

ABSTRACT
Previous work has shown that prostate cancer in a Pten-null murine model is dependent on the p110β isoform of phosphatidylinositol 3-kinase (PI3K), while breast cancer driven by either polyoma middle T antigen (MT) or HER2 is p110α dependent. Whether these differences in isoform dependence arise from tissue specificity or from the nature of the oncogenic signal activating the PI3K pathway is important, given increasing interest in using isoform-specific PI3K inhibitors in cancer therapy. To approach this question, we studied the PI3K isoform dependence of our recently constructed prostate cancer model driven by MT. Since MT activates a number of signaling pathways, we first confirmed that the MT-driven prostate cancer model was actually dependent on PI3K. A newly generated transgenic prostate line expressing an MT allele (Y315F) known to be defective for PI3K binding displayed a markedly reduced ability to drive tumor formation. We next selectively ablated expression of either p110α or p110β in mice in which wild-type MT was expressed in the prostate. We found that tumor formation driven by MT was significantly delayed by the loss of p110α expression, while ablation of p110β had no effect. Since the tumor formation driven by MT is p110α dependent in the prostate as well as in the mammary gland, our data suggest that PI3K isoform dependence is driven by the mode of PI3K pathway activation rather than by tissue type.
IMPORTANCE Middle T antigen (MT), the oncogene of polyomavirus, can drive tumor formation in a variety of cell types and tissues. Interestingly, MT has no intrinsic enzymatic activity but instead functions by binding and activating cellular signaling proteins. One of the most important of these is the lipid kinase PI3K, which was first studied in MT immunoprecipitates. Ubiquitously expressed PI3K comes in two major isoforms: p110α and p110β. Previous work in animal models showed that p110α was the key isoform in breast tumors driven by oncogenes, including MT and HER2, while p110β was key in prostate tumors driven by Pten loss. We asked the simple question of whether a prostate tumor driven by MT depends on p110α, which would suggest that the mode of activation determines p110 isoform dependence, or p110β, which would suggest that tissue type determines isoform dependence. The clear answer is that MT depends on p110α in both the prostate and breast.
INTRODUCTION
Despite significant progress, prostate cancer remains the second leading cause of cancer-related mortality in men, and identifying treatable targets of the disease is a major focus in many research laboratories. The phosphatidylinositol 3-kinase (PI3K) pathway has been recognized as a potentially valuable target in treating a broad set of human cancers and is known to be activated in prostate cancer. The class IA PI3Ks are key components of growth factor signaling pathways regulating cell proliferation, survival, and death. Class IA PI3Ks are heterodimeric lipid kinases composed of a p85 regulatory subunit and a p110 catalytic subunit. PI3K is activated in a number of manners: in response to growth factor stimulation and oncogenic activation of receptor tyrosine kinases (RTKs) or activation of G-protein-coupled receptors (GPCRs). It is also activated by oncogenes, such as Ras and polyoma middle T antigen (MT) (1–3). Upon activation, PI3K is recruited to the membrane, where the p110 catalytic subunit of PI3K then catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to form phosphatidylinositol 3,4,5-triphosphate (PIP3), subsequently activating multiple downstream signaling pathways. There are three highly homologous p110 catalytic isoforms (p110α, p110β, p110δ) encoded by three distinct genes, Pik3ca, Pik3cb, and Pik3cd, respectively, with p110α and p110β being ubiquitously expressed (1, 2, 4). The lipid phosphatase PTEN, a tumor suppressor highly relevant for prostate cancer in humans, opposes PI3K activity by converting PIP3 back to PIP2 (5). Previous work from our laboratory showed that knockout of p110β in a PTEN-null model of prostate cancer blocks transformation in the anterior lobe of the prostate, while ablation of both p110α and p110β expression is required to block transformation in the remaining lobes (6, 7). We also reported that the loss of p110α expression rescues mammary cells from the transformation induced by the activated receptor tyrosine kinase HER2 or polyomavirus MT (8).
To investigate whether these differences in isoform dependence are linked to a specific tissue type or to the nature of the signal that is activating the PI3K pathway—Pten loss in one and oncogene expression in the other—we decided to compare the same oncogene in different tissue backgrounds. Polyomavirus uses MT to induce tumors in a wide variety of tissues (3, 9). As noted earlier, MT tumorigenesis in the breast requires p110α. We have recently reported a prostate cancer model driven by wild-type MT (10). Here we demonstrate the pivotal role of the PI3K pathway by showing that the loss of PI3K signaling causes significantly delayed tumor growth. Next, we ablated expression of either p110α or p110β in mice expressing wild-type MT in the prostate. We found that in all lobes tumor formation driven by MT was significantly delayed by the loss of p110α expression, while ablation of p110β had no effect. Since tumor formation by polyomavirus MT is p110α dependent in the prostate as well as in the mammary gland, our data suggest that PI3K isoform dependence may be driven by the mode of PI3K pathway activation rather than by tissue type.
MATERIALS AND METHODS
Animal models.
(ARR)2PB-Cre (11), (ARR)2PB-MT (10), p110αL/L (LoxP/LoxP) (12), and p110βL/L (LoxP/LoxP) (7) mice were all maintained in a pure FVB/N background. These mouse strains were intercrossed to generate (ARR)2PB-MT/Cre, (ARR)2PB-MT/p110αL/L, (ARR)2PB-MT/Cre/p110αL/L, (ARR)2PB-MT/p110βL/L, and (ARR)2PB-MT/Cre/p110βL/L mice. All animals were housed, treated, and sacrificed in accordance with protocols approved by the Institutional Animal Care and Use Committees of the Dana-Farber Cancer Institute and Harvard Medical School.
Prostate dissection and wet weight measurement.
The ventral prostate (VP) and dorsolateral prostate (DLP) glands were dissected according to standard procedures. For wet weight measurement, the tissue was immediately weighed using a high-precision lab balance. Shown are mean values and standard errors of the means for at least 5 mice per genotype.
Biochemical analyses.
Prostate tissue was flash frozen in liquid nitrogen before being lysed for biochemical analyses in 1% Nonidet P-40 lysis buffer containing a protease inhibitor cocktail (Roche). After centrifugation, cleared lysates were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Western blot assays were performed with primary antibodies against MT (13), phospho-AktS473 (p-AktS473), phospho-AktT308 (p-AktT308), Akt, p110α, and p110β (all from Cell Signaling Technology). Shown are the results for representative samples, and quantitations show the mean values and standard errors of the means from 3 to 6 independent experiments. For immunoprecipitations (IPs), prostate whole-tissue lysates containing 1 mg total protein were incubated with 10 μg MT antibody (rat monoclonal antibody ab15085) overnight, and protein A/G Sepharose (30 μl 50% bead slurry; Santa Cruz) was added for 2 h. The beads were then collected by centrifugation, followed by three washes with lysis buffer. The resulting immunoprecipitates were either subjected to Western blot analysis or used in a lipid kinase assay.
Lipid kinase assay.
Lipid kinase assays were performed as previously described (8). Briefly, MT immunoprecipitates of freshly prepared cell lysates were subjected to an in vitro kinase assay using phosphatidylinositol (PI; Avanti Polar Lipids) as the substrate. The phosphorylated lipids were resolved by thin-layer chromatography (TLC) overnight. TLC plates were then exposed on a phosphor storage screen (Molecular Dynamics), which was scanned at different exposure time points, using a Storm imaging machine, to make sure that the signals were quantified within the linear range. The radioactive product of the reaction, phosphatidylinositol 3-phosphate (PIP), was quantified by ImageQuant software. In parallel, half of the same immunoprecipitates were visualized by Western blotting to ensure equal pulldown of MT. For kinase assay quantifications, the PIP signal was normalized to the immunoprecipitation efficiency (IP signal/input signal quantified with an Odyssey machine [LI-COR]). Shown are the results for representative samples; quantification plots show the mean values with standard errors of the mean for samples collected from 3 to 6 different mice.
Immunohistochemical analysis.
The individual prostate lobes of (ARR)2PB-MT/Cre, (ARR)2PB-MT/Cre/p110αL/L, and (ARR)2PB-MT/Cre/p110βL/L animals were isolated and fixed in 10% buffered formalin. Sections were stained according to a routine protocol. In brief, sections were dewaxed and rehydrated, followed by antigen retrieval by boiling in citrate buffer. Antibodies were added overnight before detection of a positive signal by diaminobenzidine (Sigma). Sections were then counterstained with Mayer's hematoxylin.
Tissue genotyping.
Frozen tissue was lysed in buffer containing proteinase K overnight at 55°C. Genomic DNA was extracted with phenol-chloroform, precipitated with 0.3 M sodium acetate and isopropanol, and subjected to PCR analysis.
RESULTS
The PI3K binding site on MT is essential for efficient transformation in a polyomavirus MT-driven prostate cancer model.
Previous work from our laboratories showed that prostate-specific expression of polyomavirus MT in the mouse leads to prostate intraepithelial neoplasia (PIN) in the dorsolateral prostate (DLP) and ventral prostate (VP) lobes. This phenotype can be observed in animals as young as 8 weeks of age and progresses throughout the animal's lifetime, ultimately leading to invasion (10).
The connection of MT to PI3K is known to be important for most, but not all, polyomavirus-induced tumors (14). To investigate whether polyomavirus MT-induced transformation is dependent on the PI3K pathway, we generated a new transgenic mouse model expressing an MT allele defective for binding of the p85 regulatory subunit of PI3K by virtue of mutation of the tyrosine at residue 315 in MT to phenylalanine [(ARR)2PB-MTY315F]. This mutation blocks binding of the SH2 domain of the p85 adaptor subunit of PI3K to MT, thus substantially reducing the downstream activation of the pathway. In this model, the mutant MT is still capable of activating the mitogen-activated protein kinase and phospholipase Cγ (PLCγ)/protein kinase C pathways (15, 16). We confirmed that animals expressing wild-type polyomavirus MT showed PIN at an early age and that it progressed to severe PIN over time (Fig. 1A, middle and right columns). Mutation of the polyomavirus MT tyrosine residue responsible for activating the PI3K pathway (Y315) significantly delayed the development of PIN (Fig. 1A, bottom row). However, as was true for a similar polyomavirus MT mutant expressed in breast (17), at the endpoint of this study at 56 weeks, (ARR)2PB-MTY315F animals still showed PIN lesions in the DLP.
FIG 1.
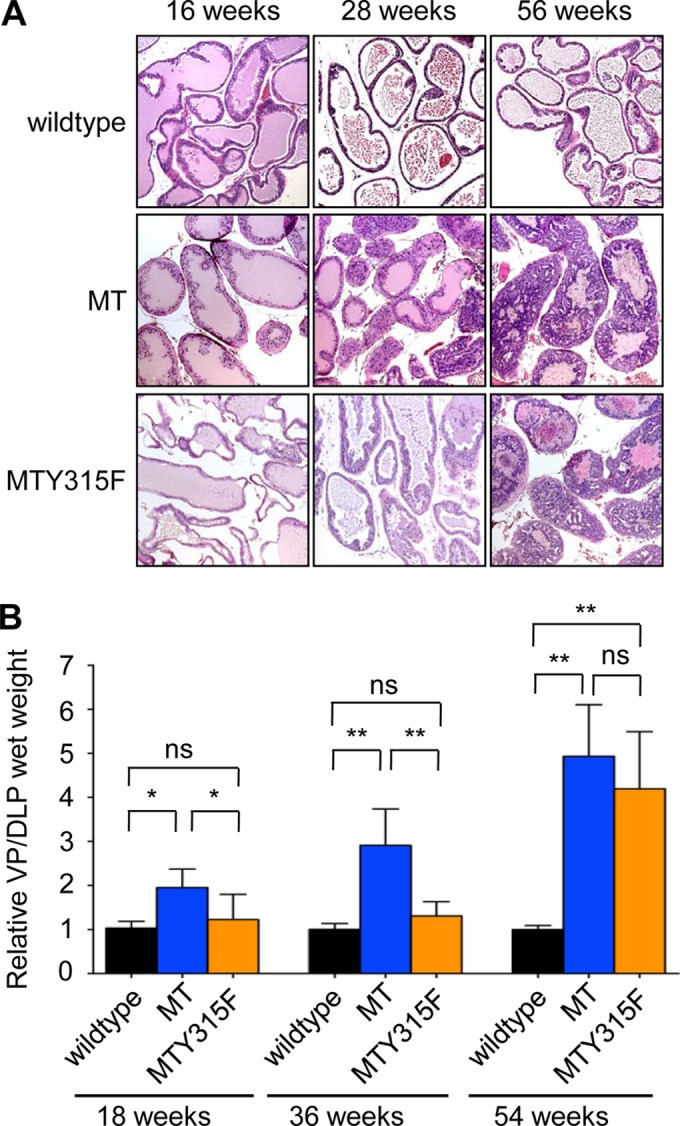
The PI3K binding site on MT is essential for efficient transformation in a polyomavirus MT-driven prostate cancer model. (A) Hematoxylin-eosin staining of formalin-fixed prostate tissue from mice with the indicated genotypes at different ages. (B) Relative wet weight of ventral and dorsolateral prostate lobes from mice with the indicated genotypes at different ages. Shown are mean values and standard errors of the means for at least 5 mice per genotype. MT, polyomavirus middle T antigen; MT Y315F, p85 binding-defective MT; ns, not statistically significant; *, P < 0.05; **, P < 0.001.
Simple measurements of tissue weight supported the interpretation of the histology. Analyses of the wet weight of VP/DLP tissue isolated from (ARR)2PB-MT as well as (ARR)2PB-MTY315F animals show significant weight gain due to PIN in the (ARR)2PB-MT animals as early as 18 weeks of age, at which time tumor growth in (ARR)2PB-MTY315F animals was minimal. However, the tumors in (ARR)2PB-MTY315F animals showed significant growth, as measured by an increased DLP/VP weight at later times (Fig. 1B).
MT Y315F should be defective in association with PI3K and in activating the PI3K pathway. To confirm that mutation of the p85 binding site on MT did in fact inhibit PI3K binding in vivo, we performed lipid kinase assays on MT immunoprecipitates using DLP/VP tissue lysates of either (ARR)2PB-MT or (ARR)2PB-MTY315F animal origin. As expected, we found a dramatically reduced ability to generate PIP from the provided substrate PI (Fig. 2A) as well as decreased downstream activation, as measured from the p-Akt abundance (Fig. 2B) at 30 to 34 weeks, a time when MT expression results in significant PIN, as shown in Fig. 1A. That there is residual activity bound to the Y315F mutant is not surprising, as it has been reported before (16).
FIG 2.
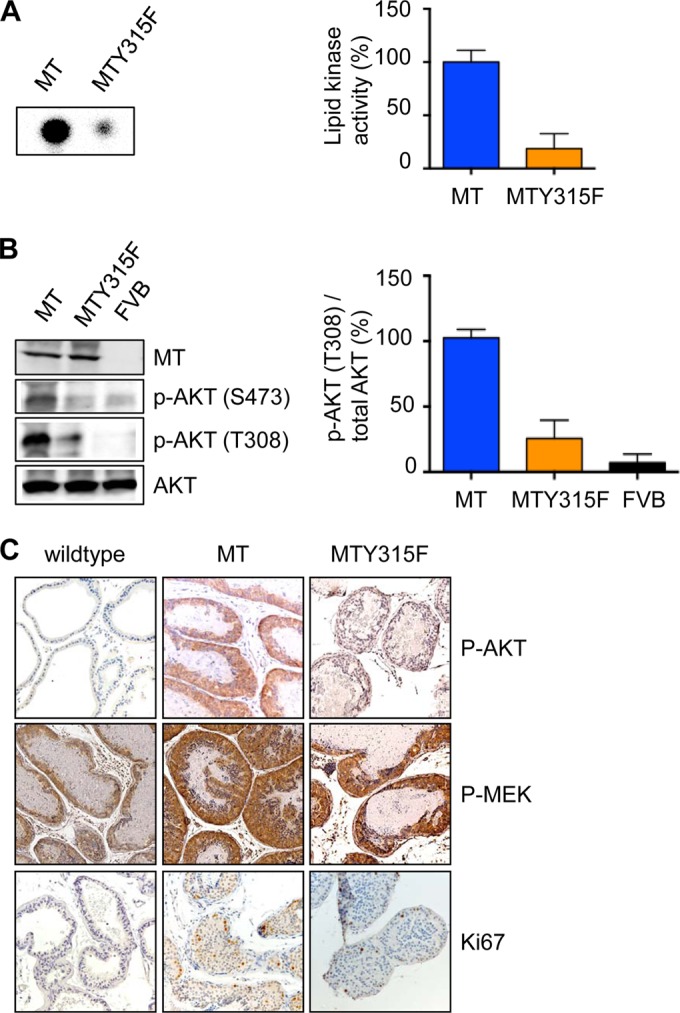
PI3K signaling is essential for efficient transformation in a polyomavirus MT-driven prostate cancer model. (A, B) Lipid kinase assay (A) and Western blot analysis (B) of prostate tissue from mice with the indicated genotypes at 30 to 34 weeks. Shown are the results of one representative experiment (left) and quantifications from 3 independent experiments with standard errors of the means (right). For kinase assay quantifications, the PIP signal was normalized to the IP efficiency (IP signal/input signal in the Western blot). (C) Immunohistochemistry of formalin-fixed prostate tissue from mice with the indicated genotypes at 30 to 34 weeks.
The cell growth seen at later times in the Y315F mutant mice is reminiscent of that seen previously in breast cancer models (17), where all mice eventually develop tumors, albeit over a much longer time period. Whether the growth observed at later times is a result of a separate pathway or the leakiness of the mutant resulting in slow, but PI3K-dependent, growth is not immediately clear. Immunohistochemical analyses of tissue sections of wild-type polyomavirus MT showed strong PI3K pathway activation, as measured by the p-Akt signal and a high proliferation index, as measured by the detection of Ki67-positive nuclei (Fig. 2C). In contrast, in the sections from (ARR)2PB-MTY315F animals, both p-Akt activation and Ki67 staining were limited (Fig. 2C). However, the phospho-MEK (p-MEK) signal was quite strong, suggesting activation of the Ras pathway. Notably, a similar pattern of strong activation of the Ras pathway has been seen in Y315F mutant MT-expressing cells in tissue culture in vitro (16).
Ablation of PI3K catalytic subunit p110α delays MT-driven transformation in the prostate, while ablation of p110β has no effect.
We previously reported that the PTEN model of prostate cancer (18) is strongly dependent on the p110β catalytic subunit of PI3K (7), while p110α expression is essential to a polyomavirus MT-driven breast cancer model (8). Here, we aimed to determine whether these differences in isoform dependencies result from tissue-specific signaling differences or from the nature of the oncogenic insult causing PI3K activation. To do this, expression of either the p110α or p110β catalytic isoform was individually ablated in our MT-driven prostate cancer model. Loss of p110β expression in the (ARR)2PB-MT/Cre/p110βL/L animals had no noticeable effect on PIN formation, whether judged by histology or by the weight of the relevant prostate lobes (Fig. 3A and B). MT-driven PIN was strongly dependent on p110α (Fig. 3A and B). Notably, this result is similar to our data from the MT-induced mammary tumor model (8). (ARR)2PB-MT/Cre/p110αL/L animals showed very few PIN lesions at 28 weeks of age (Fig. 3A, arrowheads; note the presence of normal epithelium, marked with arrows), at which point (ARR)2PB-MT/Cre animals carried a significant tumor burden. Analysis of the tissue at this time showed decreased PI3K activity associated with MT in the (ARR)2PB-MT/Cre/p110αL/L animals compared to that in the (ARR)2PB-MT/Cre/p110βL/L animals (Fig. 4A). This is consistent with the fact that p110β has a weaker kinase activity than p110α (8, 19). As would be expected from the PI3K assay results, there was also much lower Akt activation in the (ARR)2PB-MT/Cre/p110αL/L animals (Fig. 4B and C).
FIG 3.
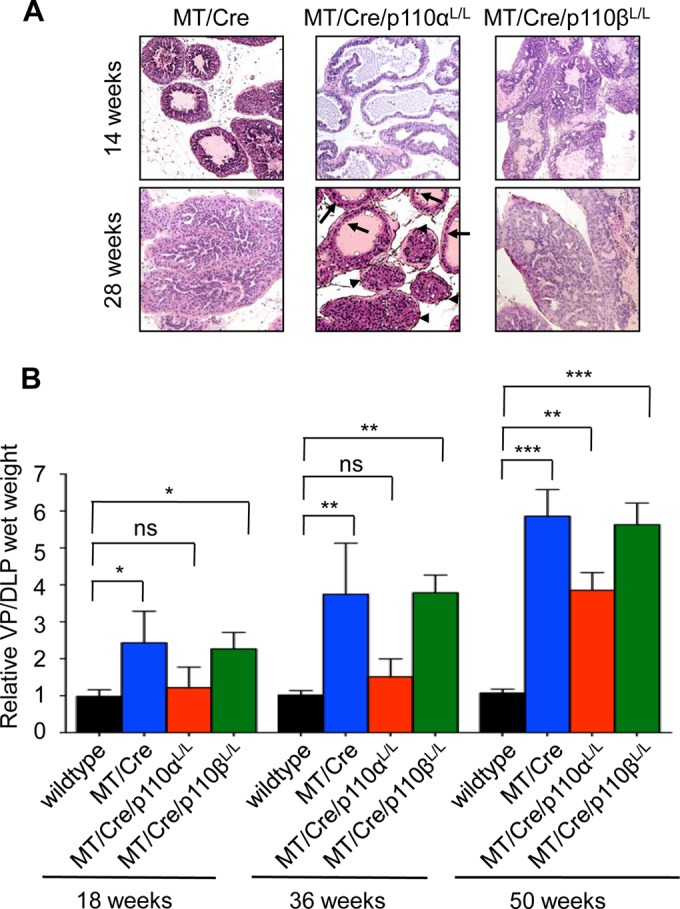
Ablation of the PI3K catalytic subunit p110α delays MT-driven transformation in the prostate. (A) Hematoxylin-eosin staining of formalin-fixed prostate tissue from mice with the indicated genotypes at different ages. Arrowheads, PIN; arrows, normal epithelium. (B) Relative wet weight of ventral and dorsolateral prostate lobes from mice with the indicated genotypes at different ages. Shown are mean values and standard errors of the mean for at least 5 mice per genotype. ns, not statistically significant; *, P < 0.05; **, P < 0.01; ***, P < 0.0001.
FIG 4.

Reduced PI3K signaling due to ablation of the catalytic subunit p110α is essential to inhibit MT-driven transformation in the prostate. (A, B) Lipid kinase assay (A) and Western blot analysis (B) of prostate tissue from mice with the indicated genotypes at 24 to 28 weeks. Shown are the results of one representative experiment (left) and quantifications from 3 independent experiments with standard errors of the means (right). For kinase assay quantifications, the lipid kinase signal was normalized to the IP efficiency (IP signal/input signal in the Western blot). (C) Immunohistochemistry staining of p-Akt in formalin-fixed prostate tissue sections from 28-week-old mice with the indicated genotypes.
PIN lesions did eventually develop in (ARR)2PB-MT/Cre/p110αL/L animals (56 weeks; Fig. 5A). As shown in Fig. 5B, the lesions stained positive for both p-Akt and Ki67. These lesions were qualitatively different from those arising from the MT Y315F animals. The MT Y315F animals showed no elevation of PI3K activity associated with MT or activation of Akt over a 44-week course (Fig. 5C). By comparison, extracts from (ARR)2PB-MT/Cre/p110αL/L animals showed increasing levels of PI3K activity and Akt activation (Fig. 5D). We hypothesized that the Cre recombination was incomplete in these animals. PCRs of tumor DNA from the older tumors revealed that p110α was incompletely deleted (Fig. 5E). The presence of late-arising p-Akt-positive cells can therefore be explained by Cre-recombinase mosaicism.
FIG 5.

Different mechanisms for delayed PIN lesions in MT-driven transformation. (A) Hematoxylin-eosin staining of formalin-fixed prostate tissue from mice with the indicated genotypes at 56 weeks. (B) Immunohistochemistry of p-Akt and Ki67 in formalin-fixed prostate tissue sections from MT/Cre/p110αL/L mice at different ages. (C, D) MT-associated lipid kinase activity (left) and Western blot analysis (middle) of prostate tissue from 3 different mice with the indicated genotypes per time point. (Right) Quantifications for 6 samples per time point. w, weeks. The lipid kinase signal was normalized to the IP efficiency (IP signal/input signal in the Western blot). n.s., not statistically significant; **, P < 0.01. (E) Genotyping of total prostate tissue lysates from mice with the indicated genotypes at 50 weeks. Upper band, floxed allele; lower band, wild-type allele. Cre excision should lead to the complete disappearance of the floxed allele (deleted allele not detectable with the primers used).
DISCUSSION
It is now abundantly clear that different tumors may depend on different class 1A PI3K species. HER2- or MT-positive breast tumors are driven by p110α (8). Prostate tumors that are PTEN deficient show dependence on p110β (7, 20). T cell acute lymphoblastic leukemia driven by Pten loss depends on p110δ and p110γ (21). Therapy targeted toward the isoform relevant to a particular tumor should limit toxicity and present a broader therapeutic window. A key question is whether isoform dependence arises from the particular tissue background or whether it arises from the initiating oncogenic event. To address this question, we have generated two new animal models of MT-driven prostate cancer. Our results now allow us to compare the effect of the same oncogenic driver, polyomavirus MT, in two different tissue backgrounds, namely, breast and prostate tissue.
First, it was first necessary to show that PI3K activity was indeed critical for MT transformation of prostate tissue, as the connection of MT to PI3K is known to be important for many, but not all, polyomavirus-induced tumors (14). Thymic tumors, for example, do not seem to be affected by an MT mutation that blocks PI3K activation. However, we showed here that MT Y315F was clearly defective in inducing prostate tumor formation. Ultimately, tumors did arise, just as has been shown previously in an MT-driven breast cancer model (17).
Having established the importance of the PI3K pathway for MT-driven prostate tumorigenesis, we subsequently tested the contribution of the individual p110 isoforms and observed that tumor formation depends strongly on p110α. This is consistent with prior data on MT-driven breast tissue transformation (8). The mechanism underpinning this isoform dependence may be similar to the model proposed by Utermark and colleagues (8). MT provides a relatively small number of pYMPM motifs required to bind and activate p85/p110 complexes, both because it is expressed at a lower level than most receptors and because only roughly 10% of MT molecules bind Src-family kinases and thus become tyrosine phosphorylated. p85 binds both p110α and p110β equally well, with the ratio of the two enzymes present on the receptor being determined largely by their relative levels of expression in a given cell type or tissue. In most cases the two enzymes have been found to be expressed at roughly comparable levels, i.e., with the major isoform making up no more than 70% of the total. Since p110α is roughly 5- to 10-fold more active on a per molecule basis, it almost always makes up the major component of signaling for the oncogene/receptor, even if p110β is expressed at slightly higher levels. Thus, according to this model, in most cases ablation of p110α will have a strong effect on PI3K pathway activation and consequent cell growth and metabolism.
Impairing PI3K pathway activation by mutating the PI3K binding site on MT (Y315) dramatically slows tumor formation but does not completely prevent it in either the breast or prostate. The same is true if we ablate the expression of p110α, the isoform responsible for carrying the majority of the PI3K signal stimulated by MT expression. Interestingly, the mechanism by which the tumors eventually grow is different in the two cases. In the Y315F mutant tumors, PI3K signaling is not elevated after a long latency. We presume that in this case tumors are driven by alternate pathways stimulated by MT. Thus, signaling via MT's interaction with SHC or PLCγ1 would seem to be a likely explanation. In the case of p110α deletion, it appears likely that the tumor formation at later times is driven by a presumably small original fraction of cells that express MT but have not completely lost expression of p110α, so PI3K and Akt are activated (Fig. 5E).
Our studies have used the ability of MT to drive tumorigenesis in many tissues to ask a fundamental question: is p110 isoform utilization in tumors dependent on the tissue of origin of the tumor or on the oncogenic lesions driving tumor formation? The answer is clear-cut for MT: tumorigenesis is dependent on p110α in two quite distinct tissues, breast and prostate, even though PTEN-driven tumors in the prostate (7), breast (22), and ovary (23) are dependent on p110β. The clear conclusion is that the oncogene/oncogenic event and not the tissue background determines the isoform specificity. We note that our genetic studies primarily address the initiation of tumors and cannot fully predict the reliance of established tumors on PI3K isoforms. Future studies with isoform-specific inhibitors will be required to address this question.
ACKNOWLEDGMENTS
We thank our lab members for helpful discussions. We thank Roderick Bronson and the Dana-Farber Cancer Institute/Harvard Cancer Center Rodent Histopathology Core for pathological analysis and helpful discussions.
This work was supported by NIH grants CA30002 (to T.M.R.), CA34722 (to B.S.S.), and CA50661 (to T.M.R. and B.S.S.).
In compliance with Harvard Medical School guidelines, we disclose that T.M.R. is a consultant for Novartis Pharmaceuticals, Inc.
Footnotes
Published ahead of print 2 July1 2014
REFERENCES
- 1.Liu P, Cheng H, Roberts TM, Zhao JJ. 2009. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8:627–644. 10.1038/nrd2926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11:329–341. 10.1038/nrm2882 [DOI] [PubMed] [Google Scholar]
- 3.Schaffhausen BS, Roberts TM. 2009. Lessons from polyoma middle T antigen on signaling and transformation: a DNA tumor virus contribution to the war on cancer. Virology 384:304–316. 10.1016/j.virol.2008.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 7:606–619. 10.1038/nrg1879 [DOI] [PubMed] [Google Scholar]
- 5.Parsons R. 2004. Human cancer, PTEN and the PI-3 kinase pathway. Semin. Cell Dev. Biol. 15:171–176. 10.1016/j.semcdb.2003.12.021 [DOI] [PubMed] [Google Scholar]
- 6.Jia S, Gao X, Lee SH, Maira SM, Wu X, Stack EC, Signoretti S, Loda M, Zhao JJ, Roberts TM. 2013. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 3:44–51. 10.1158/2159-8290.CD-12-0262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ. 2008. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454:776–779. 10.1038/nature07091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utermark T, Rao T, Cheng H, Wang Q, Lee SH, Wang ZC, Iglehart JD, Roberts TM, Muller WJ, Zhao JJ. 2012. The p110alpha and p110beta isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 26:1573–1586. 10.1101/gad.191973.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fluck MM, Schaffhausen BS. 2009. Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol. Mol. Biol. Rev. 73:542–563. 10.1128/MMBR.00009-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SH, Jia S, Zhu Y, Utermark T, Signoretti S, Loda M, Schaffhausen B, Roberts TM. 2011. Transgenic expression of polyomavirus middle T antigen in the mouse prostate gives rise to carcinoma. J. Virol. 85:5581–5592. 10.1128/JVI.02609-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, Sangiorgi FO, Maxson RE, Sucov HM, Roy-Burman P. 2001. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev. 101:61–69. 10.1016/S0925-4773(00)00551-7 [DOI] [PubMed] [Google Scholar]
- 12.Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, Roberts TM. 2006. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc. Natl. Acad. Sci. U. S. A. 103:16296–16300. 10.1073/pnas.0607899103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pallas DC, Schley C, Mahoney M, Harlow E, Schaffhausen BS, Roberts TM. 1986. Polyomavirus small t antigen: overproduction in bacteria, purification, and utilization for monoclonal and polyclonal antibody production. J. Virol. 60:1075–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freund R, Dawe CJ, Carroll JP, Benjamin TL. 1992. Changes in frequency, morphology, and behavior of tumors induced in mice by a polyoma virus mutant with a specifically altered oncogene. Am. J. Pathol. 141:1409–1425 [PMC free article] [PubMed] [Google Scholar]
- 15.Su W, Liu W, Schaffhausen BS, Roberts TM. 1995. Association of polyomavirus middle tumor antigen with phospholipase C-gamma 1. J. Biol. Chem. 270:12331–12334. 10.1074/jbc.270.21.12331 [DOI] [PubMed] [Google Scholar]
- 16.Utermark T, Schaffhausen BS, Roberts TM, Zhao JJ. 2007. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J. Virol. 81:7069–7076. 10.1128/JVI.00115-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webster MA, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton M, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ. 1998. Requirement for both Shc and phosphatidylinositol 3′ kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol. Cell. Biol. 18:2344–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. 2002. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32:148–149. 10.1002/gene.10036 [DOI] [PubMed] [Google Scholar]
- 19.Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. 2006. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733–747. 10.1016/j.cell.2006.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. 2008. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U. S. A. 105:13057–13062. 10.1073/pnas.0802655105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramaniam PS, Whye DW, Efimenko E, Chen J, Tosello V, De Keersmaecker K, Kashishian A, Thompson MA, Castillo M, Cordon-Cardo C, Dave UP, Ferrando A, Lannutti BJ, Diacovo TG. 2012. Targeting nonclassical oncogenes for therapy in T-ALL. Cancer Cell 21:459–472. 10.1016/j.ccr.2012.02.029 [DOI] [PubMed] [Google Scholar]
- 22.Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, Riddle S, Benes C, Eck M, Roberts T, Gray N, Zhao J. 2012. Functional characterization of an isoform-selective inhibitor of PI3K-p110beta as a potential anticancer agent. Cancer Discov. 2:425–433. 10.1158/2159-8290.CD-12-0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmit F, Utermark T, Zhang S, Wang Q, Von T, Roberts TM, Zhao JJ. 2014. PI3K isoform dependence of PTEN-deficient tumors can be altered by the genetic context. Proc. Natl. Acad. Sci. U. S. A. 111:6395–6400. 10.1073/pnas.1323004111 [DOI] [PMC free article] [PubMed] [Google Scholar]