

Abstract
Renal cancer metastasis may result from oncogenic forces that contribute to the primary tumor. We have recently identified microRNA-21 as an oncogenic driver of renal cancer cells. The mechanism by which miR-21 controls renal cancer cell invasion is poorly understood. We show that miR-21 directly downregulates the proapoptotic protein PDCD4 to increase migration and invasion of ACHN and 786-O renal cancer cells as a result of phosphorylation/activation of Akt and IKKβ, which activate NFκB-dependent transcription. Constitutively active (CA) Akt or CA IKKβ blocks PDCD4-mediated inhibition and restores renal cancer cell migration and invasion. PDCD4 inhibits mTORC1 activity, which was reversed by CA IKKβ. Moreover, CA mTORC1 restores cell migration and invasion inhibited by PDCD4- and dominant negative IKKβ. Moreover, PDCD4 negatively regulates mTORC2-dependent Akt phosphorylation upstream of this cascade. We show that PDCD4 forms a complex with rictor, an exclusive component of mTORC2, and that this complex formation is reduced in renal cancer cells due to increased miR-21 expression resulting in enhanced phosphorylation of Akt. Thus our results identify a previously unrecognized signaling node where high miR-21 levels reduce rictor-PDCD4 interaction to increase phosphorylation of Akt and contribute to metastatic fitness of renal cancer cells.
Keywords: Renal carcinoma, miR-21, Akt kinase, mTOR
INTRODUCTION
Kidney cancer represents one of 10 most common cancers in men and women and is often resistant to chemotherapy. About 70% of patients with renal cancer fall into the subgroup of clear cell renal cell carcinoma [1]. A quarter of these patients have metastatic disease at presentation. The VHL gene product acts as a gatekeeper to prevent clear cell renal cell carcinoma by mediating degradation of Hif2α [2]. Often VHL mutation is not sufficient to cause renal cell carcinoma. However, VHL deletion alone does not cause renal cell carcinoma [3]. Apart from VHL, alteration in PBRM1, a component of PBAF SWI/SNF complex, histone methyl transferase SETD2 and histone deubiquitinase BAP1 have been shown to contribute to development of renal cell carcinoma and its metastasis [4–6].
The short noncoding microRNAs (19 –25 nucleotides) suppress protein expression by predominantly binding with imperfect complementarity to the 3′ untranslated region of mRNAs to either repress translation or induce mRNA degradation [7]. About one third of the total protein coding transcriptome is targeted by miRNAs [8]. A firm role for multiple miRNAs has been established in renal carcinogenesis. Expression profiling of renal tumors has shown that more miRNAs are decreased than those that are increased [9–13]. In a study using normal and renal tumor tissues, among 847 miRNAs that were found to be differentially expressed, miR-21 expression was decreased [14]. Increased expression of miR-21 was recently demonstrated in a study that utilized 31 different solid tumors, suggesting a significant role of this miRNA in carcinogenesis [15]. We also detected increased miR-21 expression in renal tumor tissue and in VHL positive as well as negative renal cancer cells [16, 17]. More recently, using more than 500 clear cell renal cell carcinoma tissues for genomic analysis, expression of miR-21 was found to be increased due to altered promoter methylation [13]. While the role for miR-21 in cancer formation and progression appears to be firmly established, recent evidence suggest that this miRNA may also contribute to cancer metastasis [18–21].
miR-21 is produced as a primary transcript from the intron 11 of TMEM49 gene [22]. The pri-miR-21 is quickly processed to form 72 nucleotides long pre-miR-21 in the nucleus, which is transported to the cytoplasm and undergoes dicer-mediated cleavage to yield 22 nucleotides long mature miR-21. Although gene amplification has been proposed as a mechanism of increased miR-21 expression, transcriptional regulations by a number of transcription factors (STAT3, AP-1, SRF, p53, Ets/PU.1, Hif1α and NFκB) are established [22–29]. However, inducible expression of miR-21 has also been shown to be regulated by posttranscriptional mechanism. For example, TGFβ and BMP-specific Smads are recruited to pri-miR-21 along with the p68, a component of Drosha microprocessor complex, leading to processing to pre-miR-21 and finally mature miR-21 [30, 31]. We have recently reported a transcriptional mechanism for miR-21 expression in renal cancer cells [17]. More recently an epigenetic mechanism has been shown as a cause of miR-21 upregulation in renal tumors [13]. Although many mRNAs have been identified to be the targets of miR-21, very few have been ascribed to play specific role in tumorigenesis especially in renal cancer. The expression of one such target, PDCD4 (programmed cell death 4), is decreased in many cancers, including renal cancer [21, 26, 32–38].
PDCD4 is upregulated during apoptosis [39]. PDCD4 binds to the mRNA translation initiation factors eIF4A and eIF4G to inhibit helicase activity [40, 41]. Expression of PDCD4 is frequently inhibited in many cancers including lung, breast, hepatoma, glial, tongue and skin tumors [39]. Also inhibition of PDCD4 expression in renal tumors and in renal cancer-derived cell lines is reported albeit with discrepancy between transcript and protein levels suggesting a posttranscriptional regulation of PDCD4 expression [33, 42]. In many cancers, PDCD4 expression is regulated by different miRNAs. In the present study, we demonstrate decreased expression of PDCD4 due to direct targeting of its 3′UTR by miR-21. miR-21 increases migration and invasion of renal cancer cells by reducing PDCD4 and enhancing Akt and IKKβ phosphorylation. We also show that IKKβ downstream of PDCD4 regulates mTORC1 to control migration and invasion of renal cancer cells. Finally, we demonstrate that miR-21 reduces the association between PDCD4 and rictor, the exclusive component of mTORC2, to increase Akt phosphorylation in renal cancer cells.
MATERIALS AND METHODS
Reagents
Tissue culture materials were purchased from Invitrogen. Phospho-Akt (Ser-473), Akt, phospho-S6 kinase (Thr-389), phospho-IKKβ (Ser-180/181), IKKβ, phospho-IκBα (Ser-32), phospho-p65 (Ser-536) and rictor antibodies were obtained from Cell Signaling. Anti-HA antibody was purchased from Covance. PDCD4, IκBα, p65 and Myc antibody was from Santa Cruz Biotechnology. Nonidate P-40, Na3VO4 and FLAG and actin antibodies were obtained from Sigma. Detailed information about the antibodies is presented in Supplementary Table 1. Recombinant inactive Akt was purchased from Millipore, FuGENE HD transfection reagent was purchased from Promega Inc. HA-tagged PDCD4 expression vector was a kind gift from Dr. Kimitoshi Kohno (Department of Molecular Biology, Kyushu University, Japan) [43]. The plasmid containing PDCD4 3′UTR sequence fused to the 3′ end of firefly luciferase gene (PDCD 3′UTR-Luc) was provided by Dr. Kenneth Kosik (University of California at Santa Barbara [44]. The expression plasmids miR-21 Sponge, constitutively active Myr-Akt, constitutively active IKKβ, constitutively active mTORC1 and Myc-tagged rictor plasmids were described previously [16, 45, 46]. Pool of three siRNAs against PDCD4 was purchased from Santa Cruz Biotechnology.
Cell Culture
HK2 proximal tubular epithelial cells, VHL positive ACHN and VHL negative 786-O renal carcinoma cells were described previously and were grown as described [16, 17].
Migration and invasion assays
The migration and invasion of renal cancer cells were determined using essentially as described previously [17, 47]. Briefly, transwell chambers were used for both migration and invasion assays with 8 mm membrane. In the case of invasion assays, these filter membranes were embedded with collagen (Millipore). 2.5×104 ACHN or 786-O renal carcinoma cells were plated in trans-well chambers on the membrane. These chambers were placed in a 24-well plate and incubated for 14 hours at 37°C. The migrated/invaded cells through the membrane were stained with the reagent using a kit. The stained cells were photographed using a computer-assisted camera. After taking pictures, the stain from the membrane was eluted using a kit according to vendor’s instruction. The absorbance of the eluted stain was measured in a spectrophotometer at 590 nm. This measurement was used arbitrarily as an indicator of number of cells migrated or invaded.
Immunoblotting and immunoprecipitation
For each experiment, cells were lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1 mM Na3VO4, 1 mM NP-40, 1 mM PMSF and 0.1% protease inhibitor cocktail) at 4°C for 30 minutes as described previously [16, 17, 45]. The cell extracts were centrifuged at 10,000xg for 20 minutes at 4°C. The protein was estimated in the supernatant. Equal amounts of cell lysates were separated by SDS polyacrylamide gel electrophoresis. Separated proteins were transferred to the PVDF membrane. The transferred proteins were immunoblotted with the indicated antibodies as described previously [16, 17, 45]. The protein signals were developed using enhanced chemiluminiscence reagent. Where indicated in the figure legends independent membrane containing the same samples was used to immunoblot with different antibodies. For immunoprecipitation, the cells were lysed in IP buffer (40 mM HEPES, pH 7.5, 1 mM EDTA, 120 mM NaCl, 10 mM Pyrophosphate, 10 mM glycerophosphate, 1.5 mM Na3VO4, 50 mM NaF, 0.3% CHAPS and EDTA free protease inhibitor cocktail). The cell lysates were used to incubate with indicated antibodies. The immunoprecipitation was performed using protein G-sepharose beads essentially as described [48]. The immunoprecipitates were dissolved in SDS sample buffer followed by immunoblotting as described above. For immunecomplex kinase assay, the indicated immunoprecipitates were washed 3 times with IP buffer at 4°C. Subsequently, the immunoprecipitates were washed twice with the mTORC2immunecomplex kinase (IK) assay buffer (25 mM HEPES, pH 7.4, 100 mM potassium acetate and 1 mM MgCl2). For the kinase assay reaction, immunecomplexes were resuspended in 20 μl IK assay buffer containing 100 ng of recombinant inactive Akt as substrate and 500 μM ATP. The reaction mixture was incubated for 30 min at 37°C. The kinase reaction was terminated by adding 4X SDS sample buffer followed by immunoblot analysis with phospho-Akt (Ser-473) antibody. For control, one forth of the input recombinant inactive Akt (25 ng) was run in a separate gel and immunoblotted with Akt antibody.
Transfection
The cells were transfected with indicated expression vectors or siRNAs using FuGENE HD as described previously [16, 17, 45].
Luciferase assay
The renal cancer cells were transfected with PDCD4 3′UTR-Luc or NFκB promoter-Luc plasmids. The cell lysates were used to assay luciferase activity using an assay kit as described previously [16, 17, 45]. Mean ± SE of 6 measurements is shown. *p = 0.001 vs vector alone.
Statistics
The significance of the data was analyzed by paired t-test. For the results presented in the Supplementary Figures, the significance of the data was determined by ANOVA followed by Student-Newman-Keuls analysis as described previously [16, 17, 45]. A p value less than 0.05 was considered as significant change.
RESULTS
miR-21 regulates PDCD4 expression for migration and invasion of renal cancer cell
We and others have recently shown that the expression of miR-21 is significantly increased in patient-derived renal tumors and in cultured renal cancer cells [13, 17]. Bioinformatic analysis and experimental evidence in various cancers demonstrated PDCD4 as a functional target of miR-21 [49]. Therefore, we determined the levels of PDCD4 in VHL positive ACHN and negative 786-O renal cancer cells. Fig. 1A shows reduced levels of PDCD4 in both these renal carcinoma cells as compared to normal renal proximal tubular epithelial cell HK2 (Supplementary Fig. S1A) [33]. To test the role of miR-21 in targeting PDCD4, we used a reporter plasmid containing 3′UTR of PDCD4 mRNA. This reporter plasmid was cotransfected with a vector overexpressing miR-21 into renal cancer cells. Expression of miR-21 significantly decreased the reporter activity (Supplementary Fig. S1B). To confirm this observation, we used a miR-21 Sponge construct, which quenches the levels of endogenous miR-21[16, 17, 50, 51]. Expression of miR-21 Sponge significantly increased the PDCD4 3′UTR reporter activity in both ACHN and 786-O renal cancer cells (Fig. 1B and Supplementary Fig. S1C). Concomitantly, miR-21 Sponge increased the levels of PDCD4 protein in these cells (Fig. 1C and Supplementary Fig. S1D).
Figure 1.

miR-21 Sponge inhibits migration and invasion of renal cancer cells by modulating PDCD4 levels. (A) Expression of PDCD4 in normal and renal cancer cells. Lysates of normal proximal tubular epithelial cell HK2 and, ACHN and 786-O renal cancer cells were immunoblotted with PDCD4 and actin antibodies. Quantification of these results is shown in Supplementary Fig. S1A. (B) Effect of miR-21 Sponge on the reporter activity of PDCD4 3′UTR-Luc. The reporter plasmid was cotransfected with vector or miR-21 Sponge constructs. The luciferase activity was measured in the cell lysates. Mean ± SE of 6 measurements is shown. *p = 0.001 vs vector alone. (C) miR-21 Sponge increases PDCD4 protein levels in renal cancer cells. miR-21 Sponge was transfected into ACHN or 786-O renal cancer cells. The cell lysates were immunoblotted with PDCD4 and actin antibodies respectively. Quantification of these results is shown in Supplementary Fig. S1D. (D) miR-21-targered PDCD4 regulates renal cancer cell migration. ACHN as well as 786-O renal carcinoma cells were transfected with miR-21 Sponge and siRNAs against PDCD4 as indicated. The cell migration was examined using Boyden chamber assays as described in the Materials and Methods [17]. Quantification of these results is shown in Supplementary Fig. S2A. (E) miR-21 increases renal cancer cell invasion via downregulating PDCD4. ACHN and 786-O renal carcinoma cells were separately transfected with miR-21 Sponge and siRNAs against PDCD4 as indicated. The invasion of renal cancer cells was examined using collagen-coated membrane in Boyden chambers as described in the Materials and Methods [17]. The quantification of the invasion data is shown in Supplementary Fig. S2B. Representative expression of miR-21 Sponge and PDCD4 for panels D and E is shown in Supplementary Fig. S2A and S2B.
Next, we determined the role of PDCD4 in regulating miR-21-induced migration of renal cancer cells using transwell chamber assay. Expression of miR-21 Sponge inhibited the migration of both ACHN and 786-O renal cancer cells (Fig. 1D). Interestingly, expression of siRNAs against PDCD4 along with miR-21 Sponge reversed the miR-21 Sponge-induced inhibition of migration (Fig. 1D). Quantification of these results demonstrated significant changes in cell migration with miR-21 Sponge and siPDCD4 (Supplementary Fig. S2A). Since the initial step in metastasis consists of intravasation (local invasion), we next used invasion assay using collagen embedded membranes in transwell chambers. Vector-transfected ACHN and 786-O cells showed marked invasion (Fig. 1E). miR-21 Sponge blocked this invasion, which was prevented by co-expression of siPDCD4 with miR-21 Sponge (Fig. 1E). Quantification of these results showed significant changes in invasion (Supplementary Fig. S2B). Together these results demonstrate that miR-21 inhibits PDCD4 to facilitate invasion of VHL positive and negative renal cancer cells.
miR-21 regulates PDCD4-mediated Akt phosphorylation and activation of IKKβ
In renal cancer cells, we have recently shown that phosphorylation of Akt is significantly increased, which contributes to their proliferation and invasion [17]. Therefore, to examine the mechanism of Akt activation, we determined the effect of PDCD4 on phosphorylation of Akt. Expression of PDCD4 in ACHN and 786-O renal cancer cells inhibited phosphorylation of Akt (Fig. 2A and Supplementary Fig. S3A). These results suggest that PDCD4 may block the signal transduction pathway downstream of Akt kinase. We have recently shown activation of IKKβ in renal cancer cells downstream of Akt [16]. We examined the role of PDCD4 in phosphorylation of IKKβ. Expression of PDCD4 attenuated phosphorylation of IKKβ in renal cancer cells, resulting in inhibition of phosphorylation of its substrate IκBα, suggesting that PDCD4 inhibits IKKβ activity (Figs 2B, 2C and Supplementary Fig. S3B, S3C). As phosphorylation of p65 subunit of NFκB is also downstream of IKKβ, we found decreased phosphorylation of p65 in response to PDCD4 (Fig. 2D and Supplementary Fig. S3D). Also, expression of PDCD4 inhibited NFκB-dependent reporter transcription, which is dependent upon IKKβ activation and p65 phosphorylation in renal cancer cells (Supplementary Fig. S4 and data not shown) [16].
Figure 2.

PDCD4 regulates phosphorylation of Akt and its downstream effectors IKKβ, IκB and p65 subunit of NFκB. ACHN and 786-O renal cancer cells were transfected with a vector plasmid expressing HA-PDCD4 as indicated. The cell lysates were immunoblotted with phospho-Akt (Ser-473), Akt (panel A), phospho-IKKβ (Ser-180/181), IKKβ (panel B), phospho-IκBα (Ser-32), IκBα (panel C), phospho-p65 (Ser-536), p65 (panel D) and indicated antibodies. For panel A, same blot was used to probe with phospho-Akt, Akt and actin antibodies. Same lysates were run in parallel to probe with HA antibody to detect HA-tagged PDCD4. Quantification of these blots is shown in Supplementary Fig. S3A. For panel B, same blot was used to probe with phospho-IKKβ, IKKβ and actin antibodies. Same lysates were run in parallel to probe with HA antibody to detect HA-tagged PDCD4. Quantification of these blots is shown in Supplementary Fig. S3B. For panel C, same blot was used to probe with phospho-IκBα and actin antibodies. Same lysates were run in parallel to probe with IκBα and HA antibodies to detect IκBα and HA-tagged PDCD4. Quantification of these blots is shown in Supplementary Fig. S3A. In panel D for ACHN cells, same blot was used to probe with phospho-p65 and actin antibodies. Same lysates were run in parallel to probe with p65 and HA antibodies to detect p65 and HA-tagged PDCD4. In panel D for 786-O cells, same blot was used to probe with phospho-p65 and p65 antibodies. Same lysates were run in parallel to probe with actin and HA antibodies to detect actin and HA-tagged PDCD4. Quantification of these blots is shown in Supplementary Fig. S3D.
Above data demonstrate a role of PDCD4 in the phosphorylation of Akt and activation of IKKβ signaling in renal cancer cells. We have recently reported a role for miR-21 in regulating phosphorylation/activation of Akt [17]. We investigated the role of PDCD4 in miR-21-regulated Akt phosphorylation. As expected, expression of miR-21 Sponge inhibited Akt phosphorylation in both ACHN and 786-O renal cancer cells (Fig. 3A). Note that miR-21 Sponge increased the expression of PDCD4 (lane 2 in third part from top of Fig. 3A). siRNAs against PDCD4 reversed the inhibition of Akt phosphorylation induced by miR-21 Sponge (Fig. 3A and Supplementary Fig. S5A). Concomitantly, siPDCD4 decreased the PDCD4 to normal level (Fig. 3A, compare lane 3 with 2 in third part from top and Supplementary Figs. S5A).
Figure 3.

miR-21 increases phosphorylation of Akt and its downstream effectors by targeting PDCD4. ACHN as well as 786-O renal cancer cells were transfected with miR-21 Sponge along with siRNAs against PDCD4. The cell lysates were immunoblotted with indicated phospho-specific antibodies and other antibodies as described in the legends of Fig. 2. In panels A and C, same membranes for each panel were immunoblotted with phospho-antibodies, total antibodies of the same protein and actin antibody as indicated. Same samples for each panel were run in parallel to immunoblot with PDCD4 antibody. For panel B, same blot was used to probe with phospho-IKKβ and PDCD4 antibodies. Same lysates were run in parallel to probe with IKKβ and actin antibodies. In panel D for ACHN cells, same blot was used to probe with phospho-p65 and p65 antibodies. Same lysates were run in parallel to probe with PDCD4 and actin antibodies. For 786-O cells, same blot was used to probe with p65, PDCD4 and actin antibodies. Same lysates were run in parallel to probe with phospho-p65. Quantifications of the results and are shown in Supplementary Fig. S5. Expression of miR-21 Sponge for these results was determined in parallel experiments and is shown in Supplementary Fig. S5.
Next, we determined the involvement of PDCD4 in miR-21 regulation of IKKβ signaling in ACHN and 786-O renal cancer cells. Transfection of siPDCD4 markedly prevented the miR-21 Sponge-induced inhibition of phosphorylation of IKKβ and its substrate IκBα phosphorylation (Figs. 3B, 3C and Supplementary Figs. S5B and S5C). Concomitantly, the reduced phosphorylation of p65 by miR-21 Sponge was reversed by siPDCD4 (Fig. 3D and Supplementary Fig. S5D). siPDCD4 also blocked the inhibition of NFκB-dependent transcription by miR-21 Sponge (Supplementary Fig. S6). Collectively, these results suggest a role of PDCD4 in regulation of Akt and IKKβ activation downstream of miR-21 in renal cancer cells.
Akt kinase and IKKβ mediate PDCD4-induced inhibition of renal cancer cell migration and invasion
We have shown above that PDCD4 regulates phosphorylation/activation of Akt and IKKβ (Fig. 2). To investigate the role of these kinases in renal cancer cell migration and invasion, we first determined the effect of PDCD4 on these parameters in ACHN and 786-O renal cancer cells. Expression of PDCD4 inhibited both migration and invasion of these cells (Fig. 4). To determine the contribution of Akt in these processes, we used constitutively active Myr-Akt. Expression of Myr-Akt along with PDCD4 prevented PDCD4-induced inhibition of migration of renal cancer cells (Fig. 4A). Similarly, Myr-Akt reversed PDCD4-mediated attenuation of invasion of these cells (Fig. 4B). Quantification of migrated and invaded renal cancer cells showed significant changes (Supplementary Figs. S7A and S7B). Next, we determined the role of IKKβ. Expression of constitutively active IKKβ significantly blocked the inhibition of migration and invasion induced by PDCD4 (Figs. 4C and 4D and Supplementary Figs. S7C and S7D). Together these data indicate that Akt/IKKβ signaling contributes to PDCD4-mediated migration/invasion of renal cancer cells.
Figure 4.

PDCD4 regulates migration and invasion of renal cancer cells via Akt kinase and its downstream effector IKKβ. ACHN and 786-O renal carcinoma cells were transfected with plasmid vectors expressing HA-tagged PDCD4 and HA-tagged constitutively active (CA) HA-Myr-Akt (panels A and B) or CA FLAG-IKKβ (panels C and D) as indicated. (A and C) Migration of ACHN and 786-O renal cancer cells was determined using Boyden chamber assay as described in the Materials and Methods [17]. (B and D) Invasion of the renal cancer cells was determined using collagen-coated membrane as described in the Materials and Methods. Quantification of these results and expression of PDCD4, Myr-Akt and CA IKKβ is shown in the Supplementary Figs. S7A, S7B, S7C and S7D, respectively.
mTORC1 controls PDCD4-mediated migration and invasion of renal cancer cells
A role for mTOR, especially complex1 (mTORC1), has been established in progression of different cancers including renal cell carcinoma [17, 52]. We have recently shown that mTORC1 regulates migration and invasion of renal cancer cells [17]. Our results above demonstrate a requirement for inhibition of PDCD4 in the migration/invasion of ACHN and 786-O renal cancer cells (Fig. 4). We postulated a role of PDCD4 in regulating mTORC1 activity. To address this possibility, we assessed the phosphorylation of S6 kinase, a direct substrate of mTORC1 and an indicator of mTORC1 activation [52]. Expression of PDCD4 in ACHN and 786-O renal cancer cells inhibited phosphorylation of S6 kinase (Fig. 5A and Supplementary Fig. S8). Next, we examined the role of mTORC1 in PDCD4-induced inhibition of migration and invasion. We used a mutant mTOR with specific substitutions of four aminoacids at the C-terminus (mTOR I2017T/V2198A/L2216H/L2260P). This mutant mTOR constitutively increases mTORC1 activity (Supplemental Fig. S9) [53]. As expected, expression of PDCD4 inhibited migration as well as invasion of ACHN and 786-O renal carcinoma cells (Figs. 5B and 5C). Co-expression of constitutively active mTOR blocked the PDCD4-induced inhibition and restored renal cancer cell migration and invasion to normal levels (Figs. 5B and 5C). Quantification of these results demonstrated that the changes were significant (Supplementary Figs. S10A and S10B). These results demonstrate a role of mTORC1 in PDCD4 regulation of migration and invasion of renal carcinoma cells.
Figure 5.

PDCD4 regulates mTORC1 activity to control migration and invasion of renal cancer cells. (A) Expression of PDCD4 inhibits mTORC1 activity in renal cancer cells. ACHN and 786-O cells were transfected with PDCD4 expression vector. The cell lysates were immunoblotted with phospho-S6 kinase (Thr-389), S6 kinase and actin antibodies. Same lysates were run in parallel to immunoblot with HA antibody. Quantification of these results is shown in Supplementary Fig. S8. (B and C) Expression of CA mTORC1 reverses PDCD4-induced inhibition of migration (panel B) and invasion (panel C) of renal cancer cells. Both ACHN and 786-O renal cancer cells were separately transfected with PDCD4 and CA mTORC1 expression plasmids as indicated. Migration (panel B) and invasion (panel C) of these cells were measured as described in the Materials and Methods. The quantification of the migration and invasion data and expression of PDCD4 and CA mTORC1 shown in panels B and C is shown in Supplementary Figs. S10A and S10B, respectively.
IKKβ regulates mTORC1 activity to increase migration and invasion of renal cancer cells
Our results above show a negative regulation of mTORC1 by PDCD4 (Fig. 5A). We have also shown that IKKβ acts downstream of miR-21-PDCD4 axis in regulating migration and invasion of renal cancer cells (Figs. 4C and 4D). Therefore, we determined the role of IKKβ in PDCD4-inhibited mTORC1 activity. As expected the expression of PDCD4 markedly blocked phosphorylation of S6 kinase in ACHN and 786-O renal cancer cells (Fig. 6A). Expression of constitutively active IKKβ, however, prevented the PDCD4-induced inhibition of phosphorylation of S6 kinase in both renal cancer cells (Fig. 6A and Supplementary Fig. S11A). As mTORC1 regulates migration and invasion of renal cancer cells (Figs. 5B and 5C), we examined the connection between IKKβ and mTORC1 in these processes. Expression of kinase dead IKKβ, which confers dominant negative effect, abrogated migration and invasion of ACHN and 786-O renal cancer cells (Figs. 6B and 6C). Interestingly, expression of constitutively active mTORC1 significantly reversed the inhibition of migration and invasion of renal cancer cells induced by dominant negative IKKβ (Figs. 6B, 6C and Supplementary Figs. S11B and S11C). These results suggest a role of IKKβ and mTORC1 downstream of miR-21-targeted PDCD4 in renal carcinoma cell invasion. To confirm this hypothesis, we directly examined the involvement of IKKβ in miR-21-mediated migration and invasion. As expected, quenching of endogenous miR-21 by miR-21 Sponge blocked migration and invasion of ACHN and 786-O cells. This inhibition was restored to the normal levels by expression of constitutively active IKKβ (Figs. 6D, 6E and Supplementary Figs. S11D and S11E). We have also shown above a role for Akt in PDCD4-regulated migration and invasion of renal cancer cells (Fig. 4A). Interestingly, constitutively active Myr-Akt significantly prevented miR-21 Sponge-induced inhibition of migration and invasion of ACHN and 786-O renal cancer cells (Fig. 6F, 6G and Supplementary Fig. S11F and S11G). Similarly, constitutively active mTORC1 reversed miR-21 Sponge-inhibited migration and invasion of these cells (Fig. 6H, 6I and Supplementary Fig. S11H and S11I). Together our data support a role of miR-21 target PDCD4 in regulating renal cancer cell migration/invasion via Akt, IKKβ and mTORC1.
Figure 6.


IKKβ and mTORC1 downstream of miR-21 regulate migration and invasion of renal carcinoma cells. (A) Expression of CA IKKβ prevents PDCD4-inhibited mTORC1 activity in renal cancer cells. ACHN and 786-O renal carcinoma cells were transfected with PDCD4 and CA IKKβ as indicated. The cell lysates were immunoblotted with phospho-S6 kinase (Thr-389), S6 kinase and actin antibodies. Same lysates were run in parallel to immunoblot with HA and FLAG antibodies. Quantification of these results is shown in Supplementary Fig. S11A. (B and C) Expression of CA mTORC1 reverses dominant negative IKKβ-induced inhibition of migration (panel B) and invasion (panel C) of renal cancer cells. ACHN and 786-O cells were transfected with dominant negative (DN) IKKβ and CA mTORC1 as indicated. Migration (panel B) and invasion (panel C) of these cells were determined as described in the Materials and Methods. Expression of DN IKKβ, CA mTORC1 and quantification of these results is shown in Supplementary Fig. S11B and S11C. (D and E) IKKβ regulates miR-21-induced migration and invasion of renal cancer cells. ACHN and 786-O cells were transfected with miR-21 Sponge and CA IKKβ as indicated. Migration (panel D) and invasion (panel E) of these cells were determined as described in the Materials and Methods. Expression of CA IKKβ and miR-21 Sponge for panels D and E was examined in parallel experiments and shown in Supplemental Figs. S11D – S11E. Quantification of migration and invasion for panels D and E is shown in Supplementary Fig. S11D – S11E. (F and G) Expression of Myr-Akt prevents miR-21 Sponge-induced inhibition of migration and invasion of renal cancer cells. ACHN and 786-O cells were transfected with miR-21 Sponge and Myr-Akt as indicated. Migration (panel F) and invasion (panel G) of these cells were determined as described in the Materials and Methods. Quantification of these results, expression of Myr-Akt and expression of miR-21 Sponge are shown in Supplementary Fig. S11F and S11G. (H and I) Expression of CA mTORC1 reverses miR-21 Sponge-induced inhibition of migration (panel H) and invasion (panel I) of renal cancer cells. ACHN and 786-O cells were transfected with miR-21 Sponge and CA mTOR as indicated. Migration (panel H) and invasion (panel I) of these cells were determined as described in the Materials and Methods. Quantification of these results, expression of CA mTOR and expression of miR-21 Sponge are shown in Supplementary Fig. S11H and S11I.
miR-21 controls association of rictor with PDCD4 as a mechanism of renal cancer cell migration and invasion
Our results above provide evidence for miR-21-inititated activation of Akt via downregulation of PDCD4, which controls IKKβ-mediated mTORC1 activation leading to renal cancer cell migration and invasion. It should be noted that mTORC2 phosphorylates Akt at Ser-473 for its full activation, [54]. These results indicate that PDCD4 may regulate mTORC2 activity. To address this hypothesis, we considered association of the mTORC2 component rictor with PDCD4 that inhibits mTORC2 kinase activity towards Akt Ser-473 residue [54]. HK2 proximal tubular epithelial cells were transfected with vectors expressing HA-PDCD4 and Myc-Rictor. Immunoprecipitation of the cell lysates with HA antibody followed by immunoblotting with anti-Myc showed association of PDCD4 with rictor (Fig. 7A and Supplementary Fig. S12A). Reciprocal immunoprecipitation and immunoblotting confirmed complex formation between rictor and PDCD4 (Fig. 7B and Supplementary Fig. S12B). Since our results demonstrate that PDCD4 regulates Akt Ser-473 phosphorylation and it is associated with rictor, we determined the mTORC2 activity in HK2 proximal tubular epithelial cells in PDCD4 immunoprecipitates using immunecomplex kinase assay with recombinant inactive Akt as substrate. We could not detect any Akt Ser-473 phosphorylation (Supplementary Fig. S13) as we hypothesized that PDCD4 association with rictor inhibits mTORC2 activity. To demonstrate mTORC2 activity in these cells, we used the supernatant after PDCD4 immunoprecipitation. This supernatant was immunoprecipitated with rictor antibody. The immunoprecipitates were used in immunecomplex kinase assay using recombinant inactive Akt as substrate. Fig. 7C shows significantly reduced mTORC2 activity in the supernatant from PDCD4 overexpressing HK2 cells (Supplementary Fig. S14A). These results indicate that due to association of rictor with PDCD4, less rictor-bound mTORC2 is available in the supernatant to exhibit reduced kinase activity. Next, we compared the association PDCD4 with rictor between normal proximal tubular epithelial cells and renal cancer cells. Coimmunoprecipitation experiments revealed decreased association of PDCD4 with rictor in ACHN and 786-O renal cancer cells than in HK2 proximal tubular epithelial cells (Fig. 7D and Supplementary Fig. S14B). Reciprocally, immunoprecipitation of rictor from these cells showed increased association of rictor with PDCD4 in HK2 cells compared to renal cancer cells (Fig. 7E and Supplementary Fig. S14C). These data suggest that association of PDCD4 with rictor may constitutively inhibit mTORC2 activity. This was confirmed by reduced phosphorylation of Akt at Ser-473 in normal proximal tubular epithelial cells relative to that in the renal cancer cells (Fig. 7F and Supplementary Fig. S14D). Similarly, the mTORC2 activity as determined by immunecomplex kinase assay of rictor immunoprecipitates was significantly increased in the renal cancer cells as compared to that in normal HK2 proximal tubular epithelial cells (Fig. 7G and Supplementary Fig. S14E)
Figure 7.


miR-21 regulates association of PDCD4 with the mTORC2 component rictor to regulate phosphorylation of Akt. (A and B) Association of rictor with PDCD4 in HK2 normal proximal tubular epithelial cells. HK2 cells were transfected with HA-PDCD4 and Myc-Rictor or vector plasmids. The cell lysates were immunoprecipitated with IgG or anti-HA (panel A) or anti-Myc (panel B) antibodies. The immunoprecipitates were immunoblotted with anti-Myc and anti-HA as indicated. The bottom panels show immunoblot analysis with indicated antibodies using cell lysates. Quantification of these results is shown in Supplementary Fig. S12. (C) Measurement of mTORC2 activity in rictor immunoprecipitates from the supernatant after PDCD4 was immunoprecipitated. HK2 normal proximal tubular epithelial cells were transfected with HA-tagged PDCD4. The cell lysates were immunoprecipitated with PDCD4 antibody. The supernatants were then immunoprecipitated with IgG or rictor antibody. The immunoprecipitates were assayed for mTORC2 activity using 100 ng/ml recombinant inactive Akt as substrate. For Akt blot, 20 ng recombinant Akt was run in parallel. Bottom panels show immunoblots of the indicated proteins in the cell lysates. Quantification of these results is shown in Supplementary Fig. S14A. (D and E) Association of endogenous PDCD4 with rictor in renal cancer cells. Equal amounts of proteins from HK2 normal proximal tubular epithelial and, ACHN and 786-O renal cancer cells were immunoprecipitated with IgG or PDCD4 antibody followed by immunoblotting with rictor antibody (panel D) or immunoprecipitated with rictor antibody followed by immunoblotting with PDCD4 antibody (panel E). Bottom panels show immunoblotting of indicated proteins in the cell lysates. Quantification of these results is shown in Supplementary Fig. S14B and S14C. (F) Comparison of phosphorylation of Akt at Ser-473 among HK2, ACHN and 786-O cells. The lysates of these cells were immunoblotted with phospho-Akt (Ser-473) and Akt antibodies, respectively. Quantification of these results is shown in Supplementary Fig. S14D. (G) Comparison of in vitro mTORC2 activity among HK2, ACHN and 786-O cells. Cell lysates were immunoprecipitated with rictor antibody. The immunoprecipitates were used in immunecomplex kinase assay using 100 ng/ml recombinant inactive Akt as substrate. For Akt blot, 20 ng recombinant Akt was run in parallel. Quantification of these results is shown in Supplementary Fig. S14E.
We have shown above that increased expression of miR-21 in renal cancer cells downregulates PDCD4 levels to regulate Akt phosphorylation (Fig. 3A). Therefore, we examined the role of miR-21 in regulating association of PDCD4 with rictor. miR-21 Sponge was transfected into ACHN and 786-O renal cancer cells. Coimmunoprecipitation experiments showed increased association of PDCD4 with rictor in miR-21 Sponge-transfected renal cancer cells (Fig. 8A and Supplemental Fig. S15A). Reciprocal experiment showed similar results (Fig. 8B and Supplemental Fig. S15B). These data conclusively demonstrate miR-21 regulation of the association between PDCD4 and Rictor, which contributes to regulation of Akt phosphorylation and hence downstream signal transduction, leading to renal cancer cell invasion.
Figure 8.

Inhibition of miR-21 increases association of rictor with PDCD4 in renal cancer cells. ACHN and 786-O cells were transfected with miR-21 Sponge. The cell lysates were immunoprecipitated with IgG or PDCD4 antibody followed by immunoblotting with rictor and PDCD4 antibodies (panel A). In panel B, reciprocal immunoprecipitation and immunoblotting were performed. The bottom panels show immunoblotting of the indicated proteins in the cell lysates. Quantification of these results and expression of miR-21 Sponge is shown in Supplementary Fig. S15A and S15B.
DISCUSSION
PDCD4 was originally identified as a proapoptotic protein in mouse cell line and latter isolated from human glioma [55, 56]. Its role in cancer is established. For example, PDCD4-deficient mice develop lymphoid tumors [57] and mice overexpressing PDCD4 display resistance to tumorigenesis [58]. Interestingly, delivery of PDCD4 inhibits cell proliferation and angiogenesis and induces apoptosis of tumor cells in a mouse model of non-small-cell lung cancer [59]. Also, its role in invasion of several solid tumors has been reported [21, 34, 38, 39, 49, 60–62]. More recently, decreased expression of PDCD4 has been reported in renal tumors [33]. Transcriptional and epigenetic regulations represent major mechanisms for PDCD4 expression [63–65]. Recent reports also indicate that downregulation of PDCD4 in many cancers is due to upregulation of different miRNAs including miR-21 [39, 49, 66, 67]. However, their relationship has not been examined in renal cancer. In the present study, we demonstrate decreased expression of PDCD4 in renal cancer cells irrespective of the VHL status. In these cells, and in renal tumors, we and others have shown recently increased expression of miR-21 [13, 17]. Thus a reciprocal relationship exists between miR-21 and PDCD4 levels in renal cancer cells. Our results demonstrate that PDCD4 regulates Akt and IKKβ activation, which contribute to activation of mTORC1 necessary for renal cancer cell migration and invasion. We show that IKKβ, downstream of miR-21 and Akt, regulates migration and invasion of renal cancer cells. Finally, we provide the first evidence for decreased association between PDCD4 and rictor, the exclusive mTORC2 component, in renal cancer cells as a mechanism of increased Akt activity. These results are summarized in Fig. 9.
Figure 9.
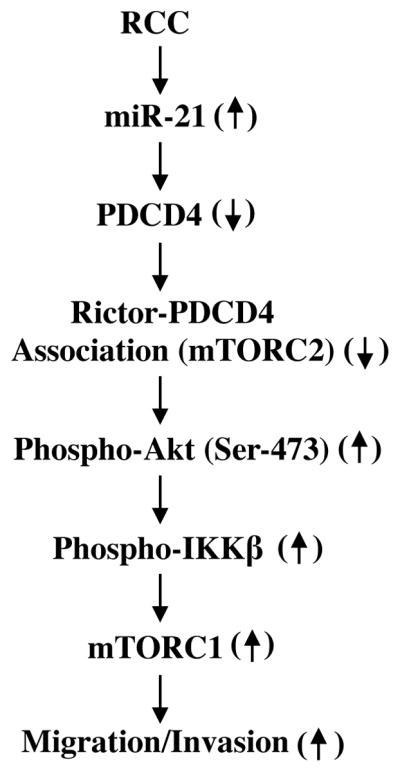
Schema describing the results presented in this paper.
miR-21 is abundantly expressed in the renal proximal tubular epithelial cells and its expression is significantly increased in fibrotic diseases of kidney [50, 51, 68–74]. Moreover, profiling studies demonstrated increased miR-21 expression in both clear cell and papillary renal carcinomas [10, 75, 76]. These results support the notion of miR-21 as an oncomiR as suggested by its upregulation in many other cancers [15, 77]. In fact, mice overexpressing miR-21 show increased lung tumorigenesis while ablation of this overexpression protects against tumor formation [78]. miR-21 deficient mice show normal development but decreased eosinophil progenitors [78, 79]. Also, deletion of miR-21 results in reduced tumorigenesis in a mouse skin carcinogenesis model [80]. In a separate study, it was shown that overexpression of miR-21 in a transgenic mouse model leads to hematological malignancies with lymphoma, which completely regressed after inactivating the miR-21 expression, indicating a single gene effect of miR-21 in development of the tumor [81]. Interestingly, PDCD4 deletion in mice also shows lymphoma formation, suggesting a possible concerted role of miR-21 and PDCD4 in tumorigenesis [57]. These studies provide evidence for the phenomena of oncomiR addiction in which overexpression of miR-21 maintains the tumor phenotype possibly through downregulation of PDCD4. In renal cancer, we and others have shown expression of miR-21, which inhibits PDCD4 expression (Fig. 1) [17].
Although miR-21 was identified as an inhibitor of apoptosis in various cell lines, its role in stem cell fate determination is established [82]. miR-21 regulates self-renewal of mouse spermatogonial stem cells [83]. A recent study identified increased miR-21 expression to regulate stem cells during embryonic brain development [84]. This expression of miR-21 correlates with Sox2 expression. Along with its role in regulation of embryonic stem cells, miR-21 regulates invasion of breast, hepatocellular and glioblastoma stem-like cells [85–88]. Furthermore, miR-21-mediated Sox2 expression was observed in human glioma cells, indicating a role of this miRNA in determining cancer cell stemness, which regulate tumor invasion. We have recently shown involvement of miR-21 in renal cancer cell migration and invasion [17]. In the present report, we identify PDCD4 as a target of miR-21, which contributes to renal cancer cell migration and invasion (Fig. 1).
Role of PI 3 kinase/Akt signaling in cancer cell proliferation and invasion is established. Activating mutations are found in both these enzymes in many cancers [89, 90]. Alteration in both PI 3 kinase and Akt genes was observed in a recent rigorous study on 417 renal tumor samples [13]. We have recently shown a role of Akt in migration and invasion of renal cancer cells [17]. But, in the absence of direct mutation in this pathway, activation of Akt can occur due to altered expression of the tumor suppressor protein PTEN, which contributes to the regulation of Akt kinase activity [91]. In fact, we have shown the involvement of PTEN in renal cancer cell proliferation [17]. Interestingly, ACHN renal cancer cells contain wild type PTEN. On the other hand, 786-O cells are PTEN negative [92]. Therefore, PTEN may not completely regulate Akt phosphorylation in these renal cancer cell lines. Interestingly, we find PDCD4 negatively regulates phosphorylation of Akt and migration and invasion in VHL positive and VHL negative renal cancer cells (Figs. 2A, 4A and 4B). Also, our data show that miR-21-mediated downregulation of PDCD4 contributes to phosphorylation of Akt (Fig. 3A). Furthermore, we identify Akt kinase as a negative regulatory target for PDCD4-induced inhibition of migration and invasion of renal cancer cells (Figs. 4A and 4B).
Phosphorylation of IKKβ upstream of canonical NFκB activation contributes to all steps of tumorigenesis including metastasis [93–96]. 70% of renal tumors contain activated NFκB [97]. Furthermore, NFκB has been shown to correlate with renal tumor grade and metastasis [98]. In renal cancer cells, we identified a positive correlation between increased miR-21 and NFκB activation due to increased phosphorylation of IKKβ [16]. We showed that phosphorylated IKKβ and NFκB, and expression of miR-21 constitute a positive feedback loop in renal cancer cells [16, 17]. Previously, Yang et al reported that the PDCD4 activates NFκB [99]. Although PDCD4 is a proapoptotic protein that activates NFκB, the latter either acts as a survival or apoptotic transcription factor in a context dependent manner [100–103]. Using PDCD4 deficient mice, Sheedy et al demonstrated the requirement of NFκB for resistance to death of these mice in response to lipopolysaccharide [104]. These authors highlighted the requirement of PDCD4 for activation of NFκB independent of IKKβ. In contrast to these results, we demonstrate a negative regulatory role of PDCD4 on phosphorylation of IKKβ and NFκB activation (Figs 2B –2D). Also, we conclusively show a negative role of PDCD4 in miR-21-induced phosphorylation of IKKβ and NFκB activation (Figs. 3B – 3D). Furthermore, our data provide the first evidence for involvement of IKKβ in PDCD4 regulation of renal cancer cell migration and invasion (Figs. 4C and 4D).
Due to alteration in PI 3 kinase, Akt and loss/downregulation of PTEN and p53 mutation, mTOR signaling is deregulated in many cancers including renal cell carcinoma [52, 105–107]. Two complexes of mTOR (mTORC1 and mTORC2) exist. An exclusive set of associated proteins defines each complex. For example, raptor is only present in mTORC1 while rictor and Sin1 are present in mTORC2. mTORC1 increases biosynthesis of macromolecules including, lipids and nucleic acids by regulating transcription factors SREBPs, PPARγ, STAT3, YY1-PGC1α, TFEB, STAT3 and RNA polymerases 1 and III to promote tumor cell proliferation [108, 109]. Furthermore, mTORC1 also regulates 4EBP/eIF4E axis to increase translation of specific mRNAs coding for oncogenic proteins that regulate cell cycle, metabolism, cell survival, angiogenesis and metastasis [110]. Also a significant role of mTORC2 in cancer cells via activation of PI 3 kinase is reported. mTORC2 contributes to proliferation, survival and invasion of various cancer cells mainly by phosphorylation of Akt at Ser-473 [52]. For example, the positive regulatory component of mTORC2, rictor, has been shown to be upregulated in glioma and specific activation of mTORC2 increased cancer cell proliferation and invasion [111, 112]. Rapamycin and its analogs which predominantly block mTORC1 activity showed some success against many cancers including patients with renal cell carcinoma; however, they show only modest efficacy due to rapamycin resistant kinase activity of mTORC1 and for the presence of negative feedback action of this kinase to the phosphorylation of Akt [110, 113–118]. Inhibitors that block both mTORC1 and mTORC2 activities towards phosphorylation of all known downstream targets of these kinase attenuate cell growth in vitro and in vivo with much greater efficiency than rapalogs [52, 110]. This may be due to lack of mTORC2-mediated phosphorylation of Akt at Ser-473, which is necessary for proliferation and invasion as well as inhibition of rapamycin resistant functions of mTORC1 [54, 110].
Signaling mechanism by which mTORC1 is activated involves Akt-mediated phosphorylation of PRAS40 and tuberin [119–121]. mTORC2 downstream of PI 3 kinase phosphorylates Akt at Ser-473 for its full activation [54, 122]. We showed that mTORC2 regulates mTORC1 activity in renal epithelial cells [45]. mTORC1 inhibition attenuates cancer cell invasion and metastasis of tumor cells in mouse models, including lung metastasis of human renal cancer cells [123, 124]. More recently, we have shown a role of miR-21 in the activation of Akt and mTORC1, which regulate migration and invasion of renal cancer cells [17]. Our results in this paper now provide evidence for a role of PDCD4 downstream of miR-21 to activate mTORC1 (Fig. 5A). Also, we demonstrate that mTORC1 regulates PDCD4-induced migration and invasion of renal cancer cells (Figs. 5B and 5C).
Alternative to those mechanisms described above has also been described for activation of mTORC1. For example, Dan et al showed an Akt-dependent association of IKKα with mTOR to increase the activity of both these kinases for transcriptional activation of NFκB [125]. In contrast to these results, Akt kinase independent requirement of IKKβ was shown to be required for mTORC1 activation. This was mediated by direct phosphorylation/inactivation of TSC1 [126, 127]. Contrary to these results, our data indicate a requirement of miR-21-dependent Akt downstream of reduced levels of PDCD4 in activation of mTORC1 in renal cancer cells (Fig. 5A) [17]. We provide evidence that IKKβ contributes to PDCD4 regulation of mTORC1 in the renal cancer cells (Fig. 6A). Moreover, our data demonstrate a conclusive role for the miR-21-IKKβ-mTORC1 axis in renal cancer cell migration and invasion (Figs. 6B – 6I).
Regulation PDCD4 is complex. PDCD4 protein contains one RNA binding domain in its C-terminus followed by two MA-3 domains. It also has two nuclear localization signals at N- and C-terminus. MA-3 domains present in eIF4Gs interact with eIF4A RNA helicase to facilitate the initiation phase of mRNA translation [128]. PDCD4 binds to eIF4A through high affinity binding of its two MA-3 domains to inhibit the function of eIF4A [39]. Additionally, phosphorylation of PDCD4 has been reported. In fact, in cancer cells, it is shown that S6 kinase downstream of Akt/mTORC1 phosphorylates PDCD4 at Ser-67 to induce its degradation by βTrCP pathway [129]. We have shown that in renal cancer cells, mTORC1 activity is significantly increased [17]. Therefore, we cannot rule out the possibility of reduced expression of PDCD4 in renal cancer cells by mTORC1-activated S6 kinase-dependent phosphorylation/degradation of this protein. However, our results show a preference for miR-21-dependent mechanism for decrease in PDCD4 levels that regulate phosphorylation of Akt and increase IKKβ-dependent activation of mTORC1. We detected complex formation between rictor, the exclusive component of mTORC2, and PDCD4, which may block mTORC2 activity (Figs. 7A and 7B). Moreover, the abundance of this complex in renal cancer cells is less than in normal proximal tubular epithelial cells (Figs. 7D and 7E). These results indicate a possible interference of mTORC2 activity in the presence of PDCD4 to regulate phosphorylation of Akt at Ser-473. These data may explain the increased phosphorylation of Akt and mTORC2 activity in the renal cancer cells (Figs. 7F and 7G). Furthermore, inhibition of miR-21 increases association of PDCD4 with rictor (Figs. 8A and 8B). We propose hyperactivation of mTORC2 due to increased miR-21 and decreased PDCD4 resulting in reduced complex formation with rictor, leading to activation of Akt, IKKβ and mTORC1 for induction of invasion of renal cancer cells (Fig. 9). Collectively, our observations suggest that by identifying rictor as an effector of PDCD4 downstream of miR-21, it may be possible to develop therapy for prevention of mTORC2 activity and renal cancer cell invasion by targeting this axis.
Supplementary Material
Highlights.
iR-21 targets PDCD4 to activate Akt and IKKβ for renal cancer cell invasion.
PDCD4 inhibits mTORC1 to prevent renal cancer cell migration and invasion.
IKKβ acts downstream of PDCD4 to activate mTORC1.
miR-21 regulates IKKβ to increase migration and invasion of renal cancer cells.
PDCD4 associates with rictor to inhibit mTORC2 activity.
Acknowledgments
The results described in this manuscript are the work supported by VA Research Service Merit Review 5I01BX000926 and NIH RO1 DK50190 grants to GGC. GGC is a recipient of VA Senior Research Career Scientist Award. BSK and HEA are supported by NIH and VA Research Service grants (BSK and HEA). NGC is supported by VA Research Service Merit Review grant and a pilot grant from Cancer Therapy and Research Center at San Antonio.
Footnotes
DISCLOSURE
The authors declare no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERNCES
- 1.Novick AC. Kidney cancer: past, present, and future. Urol Oncol. 2007;25:188–195. doi: 10.1016/j.urolonc.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG., Jr Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115:2262–2272. doi: 10.1002/cncr.24232. [DOI] [PubMed] [Google Scholar]
- 3.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O’Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, Jia W, Li Z, He M, Sun L, Song P, Sun X, Zhao X, Yang S, Liang C, Wan S, Zhou F, Chen C, Zhu J, Li X, Jian M, Zhou L, Ye R, Huang P, Chen J, Jiang T, Liu X, Wang Y, Zou J, Jiang Z, Wu R, Wu S, Fan F, Zhang Z, Liu L, Yang R, Wu H, Yin W, Liu Y, Peng H, Jiang B, Feng Q, Li C, Xie J, Lu J, Kristiansen K, Li Y, Zhang X, Li S, Wang J, Yang H, Cai Z. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2012;44:17–19. doi: 10.1038/ng.1014. [DOI] [PubMed] [Google Scholar]
- 6.Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, Bignell G, Butler A, Cho J, Dalgliesh GL, Galappaththige D, Greenman C, Hardy C, Jia M, Latimer C, Lau KW, Marshall J, McLaren S, Menzies A, Mudie L, Stebbings L, Largaespada DA, Wessels LF, Richard S, Kahnoski RJ, Anema J, Tuveson DA, Perez-Mancera PA, Mustonen V, Fischer A, Adams DJ, Rust A, Chan-on W, Subimerb C, Dykema K, Furge K, Campbell PJ, Teh BT, Stratton MR, Futreal PA. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–1784. doi: 10.1126/science.319.5871.1782. [DOI] [PubMed] [Google Scholar]
- 9.Huang Y, Dai Y, Yang J, Chen T, Yin Y, Tang M, Hu C, Zhang L. Microarray analysis of microRNA expression in renal clear cell carcinoma. Eur J Surg Oncol. 2009;35:1119–1123. doi: 10.1016/j.ejso.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C, Ganesan S, Bhanot G, Liou LS. Identification of a microRNA panel for clear-cell kidney cancer. Urology. 2010;75:835–841. doi: 10.1016/j.urology.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 11.Nakada C, Matsuura K, Tsukamoto Y, Tanigawa M, Yoshimoto T, Narimatsu T, Nguyen LT, Hijiya N, Uchida T, Sato F, Mimata H, Seto M, Moriyama M. Genome-wide microRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J Pathol. 2008;216:418–427. doi: 10.1002/path.2437. [DOI] [PubMed] [Google Scholar]
- 12.Petillo D, Kort EJ, Anema J, Furge KA, Yang XJ, Teh BT. MicroRNA profiling of human kidney cancer subtypes. Int J Oncol. 2009;35:109–114. doi: 10.3892/ijo_00000318. [DOI] [PubMed] [Google Scholar]
- 13.TCGAR Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yi Z, Fu Y, Zhao S, Zhang X, Ma C. Differential expression of miRNA patterns in renal cell carcinoma and nontumorous tissues. J Cancer Res Clin Oncol. 2010;136:855–862. doi: 10.1007/s00432-009-0726-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volinia S, Galasso M, Costinean S, Tagliavini L, Gamberoni G, Drusco A, Marchesini J, Mascellani N, Sana ME, Abu Jarour R, Desponts C, Teitell M, Baffa R, Aqeilan R, Iorio MV, Taccioli C, Garzon R, Di Leva G, Fabbri M, Catozzi M, Previati M, Ambs S, Palumbo T, Garofalo M, Veronese A, Bottoni A, Gasparini P, Harris CC, Visone R, Pekarsky Y, de la Chapelle A, Bloomston M, Dillhoff M, Rassenti LZ, Kipps TJ, Huebner K, Pichiorri F, Lenze D, Cairo S, Buendia MA, Pineau P, Dejean A, Zanesi N, Rossi S, Calin GA, Liu CG, Palatini J, Negrini M, Vecchione A, Rosenberg A, Croce CM. Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010;20:589–599. doi: 10.1101/gr.098046.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bera A, Ghosh-Choudhury N, Dey N, Das F, Kasinath BS, Abboud HE, Choudhury GG. NFkappaB-mediated cyclin D1 expression by microRNA-21 influences renal cancer cell proliferation. Cell Signal. 2013;25:2575–2586. doi: 10.1016/j.cellsig.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dey N, Das F, Ghosh-Choudhury N, Mandal CC, Parekh DJ, Block K, Kasinath BS, Abboud HE, Choudhury GG. microRNA-21 governs TORC1 activation in renal cancer cell proliferation and invasion. PLoS One. 2012;7:e37366. doi: 10.1371/journal.pone.0037366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, Croce CM. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–4684. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 19.Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–359. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
- 20.Liu ZL, Wang H, Liu J, Wang ZX. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol Cell Biochem. 2013;372:35–45. doi: 10.1007/s11010-012-1443-3. [DOI] [PubMed] [Google Scholar]
- 21.Zhou L, Yang ZX, Song WJ, Li QJ, Yang F, Wang DS, Zhang N, Dou KF. MicroRNA-21 regulates the migration and invasion of a stem-like population in hepatocellular carcinoma. Int J Oncol. 2013;43:661–669. doi: 10.3892/ijo.2013.1965. [DOI] [PubMed] [Google Scholar]
- 22.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, Wu TD, Cavet G, Zhang Z, Chant J. High-resolution genomic and expression analyses of copy number alterations in breast tumors. Genes Chromosomes Cancer. 2008;47:530–542. doi: 10.1002/gcc.20558. [DOI] [PubMed] [Google Scholar]
- 24.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 25.Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–1333. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 26.Mudduluru G, George-William JN, Muppala S, Asangani IA, Kumarswamy R, Nelson LD, Allgayer H. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. 2011;31:185–197. doi: 10.1042/BSR20100065. [DOI] [PubMed] [Google Scholar]
- 27.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ribas J, Lupold SE. The transcriptional regulation of miR-21, its multiple transcripts, and their implication in prostate cancer. Cell Cycle. 2010;9:923–929. doi: 10.4161/cc.9.5.10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mace TA, Collins AL, Wojcik SE, Croce CM, Lesinski GB, Bloomston M. Hypoxia induces the overexpression of microRNA-21 in pancreatic cancer cells. J Surg Res. 2013;184:855–860. doi: 10.1016/j.jss.2013.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2011;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferraro A, Kontos C, Boni T, Bantounas I, Siakouli D, Kosmidou V, Vlassi M, Spyridakis Y, Tsipras I, Zografos G, Pintzas A. Epigenetic regulation of miR-21 in colorectal cancer: ITGB4 as a novel miR-21 target and a three-gene network (miR-21-ITGBeta4-PCDC4) as predictor of metastatic tumor potential. Epigenetics. 2013;9 doi: 10.4161/epi.26842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Xin S, Yang D, He Z, Che X, Wang J, Chen F, Wang X, Song X. Down-regulation of PDCD4 expression is an independent predictor of poor prognosis in human renal cell carcinoma patients. J Cancer Res Clin Oncol. 2012;138:529–535. doi: 10.1007/s00432-011-1121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 35.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 36.Baffa R, Fassan M, Volinia S, O’Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, Rosenberg A. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J Pathol. 2009;219:214–221. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 37.Hiyoshi Y, Kamohara H, Karashima R, Sato N, Imamura Y, Nagai Y, Yoshida N, Toyama E, Hayashi N, Watanabe M, Baba H. MicroRNA-21 regulates the proliferation and invasion in esophageal squamous cell carcinoma. Clin Cancer Res. 2009;15:1915–1922. doi: 10.1158/1078-0432.CCR-08-2545. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Liu W, Chao T, Zhang Y, Yan X, Gong Y, Qiang B, Yuan J, Sun M, Peng X. MicroRNA-21 down-regulates the expression of tumor suppressor PDCD4 in human glioblastoma cell T98G. Cancer Lett. 2008;272:197–205. doi: 10.1016/j.canlet.2008.06.034. [DOI] [PubMed] [Google Scholar]
- 39.Lankat-Buttgereit B, Goke R. The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell. 2009;101:309–317. doi: 10.1042/BC20080191. [DOI] [PubMed] [Google Scholar]
- 40.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004;24:3894–3906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zakowicz H, Yang HS, Stark C, Wlodawer A, Laronde-Leblanc N, Colburn NH. Mutational analysis of the DEAD-box RNA helicase eIF4AII characterizes its interaction with transformation suppressor Pdcd4 and eIF4GI. RNA. 2005;11:261–274. doi: 10.1261/rna.7191905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen AP, Camalier CE, Stark C, Colburn NH. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol Cancer Ther. 2004;3:103–110. [PubMed] [Google Scholar]
- 43.Shiota M, Izumi H, Tanimoto A, Takahashi M, Miyamoto N, Kashiwagi E, Kidani A, Hirano G, Masubuchi D, Fukunaka Y, Yasuniwa Y, Naito S, Nishizawa S, Sasaguri Y, Kohno K. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res. 2009;69:3148–3156. doi: 10.1158/0008-5472.CAN-08-2334. [DOI] [PubMed] [Google Scholar]
- 44.Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008;68:8164–8172. doi: 10.1158/0008-5472.CAN-08-1305. [DOI] [PubMed] [Google Scholar]
- 45.Das F, Ghosh-Choudhury N, Dey N, Mandal CC, Mahimainathan L, Kasinath BS, Abboud HE, Choudhury GG. Unrestrained mammalian target of rapamycin complexes 1 and 2 increase expression of phosphatase and tensin homolog deleted on chromosome 10 to regulate phosphorylation of Akt kinase. J Biol Chem. 2012;287:3808–3822. doi: 10.1074/jbc.M111.246397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das F, Ghosh-Choudhury N, Venkatesan B, Li X, Mahimainathan L, Choudhury GG. Akt kinase targets association of CBP with SMAD 3 to regulate TGFbeta-induced expression of plasminogen activator inhibitor-1. J Cell Physiol. 2008;214:513–527. doi: 10.1002/jcp.21236. [DOI] [PubMed] [Google Scholar]
- 47.Mandal CC, Ghosh-Choudhury N, Yoneda T, Choudhury GG. Simvastatin prevents skeletal metastasis of breast cancer by an antagonistic interplay between p53 and CD44. J Biol Chem. 2011;286:11314–11327. doi: 10.1074/jbc.M110.193714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das F, Ghosh-Choudhury N, Mahimainathan L, Venkatesan B, Feliers D, Riley DJ, Kasinath BS, Choudhury GG. Raptor-rictor axis in TGFbeta-induced protein synthesis. Cell Signal. 2008;20:409–423. doi: 10.1016/j.cellsig.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 49.Allgayer H. Pdcd4, a colon cancer prognostic that is regulated by a microRNA. Crit Rev Oncol Hematol. 2010;73:185–191. doi: 10.1016/j.critrevonc.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Dey N, Das F, Mariappan MM, Mandal CC, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. MicroRNA-21 orchestrates high glucose-induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J Biol Chem. 2011;286:25586–25603. doi: 10.1074/jbc.M110.208066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dey N, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. TGFbeta-stimulated microRNA-21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS One. 2012;7:e42316. doi: 10.1371/journal.pone.0042316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohne Y, Takahara T, Hatakeyama R, Matsuzaki T, Noda M, Mizushima N, Maeda T. Isolation of hyperactive mutants of mammalian target of rapamycin. J Biol Chem. 2008;283:31861–31870. doi: 10.1074/jbc.M801546200. [DOI] [PubMed] [Google Scholar]
- 54.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 55.Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, Honjo T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene. 1995;166:297–301. doi: 10.1016/0378-1119(95)00607-9. [DOI] [PubMed] [Google Scholar]
- 56.Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N, Colburn NH. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol. 2003;23:26–37. doi: 10.1128/MCB.23.1.26-37.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hilliard A, Hilliard B, Zheng SJ, Sun H, Miwa T, Song W, Goke R, Chen YH. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol. 2006;177:8095–8102. doi: 10.4049/jimmunol.177.11.8095. [DOI] [PubMed] [Google Scholar]
- 58.Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65:6034–6041. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- 59.Jin H, Kim TH, Hwang SK, Chang SH, Kim HW, Anderson HK, Lee HW, Lee KH, Colburn NH, Yang HS, Cho MH, Cho CS. Aerosol delivery of urocanic acid-modified chitosan/programmed cell death 4 complex regulated apoptosis, cell cycle, and angiogenesis in lungs of K-ras null mice. Mol Cancer Ther. 2006;5:1041–1049. doi: 10.1158/1535-7163.MCT-05-0433. [DOI] [PubMed] [Google Scholar]
- 60.Yao Q, Xu H, Zhang QQ, Zhou H, Qu LH. MicroRNA-21 promotes cell proliferation and down-regulates the expression of programmed cell death 4 (PDCD4) in HeLa cervical carcinoma cells. Biochem Biophys Res Commun. 2009;388:539–542. doi: 10.1016/j.bbrc.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 61.Wen YH, Shi X, Chiriboga L, Matsahashi S, Yee H, Afonja O. Alterations in the expression of PDCD4 in ductal carcinoma of the breast. Oncol Rep. 2007;18:1387–1393. [PubMed] [Google Scholar]
- 62.Zhang H, Ozaki I, Mizuta T, Hamajima H, Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K, Matsuhashi S. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene. 2006;25:6101–6112. doi: 10.1038/sj.onc.1209634. [DOI] [PubMed] [Google Scholar]
- 63.Gao F, Wang X, Zhu F, Wang Q, Zhang X, Guo C, Zhou C, Ma C, Sun W, Zhang Y, Chen YH, Zhang L. PDCD4 gene silencing in gliomas is associated with 5′CpG island methylation and unfavourable prognosis. J Cell Mol Med. 2009;13:4257–4267. doi: 10.1111/j.1582-4934.2008.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan H, Zhao Z, Quan Y, Xu J, Zhang J, Xie W. DNA methyltransferase 1 knockdown induces silenced CDH1 gene reexpression by demethylation of methylated CpG in hepatocellular carcinoma cell line SMMC-7721. Eur J Gastroenterol Hepatol. 2007;19:952–961. doi: 10.1097/MEG.0b013e3282c3a89e. [DOI] [PubMed] [Google Scholar]
- 65.Appl H, Klempnauer KH. Targeted disruption of c-myb in the chicken pre B-cell line DT40. Oncogene. 2002;21:3076–3081. doi: 10.1038/sj.onc.1205427. [DOI] [PubMed] [Google Scholar]
- 66.Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther. 2010;10:1224–1232. doi: 10.4161/cbt.10.12.14252. [DOI] [PubMed] [Google Scholar]
- 67.Wang YQ, Guo RD, Guo RM, Sheng W, Yin LR. MicroRNA-182 promotes cell growth, invasion, and chemoresistance by targeting programmed cell death 4 (PDCD4) in human ovarian carcinomas. J Cell Biochem. 2013;114:1464–1473. doi: 10.1002/jcb.24488. [DOI] [PubMed] [Google Scholar]
- 68.Denby L, Ramdas V, McBride MW, Wang J, Robinson H, McClure J, Crawford W, Lu R, Hillyard DZ, Khanin R, Agami R, Dominiczak AF, Sharpe CC, Baker AH. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179:661–672. doi: 10.1016/j.ajpath.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci U S A. 2010;107:14339–14344. doi: 10.1073/pnas.0912701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kato M, Arce L, Natarajan R. MicroRNAs and their role in progressive kidney diseases. Clin J Am Soc Nephrol. 2009;4:1255–1266. doi: 10.2215/CJN.00520109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zarjou A, Yang S, Abraham E, Agarwal A, Liu G. Identification of a microRNA signature in renal fibrosis: role of miR-21. Am J Physiol Renal Physiol. 2011;301:F793–801. doi: 10.1152/ajprenal.00273.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato M, Natarajan R. MicroRNA circuits in transforming growth factor-beta actions and diabetic nephropathy. Semin Nephrol. 2012;32:253–260. doi: 10.1016/j.semnephrol.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saal S, Harvey SJ. MicroRNAs and the kidney: coming of age. Curr Opin Nephrol Hypertens. 2009;18:317–323. doi: 10.1097/MNH.0b013e32832c9da2. [DOI] [PubMed] [Google Scholar]
- 74.Sataranatarajan K, Feliers D, Mariappan MM, Lee HJ, Lee MJ, Day RT, Yalamanchili HB, Choudhury GG, Barnes JL, Van Remmen H, Richardson A, Kasinath BS. Molecular events in matrix protein metabolism in the aging kidney. Aging Cell. 2012;11:1065–1073. doi: 10.1111/acel.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neal CS, Michael MZ, Rawlings LH, Van der Hoek MB, Gleadle JM. The VHL-dependent regulation of microRNAs in renal cancer. BMC Med. 2010;8:64. doi: 10.1186/1741-7015-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powers MP, Alvarez K, Kim HJ, Monzon FA. Molecular classification of adult renal epithelial neoplasms using microRNA expression and virtual karyotyping. Diagn Mol Pathol. 2011;20:63–70. doi: 10.1097/PDM.0b013e3181efe2a9. [DOI] [PubMed] [Google Scholar]
- 77.Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol. 2011;8:706–713. doi: 10.4161/rna.8.5.16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E, Olson EN. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell. 2010;18:282–293. doi: 10.1016/j.ccr.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu TX, Lim EJ, Itskovich S, Besse JA, Plassard AJ, Mingler MK, Rothenberg JA, Fulkerson PC, Aronow BJ, Rothenberg ME. Targeted ablation of miR-21 decreases murine eosinophil progenitor cell growth. PLoS One. 2013;8:e59397. doi: 10.1371/journal.pone.0059397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma X, Kumar M, Choudhury SN, Becker Buscaglia LE, Barker JR, Kanakamedala K, Liu MF, Li Y. Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proc Natl Acad Sci U S A. 2011;108:10144–10149. doi: 10.1073/pnas.1103735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 82.Mei Y, Bian C, Li J, Du Z, Zhou H, Yang Z, Zhao RC. miR-21 modulates the ERK-MAPK signaling pathway by regulating SPRY2 expression during human mesenchymal stem cell differentiation. J Cell Biochem. 2013;114:1374–1384. doi: 10.1002/jcb.24479. [DOI] [PubMed] [Google Scholar]
- 83.Niu Z, Goodyear SM, Rao S, Wu X, Tobias JW, Avarbock MR, Brinster RL. MicroRNA-21 regulates the self-renewal of mouse spermatogonial stem cells. Proc Natl Acad Sci U S A. 2011;108:12740–12745. doi: 10.1073/pnas.1109987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Polajeva J, Swartling FJ, Jiang Y, Singh U, Pietras K, Uhrbom L, Westermark B, Roswall P. miRNA-21 is developmentally regulated in mouse brain and is co-expressed with SOX2 in glioma. BMC Cancer. 2012;12:378. doi: 10.1186/1471-2407-12-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han M, Liu M, Wang Y, Chen X, Xu J, Sun Y, Zhao L, Qu H, Fan Y, Wu C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS One. 2012;7:e39520. doi: 10.1371/journal.pone.0039520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang S, Wan Y, Pan T, Gu X, Qian C, Sun G, Sun L, Xiang Y, Wang Z, Shi L. MicroRNA-21 inhibitor sensitizes human glioblastoma U251 stem cells to chemotherapeutic drug temozolomide. J Mol Neurosci. 2012;47:346–356. doi: 10.1007/s12031-012-9759-8. [DOI] [PubMed] [Google Scholar]
- 87.Han M, Liu M, Wang Y, Mo Z, Bi X, Liu Z, Fan Y, Chen X, Wu C. Re-expression of miR-21 contributes to migration and invasion by inducing epithelial-mesenchymal transition consistent with cancer stem cell characteristics in MCF-7 cells. Mol Cell Biochem. 2012;363:427–436. doi: 10.1007/s11010-011-1195-5. [DOI] [PubMed] [Google Scholar]
- 88.Han M, Wang Y, Liu M, Bi X, Bao J, Zeng N, Zhu Z, Mo Z, Wu C, Chen X. MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1alpha expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci. 2012;103:1058–1064. doi: 10.1111/j.1349-7006.2012.02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castaneda CA, Cortes-Funes H, Gomez HL, Ciruelos EM. The phosphatidyl inositol 3-kinase/AKT signaling pathway in breast cancer. Cancer Metastasis Rev. 2010;29:751–759. doi: 10.1007/s10555-010-9261-0. [DOI] [PubMed] [Google Scholar]
- 90.Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4 doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- 91.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 92.Petrella BL, Brinckerhoff CE. PTEN suppression of YY1 induces HIF-2 activity in von-Hippel-Lindau-null renal-cell carcinoma. Cancer Biol Ther. 2009;8:1389–1401. doi: 10.4161/cbt.8.14.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aggarwal BB, Sung B. NF-kappaB in cancer: a matter of life and death. Cancer Discov. 2011;1:469–471. doi: 10.1158/2159-8290.CD-11-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 95.Hiraga T, Myoui A, Hashimoto N, Sasaki A, Hata K, Morita Y, Yoshikawa H, Rosen CJ, Mundy GR, Yoneda T. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res. 2012;72:4238–4249. doi: 10.1158/0008-5472.CAN-11-3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li S, Kendall SE, Raices R, Finlay J, Covarrubias M, Liu Z, Lowe G, Lin YH, Teh YH, Leigh V, Dhillon S, Flanagan S, Aboody KS, Glackin CA. TWIST1 associates with NF-kappaB subunit RELA via carboxyl-terminal WR domain to promote cell autonomous invasion through IL8 production. BMC Biol. 2012;10:73. doi: 10.1186/1741-7007-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matusan-Ilijas K, Damante G, Fabbro D, Dordevic G, Hadzisejdic I, Grahovac M, Maric I, Spanjol J, Grahovac B, Jonjic N, Lucin K. Osteopontin expression correlates with nuclear factor-kappaB activation and apoptosis downregulation in clear cell renal cell carcinoma. Pathol Res Pract. 2011;207:104–110. doi: 10.1016/j.prp.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 98.Oya M, Ohtsubo M, Takayanagi A, Tachibana M, Shimizu N, Murai M. Constitutive activation of nuclear factor-kappaB prevents TRAIL-induced apoptosis in renal cancer cells. Oncogene. 2001;20:3888–3896. doi: 10.1038/sj.onc.1204525. [DOI] [PubMed] [Google Scholar]
- 99.Yang HS, Jansen AP, Nair R, Shibahara K, Verma AK, Cmarik JL, Colburn NH. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene. 2001;20:669–676. doi: 10.1038/sj.onc.1204137. [DOI] [PubMed] [Google Scholar]
- 100.Heimberg H, Heremans Y, Jobin C, Leemans R, Cardozo AK, Darville M, Eizirik DL. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes. 2001;50:2219–2224. doi: 10.2337/diabetes.50.10.2219. [DOI] [PubMed] [Google Scholar]
- 101.Kim S, Millet I, Kim HS, Kim JY, Han MS, Lee MK, Kim KW, Sherwin RS, Karin M, Lee MS. NF-kappa B prevents beta cell death and autoimmune diabetes in NOD mice. Proc Natl Acad Sci U S A. 2007;104:1913–1918. doi: 10.1073/pnas.0610690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chang I, Kim S, Kim JY, Cho N, Kim YH, Kim HS, Lee MK, Kim KW, Lee MS. Nuclear factor kappaB protects pancreatic beta-cells from tumor necrosis factor-alpha-mediated apoptosis. Diabetes. 2003;52:1169–1175. doi: 10.2337/diabetes.52.5.1169. [DOI] [PubMed] [Google Scholar]
- 103.Giannoukakis N, Rudert WA, Trucco M, Robbins PD. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem. 2000;275:36509–36513. doi: 10.1074/jbc.M005943200. [DOI] [PubMed] [Google Scholar]
- 104.Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LA. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. 2010;11:141–147. doi: 10.1038/ni.1828. [DOI] [PubMed] [Google Scholar]
- 105.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 106.Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl 2):S43–51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans. 2013;41:906–912. doi: 10.1042/BST20130041. [DOI] [PubMed] [Google Scholar]
- 109.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hietakangas V, Cohen SM. TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer. 2008;8:282. doi: 10.1186/1471-2407-8-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 113.Atkins MB, Hidalgo M, Stadler WM, Logan TF, Dutcher JP, Hudes GR, Park Y, Liou SH, Marshall B, Boni JP, Dukart G, Sherman ML. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–918. doi: 10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 114.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 115.Traynor K. New oral treatment for kidney cancer approved. Am J Health Syst Pharm. 2009;66:788. doi: 10.2146/news090037. [DOI] [PubMed] [Google Scholar]
- 116.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 119.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 120.Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29:32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 121.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 122.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 123.Luan FL, Ding R, Sharma VK, Chon WJ, Lagman M, Suthanthiran M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney Int. 2003;63:917–926. doi: 10.1046/j.1523-1755.2003.00805.x. [DOI] [PubMed] [Google Scholar]
- 124.Zhou H, Huang S. mTOR signaling in cancer cell motility and tumor metastasis. Crit Rev Eukaryot Gene Expr. 2010;20:1–16. doi: 10.1615/critreveukargeneexpr.v20.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 127.Dan HC, Baldwin AS. Differential involvement of IkappaB kinases alpha and beta in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J Immunol. 2008;180:7582–7589. doi: 10.4049/jimmunol.180.11.7582. [DOI] [PubMed] [Google Scholar]
- 128.Ponting CP. Novel eIF4G domain homologues linking mRNA translation with nonsense-mediated mRNA decay. Trends Biochem Sci. 2000;25:423–426. doi: 10.1016/s0968-0004(00)01628-5. [DOI] [PubMed] [Google Scholar]
- 129.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.