google Scholar]
- Redondo RL, Morris RG 2011. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci 12: 17–30 [DOI] [PubMed] [Google Scholar]
- Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA 2005. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol 15: 1156–1163 [DOI] [PubMed] [Google Scholar]
- Renoux AJ, Sala-Hamrick KJ, Carducci NM, Frazer M, Halsey KE, Sutton MA, Dolan DF, Murphy GG, Todd PK 2014. Impaired sensorimotor gating in Fmr1 knock out and Fragile X premutation model mice. Behav Brain Res 267C: 42–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M 2005. Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci 102: 11557–11562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rihs TA, Tomescu MI, Britz J, Rochas V, Custo A, Schneider M, Debbane M, Eliez S, Michel CM 2013. Altered auditory processing in frontal and left temporal cortex in 22q11.2 deletion syndrome: a group at high genetic risk for schizophrenia. Psychiatry Res 212: 141–149 [DOI] [PubMed] [Google Scholar]
- Rogoz Z, Skuza G 2011. Anxiolytic-like effects of olanzapine, risperidone and fluoxetine in the elevated plus-maze test in rats. Pharmacol Rep 63: 1547–1552 [DOI] [PubMed] [Google Scholar]
- Roman G, Davis RL 2001. Molecular biology and anatomy of Drosophila olfactory associative learning. Bioessays 23: 571–581 [DOI] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, Huber KM 2012. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 15: 431–440, S431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotschafer SE, Trujillo MS, Dansie LE, Ethell IM, Razak KA 2012. Minocycline treatment reverses ultrasonic vocalization production deficit in a mouse model of Fragile X Syndrome. Brain Res 1439: 7–14 [DOI] [PubMed] [Google Scholar]
- Roy S, Watkins N, Heck D 2012. Comprehensive analysis of ultrasonic vocalizations in a mouse model of fragile X syndrome reveals limited, call type specific deficits. PLoS One 7: e44816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Segal M 2014. Dendritic spines: the locus of structural and functional plasticity. Physiol Rev 94: 141–188 [DOI] [PubMed] [Google Scholar]
- Sanchez-Morla EM, Santos JL, Aparicio A, Garcia-Jimenez MA, Villanueva C, Martinez-Vizcaino V, Arango C 2009. Antipsychotic effects on auditory sensory gating in schizophrenia patients. Eur Neuropsychopharmacol 19: 905–909 [DOI] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A 2003. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron 38: 887–898 [DOI] [PubMed] [Google Scholar]
- Sehgal A, Joiner W, Crocker A, Koh K, Sathyanarayanan S, Fang Y, Wu M, Williams JA, Zheng X 2007. Molecular analysis of sleep: wake cycles in Drosophila. Cold Spring Harb Symp Quant Biol 72: 557–564 [DOI] [PubMed] [Google Scholar]
- Sekine T, Yamaguchi T, Hamano K, Siomi H, Saez L, Ishida N, Shimoda M 2008. Circadian phenotypes of Drosophila fragile x mutants in alternative genetic backgrounds. Zoolog Sci 25: 561–571 [DOI] [PubMed] [Google Scholar]
- Sethna F, Moon C, Wang H 2013. From FMRP function to potential therapies for fragile X syndrome. Neurochem Res. 39: 1016–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SL 2000. Premature ovarian failure in the fragile X syndrome. Am J Med Genet 97: 189–194 [DOI] [PubMed] [Google Scholar]
- Siegel C, Waldo M, Mizner G, Adler LE, Freedman R 1984. Deficits in sensory gating in schizophrenic patients and their relatives. Evidence obtained with auditory evoked responses. Arch Gen Psychiatry 41: 607–612 [DOI] [PubMed] [Google Scholar]
- Siller SS, Broadie K 2011. Neural circuit architecture defects in a Drosophila model of Fragile X syndrome are alleviated by minocycline treatment and genetic removal of matrix metalloproteinase. Dis Model Mech 4: 673–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola O, Sundram V, Ng F, Kleyner Y, Morales J, Botas J, Jackson FR, Nelson DL 2008. The Drosophila FMRP and LARK RNA-binding proteins function together to regulate eye development and circadian behavior. J Neurosci 28: 10200–10205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R 2005. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav 4: 420–430 [DOI] [PubMed] [Google Scholar]
- Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL, Paylor R 2006. Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet 15: 1984–1994 [DOI] [PubMed] [Google Scholar]
- Sun MK, Hongpaisan J, Lim C, Alkon DL 2014. Bryostatin-1 restores hippocampal synapses and spatial learning and memory in adult fragile X mice. J Pharmacol Exp Ther 349: 393–401 [DOI] [PubMed] [Google Scholar]
- Suryavanshi PS, Ugale RR, Yilmazer-Hanke D, Stairs DJ, Dravid SM 2014. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br J Pharmacol 171: 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauber JM, Vanlandingham PA, Zhang B 2011. Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS One 6: e27100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier CR, Broadie K 2008. Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135: 1547–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Bui N, Perkins JR, Yuva-Paylor LA, Paylor R 2012. Group I metabotropic glutamate receptor antagonists alter select behaviors in a mouse model for fragile X syndrome. Psychopharmacology 219: 47–58 [DOI] [PubMed] [Google Scholar]
- Torrioli MG, Vernacotola S, Peruzzi L, Tabolacci E, Mila M, Militerni R, Musumeci S, Ramos FJ, Frontera M, Sorge G, et al. 2008. A double-blind, parallel, multicenter comparison of L-acetylcarnitine with placebo on the attention deficit hyperactivity disorder in fragile X syndrome boys. Am J Med Genet A 146: 803–812 [DOI] [PubMed] [Google Scholar]
- Tully T 1984. Drosophila learning: behavior and biochemistry. Behav Genet 14: 527–557 [DOI] [PubMed] [Google Scholar]
- Tully T, Quinn WG 1985. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A 157: 263–277 [DOI] [PubMed] [Google Scholar]
- Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. 2013. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 19: 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utari A, Chonchaiya W, Rivera SM, Schneider A, Hagerman RJ, Faradz SM, Ethell IM, Nguyen DV 2010. Side effects of minocycline treatment in patients with fragile X syndrome and exploration of outcome measures. Am J Intellect Dev Disabil 115: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Alphen B, Yap MH, Kirszenblat L, Kottler B, van Swinderen B 2013. A dynamic deep sleep stage in Drosophila. J Neurosci 33: 6917–6927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Hauben E, Reyniers E, Gantois I, Bakker CE, Oostra BA, Kooy RF, De Deyn PP 2000. Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav Brain Res 117: 127–136 [DOI] [PubMed] [Google Scholar]
- Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R 2012. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS). Behav Brain Res 228: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S 2004. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav Pharmacol 15: 433–442 [DOI] [PubMed] [Google Scholar]
- Verheij C, Bakker CE, de Graaff E, Keulemans J, Willemsen R, Verkerk AJ, Galjaard H, Reuser AJ, Hoogeveen AT, Oostra BA 1993. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363: 722–724 [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914 [DOI] [PubMed] [Google Scholar]
- Vinueza Veloz MF, Buijsen RA, Willemsen R, Cupido A, Bosman LW, Koekkoek SK, Potters JW, Oostra BA, De Zeeuw CI 2012. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav 11: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltes R, Duketis E, Knapp M, Anney RJ, Huguet G, Schlitt S, Jarczok TA, Sachse M, Kampfer LM, Kleinbock T, et al. 2014. Common variants in genes of the postsynaptic FMRP signalling pathway are risk factors for autism spectrum disorders. Hum Genet. 133: 781–792 [DOI] [PubMed] [Google Scholar]
- Wan L, Dockendorff TC, Jongens TA, Dreyfuss G 2000. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol 20: 8536–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Lin ML, Lin SJ, Li YC, Li SY 1997. Novel point mutation within intron 10 of FMR-1 gene causing fragile X syndrome. Hum Mutat 10: 393–399 [DOI] [PubMed] [Google Scholar]
- Wang DO, Kim SM, Zhao Y, Hwang H, Miura SK, Sossin WS, Martin KC 2009. Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science 324: 1536–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Bray SM, Warren ST 2012. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev 22: 256–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, et al. 2004. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci 101: 17504–17509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Westmark PR, O'Riordan KJ, Ray BC, Hervey CM, Salamat MS, Abozeid SH, Stein KM, Stodola LA, Tranfaglia M, et al. 2011. Reversal of fragile X phenotypes by manipulation of AbetaPP/Abeta levels in Fmr1KO mice. PLoS One 6: e26549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Bogert BA, Li W, Su K, Lee A, Gao FB 2004. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr Biol 14: 1025–1034 [DOI] [PubMed] [Google Scholar]
- Yamamoto D, Koganezawa M 2013. Genes and circuits of courtship behaviour in Drosophila males. Nat Rev Neurosci 14: 681–692 [DOI] [PubMed] [Google Scholar]
- Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP 2004. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 3: 337–359 [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP 2005. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49: 1053–1066 [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS 2010. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol 79: 632–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnescu DC, Shan G, Warren ST, Jin P 2005. Come FLY with us: toward understanding fragile X syndrome. Genes Brain Behav 4: 385–392 [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K 2001. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107: 591–603 [DOI] [PubMed] [Google Scholar]
- Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, Albrecht U, Tamanini F, Meijer JH, Oostra BA, et al. 2008. Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am J Hum Genet 83: 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang D, Wang Q, Rodal AA, Zhang YQ 2013. Drosophila cyfip regulates synaptic development and endocytosis by suppressing filamentous actin assembly. PLoS Genet 9: e1003450. [DOI] [PMC free article] [PubMed] [Google Scholar]