

Abstract
Histone acetylation modulates alternative splicing of several hundred genes. Here, we tested the role of the histone acetyltransferase p300 in alternative splicing and showed that knockdown of p300 promotes inclusion of the fibronectin (FN1) alternative EDB exon. p300 associates with CRE sites in the promoter via the CREB transcription factor. We created mini-gene reporters driven by an artificial promoter containing CRE sites. Both deletion and mutation of the CRE site affected EDB alternative splicing in the same manner as p300 knockdown. Next we showed that p300 controls histone H4 acetylation along the FN1 gene. Consistently, p300 depletion and CRE deletion/mutation both reduced histone H4 acetylation on mini-gene reporters. Finally, we provide evidence that the effect of CRE inactivation on H4 acetylation and alternative splicing is counteracted by the inhibition of histone deacetylases. Together, these data suggest that histone acetylation could be one of the mechanisms how promoter and promoter binding proteins influence alternative splicing.
Keywords: alternative splicing, fibronectin, p300, histone acetylation, promoter
Introduction
Most eukaryotic cells use alternative splicing as a tool to increase the coding potential of their genomes. When alternative splicing was discovered it was considered a rare event, but high-throughput technologies have shown that 95% of multi-exon genes undergo alternative splicing.1-3 The basic regulators of alternative splicing are cis-elements in pre-mRNA and trans-acting factors, which recognize cis-elements and determine alternative splicing outcomes.4,5
RNA polymerase II (Pol II) synthesizes 3000 to 5000 nucleotides per minute and transcription of most genes is accomplished within a couple of minutes.6-8 In vivo splicing is achieved within tens of seconds suggesting most introns are spliced out while RNA is still attached to Pol ll and the DNA template.9-11 Consistent with this data, splicing factors have been shown to be recruited to the site of pre-mRNA synthesis and bind the nascent pre-mRNA12-14 and 80% of active spliceosomes are attached to chromatin.15 This coupling of pre-mRNA splicing and Pol II transcription brings another layer of regulation.16-20 The C-terminal domain of Pol ll is necessary for proper pre-mRNA processing21-24 and there are several lines of evidence indicating Pol II elongation rate affects splice site choice.25-29
The fact that most introns are removed co-transcriptionally means splicing occurs while pre-mRNA is close to chromatin, which may further influence splicing decisions. Indeed, chromatin structure and modifications have been found to influence alternative splicing.30-35 Several methylation marks were shown to regulate alternative splicing or affect splicing efficiency.36-38 The close relationship between splicing and chromatin is further documented by splicing dependent methylation of H3K3639,40 or a feedback loop between H3K4me3, splicing and transcription.41
In addition to histone methylation, histone acetylation has also been shown to affect alternative splicing.42,43 In human cells, higher level of histone acetylation leads to higher Pol II elongation rate and recruitment of different splicing factors to pre-mRNA.42,43 Specifically, H3K9 hyperacetylation regulates NCAM alternative splicing during membrane depolarization of neuronal cell.44
Finally, findings that connect the promoter with alternative splicing regulation strengthened the relationship between transcription and pre-mRNA splicing.45-47 Alternative splicing regulation was apparently not dependent on promoter strength or the amount of mRNA transcribed from individual promoters.47 Experiments with steroid-sensitive promoters showed steroid hormones affected alternative splicing only in the case of steroid-sensitive promoters.48 These results pointed to the possibility that factors regulating alternative splicing somehow act through specific promoter occupancy. Indeed, different promoter-associated transcriptional co-activators affect alternative splicing and the thermogenic coactivator PGC-1 influenced alternative splicing only when tethered to the promoter.49-51 Recently, we showed the acetylated histone binding protein Brd2, which regulates the alternative splicing of several hundred genes, is also preferentially found at promoters of target genes.52 However, the molecular mechanism of how promoters regulate alternative splicing is missing.
Here, we concentrated on histone acetyltransferase p300, which associates with CRE sites in promoters. To analyze the relationship between promoter and alternative splicing we utilized a mini-gene alternative splicing reporter, which can be easily manipulated. We combined promoter mutagenesis with chromatin immunoprecipitation and alternative splicing analysis to test whether p300 association with the promoter regulates alternative splicing via histone acetylation.
Results
p300 knockdown changes alternative splicing
In our previous work we have shown that alternative splicing of the EDB (EDIII, extra domain B) exon of fibronectin (FN1) is sensitive to changes in histone acetylation and that histone deacetylase (HDAC) inhibition or depletion promoted EDB exon exclusion.42 We then searched for histone acetyltransfereses which would affect alternative splicing of EDB in the opposite manner. First, we focused on the protein p300, a transcription co-activator that possesses histone acetyltransferase activity and adds acetyl groups on to histone H2B (K12/K15), H3 (K12/K18) and H4 (K5/K8).53 We knocked down p300 by RNAi and analyzed the splicing pattern of the endogenous EDB exon by classical and quantitative RT-PCR (Fig. 1A). Upon p300 depletion we observed an increase in EDB exon inclusion, which was the opposite effect of HDAC inhibition.42 To test the specificity of the knockdown we utilized three different siRNAs and observed a similar effect on EDB splicing (Figure S1A, S1B). In addition, the knockdown of another histone acetyltransferase, CBP, showed no effect on EDB alternative splicing (Figure S2A), suggesting that the effect of p300 is specific. Because EDB exon inclusion is regulated by SR proteins,54 we checked the levels of splicing regulators after p300 knockdown (Fig. 1B and Figure S1C). We did not observe any significant change in the expression of EDB splicing regulators SRSF1 and SRSF5. It should be noted that we did not detect any global changes of acetylated histone H4 either. These data suggested that p300 is not the major cellular histone acetyltransferase but rather, p300 acts locally at selected sites.

Figure 1. p300 knockdown affects alternative splicing of EDB alternative exon. (A) Splicing pattern of endogenous alternative EDB exon was analyzed by classical RT-PCR and by RT-qPCR after p300 knockdown (black box – EDB exon, gray boxes – constitutive exons). The graph shows the ratio of endogenous EDB included or excluded after p300 knockdown normalized to NC siRNA. The average of four experiments is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to cells treated with NC siRNA. (B) Western blots showing levels of splicing factors after p300 knockdown. It also shows the p300 knockdown efficiency and global H4 acetylation after p300 knockdown, GAPDH served as a loading control. Expression level of SRSF1 after p300 knockdown was monitored in a HeLa cell line stably expressing SRSF1-GFP from bacterial artificial chromosome that preserves endogenous SRSF1 promoter as well as its exon-intron structure. Parental HeLa cells were used as a negative control and GAPDH as a loading control. (C) Analysis of the alternative EDB exon splicing transcribed from the mini-gene reporter driven by endogenous FN1 promoter after p300 knockdown. (D, E) Analysis of splicing pattern of EDB alternative exon from the Tet driven mini-gene reporter (D) or the CMV driven mini-gene reporter (E) by classical RT-PCR and RT-qPCR after p300 knockdown. Graphs show the ratio of EDB included or excluded after p300 knockdown normalized to NC siRNA. The average of three experiments is shown including SEM, ** indicates P ≤ 0.01 of the t test with respect to cells treated with NC siRNA. In all cases mRNAs originated from mini-gene reporters were detected by reverse primer specific for mini-gene reporter.
CRE site controls EDB alternative splicing
p300 associates with promoters via the CREB protein, by binding a short 8 nucleotide sequence (TGACGTCA) called cAMP response element (CRE). The CRE site is present in the promoters of many genes including FN1.55 We tested alternative splicing of six additional genes containing CRE sites in their promoters. Three of these genes produced two splicing variants in HeLa cells and the ratio of these splicing variants changed upon p300 knockdown (Figure S2B). To examine whether the CRE site is important for p300 regulation of alternative splicing, we created a splicing reporter comprised of the alternative EDB exon, the surrounding introns and a partial section of the neighboring exons, all under the control of the endogenous FN1 promoter. Unfortunately, this splicing reporter produced only one mRNA variant with included EDB exon (Fig. 1C). Because p300 knockdown promoted inclusion of the EDB exon we could not use this construct for further experiments. Therefore we substituted the FN1 promoter in the splicing reporter by the CMV promoter, which possesses four CRE sites, or Tet responsive promoter, which is leaky in HeLa cells and does not contain any CRE site. Both reporters generated two splicing variants (Fig. 1D and E), but only the CMV driven reporter was sensitive to p300 depletion and exhibited elevated EDB exon inclusion after p300 knockdown. This alternative splicing change of the CMV reporter corresponded to alternative splicing changes of the endogenous EDB exon.
These data suggest that the CRE site is important for p300-mediated splicing regulation. To confirm the importance of CRE sites in alternative splicing regulation we deleted all CRE sites in the CMV promoter (Δ all) and tested EDB splicing. Removal of all CRE sites had a similar effect as p300 depletion and promoted EDB inclusion, but the inclusion was less pronounced than in the case of p300 knockdown (Fig. 2B). Next, we analyzed whether individual CRE sites controlled alternative splicing in a similar way. We deleted various CRE sites and found that only the removal of the first CRE site (Δ 1) promoted EDB inclusion, while deletion of other CRE sites had only a minimal effect (Fig. 2B and Figure S3). Surprisingly, deletion of the first CRE site showed a stronger effect than deletion of all four CRE sites, suggesting additional sequences in the promoter might counterbalance the first CRE. To further demonstrate that the first CRE site is important for EDB alternative splicing regulation, we mutated two nucleotides essential for binding of the CREB protein, which interacts with p300 and navigates it to the promoter.56 Mutation of the first CRE site (mut 1) had a similar effect on alternative splicing as its deletion and promoted inclusion of the EDB exon (Fig. 2B). To confirm that CRE site mutations abolished targeting of p300 to this site we performed chromatin immunoprecipitation (ChIP) with p300 and found significantly reduced p300 association with the promoter containing the mutated CRE site (Fig. 2C). To test whether mutations in the promoter affected transcription of the reporter we compared expression of the splicing reporter and the ampicillin resistance gene. Both genes are transcribed from the same plasmid57 and allow normalization of mini-gene reporter transcription to transfection efficiency. We observed the same amount of transcribed reporter RNA from different promoters (Fig. 2D), suggesting that promoter modifications did not significantly affect total amounts of reporter RNA.

Figure 2. The first CRE site in the promoter influences alternative splicing of the mini-gene reporter. (A) Schematic interpretation of splicing reporters used in this study. White boxes – promoter with CRE sites, gray boxes – constitutive exons, black lines – introns, black box – alternative EDB exon, light gray lines above scheme represent primers used for RT-PCR. The mutated first CRE site and mutated nucleotides are marked by a star. (B) Analysis of splicing pattern of EDB exon after manipulation with CMV promoter by RT-PCR and RT-qPCR (black box – EDB exon, gray boxes – surrounding exons, * marks an unspecific band). The graph shows the ratio of endogenous EDB included or excluded mRNAs originated from individual reporters. The average of three to six experiments (normalized to wt reporter) is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to wt reporter. (C) p300 binding to first CRE site analyzed by ChIP. The average of three experiments is shown including SEM, * indicates P ≤ 0.05 of the t test with respect to wt reporter. (D) Relative expression of FN1 cassette from individual reporters was measured by comparing total FN1 reporter mRNA to mRNA of ampicillin resistance gene on same plasmids. The average of three experiments is shown including SEM.
CRE site and p300 affect H4 acetylation
The previous data showed that the mutation of the CRE site in the CMV promoter reduced binding of histone acetyltransferase p300 (Fig. 2C). To test whether decreased p300 association affected histone acetylation along the mini-gene reporter we analyzed histone H4 acetylation by native chromatin immunoprecipitation (nChIP) at two positions unique to the reporters – the promoter itself and the sequence downstream of the FN1 cassette. We focused on H4 acetylation, because we previously observed the highest changes in H4 acetylation after HDAC inhibition at the FN1 gene.52 We normalized the H4 acetylation signal to the total H3 signal to rule out whether the increased H4 acetylation signal was being caused by an increase in nucleosome occupancy. We observed reduced H4 acetylation at promoters with deleted CRE sites (Fig. 3A). Surprisingly, we observed even greater differences in H4 acetylation on the region downstream of the FN1 cassette, suggesting that proteins bound to the CRE site in the promoter region influenced histone acetylation on regions several kilobases downstream of that promoter (Fig. 3A). The wt reporter, which preferentially excludes the EDB exon, manifested the highest level of H4 acetylation compared with other reporters. Conversely, the Δ1 reporter, which largely includes the EDB exon, exhibited the lowest H4 acetylation. To confirm that p300 depletion reduced histone acetylation we used nChIP to analyze histone acetylation on the endogenous FN1 gene and the wt reporter after p300 knockdown (Fig. 3B and C).

Figure 3. Mutation or deletion of the first CRE site and p300 knockdown reduce H4 acetylation. (A) Native ChIP showing histone H4 acetylation at the promoter and downstream of the FN1 cassette of reporters with different promoters. Grey lines under reporter schemes show primer positions. Nonspecific IgG signal was below 1% of input in all cases. The average of three experiments (normalized to H3 signal and wt reporter) is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to wt reporter. (B) Decrease of histone H4 acetylation after p300 knockdown on wt reporter was determined by nChIP. Nonspecific IgG signal was below 1% of input in all cases. The average of four experiments normalized to H3 and NC siRNA treated cells is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to NC siRNA treated cells. (C) Significant changes of H4 acetylation along the endogenous FN1 gene after p300 depletion were determined by nChIP. The average of three experiment is shown including SEM, * indicates P ≤ 0.05 and ** indicates P ≤ 0.01 of the paired t test with respect to NC siRNA treated cells. (D) Pol II elongation rate on the FN1 endogenous gene was determined as a ratio of two pre-mRNA sequences (downstream/upstream, B/A) which were positioned within an intron located either upstream or downstream of EDB exon. The average of three experiment is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to NC siRNA treated cells.
It was previously described that the level of histone acetylation affected Pol II elongation rate.58 Therefore, we analyzed the elongation rate of Pol II on the endogenous FN1 gene sequences around the alternative EDB exon. Pol II elongation rate was measured by calculating the ratio of two regions (A and B) within one intron as previously described.44 We observed a partial reduction of Pol II elongation rate (the lower B:A ratio the lower Pol II elongation rate) after p300 knockdown at both introns upstream and downstream of the EDB exon (Fig. 3D). Those findings correlate with our previous results showing that an increase in H4 acetylation both promotes EDB exon exclusion and enhances Pol II elongation rate.42
HDAC inhibition counteracts the deletion/mutation of CRE sites
Our data showed a negative correlation between histone acetylation and alternative exon inclusion (Fig. 2 and 3). Next we tested whether the effect of a CRE mutation/deletion can be overridden by an increase in histone acetylation. We transfected cells with individual reporters and 24 h after transfection treated cells with sodium butyrate (NaB), a potent HDAC inhibitor, to increase histone acetylation. Inhibition of HDACs was shown by both a global increase in H4 acetylation as monitored by western blot using an antibody against acetylated H4 (Fig. 4A) and a specific increase of H4 acetylation on splicing reporters by nChIP (Figure S4). Next, we analyzed the splicing pattern of mini-gene reporters by classical RT-PCR and observed that higher histone acetylation induced by NaB treatment promoted EDB exclusion. The only exception was the wt reporter, which was not affected by HDAC inhibition, likely due to the fact that the EDB exon was already largely excluded in untreated control cells (Fig. 4B,C). These data are consistent with the model that dynamic interplay between histone acetylases and deacetylases modulate alternative splicing.
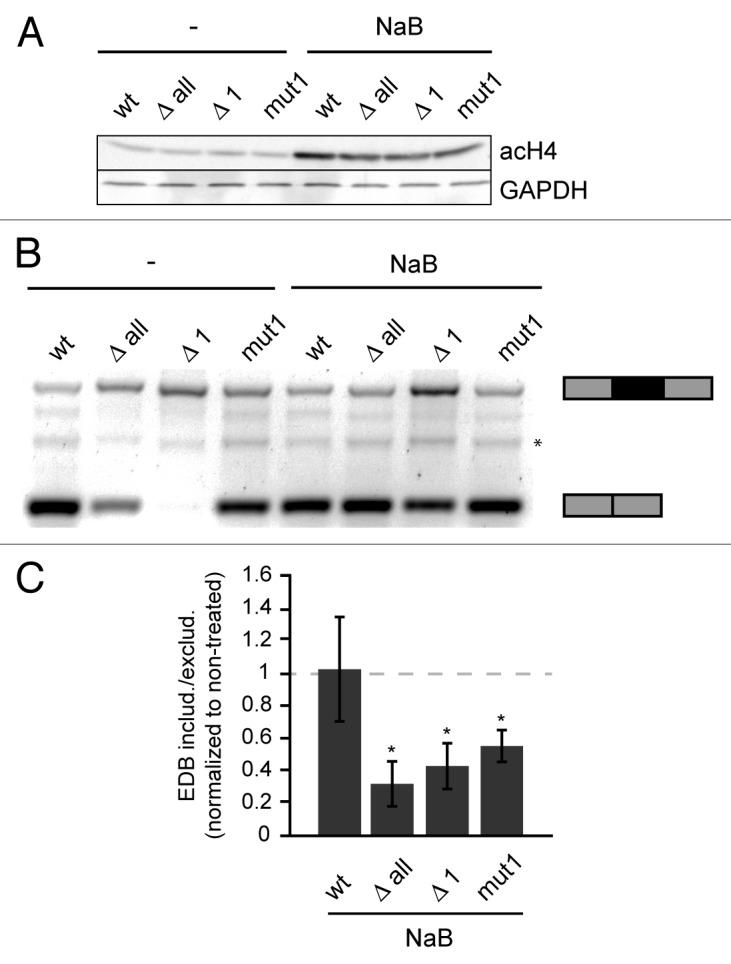
Figure 4. Inhibition of HDACs promotes EDB exon exclusion. (A) Global histone acetylation of H4 analyzed by western blotting before and after NaB treatment. GAPDH was used as a loading control. (B) RT-PCR analysis of alternative splicing of EDB exon from reporters in non-treated and NaB treated cells (* marks an unspecific band). (C) Ratio of EDB inclusion or skipping analyzed by colorimetric analysis using ImageJ software and normalized to non-treated cells. The gray dashed line represent ratio of EDB included/excluded in non-treated cells. The average of three experiments is shown including SEM, * indicates P ≤ 0.05 of the paired t test with respect to non-treated reporter.
Discussion
For more than 15 y it has been known that promoter identity affects alternative splicing decisions (for review see ref.59). However, the molecular mechanisms that underlay this regulation have been elusive. Here, we provide evidence that promoter controlled histone acetylation correlates with alternative splicing changes, suggesting the promoter modulates alternative splicing via chromatin acetylation. Our data show that a deletion or mutation of the CRE site in the promoter reduces the interaction of the promoter with histone acetyltransferase p300, which in turn lowers histone acetylation at both the promoter and sequences several kilobases downstream of the promoter. A similar promoter modulation of downstream histone positioning was recently shown in yeast.60 This suggests that promoter occupancy regulates events that occur on chromatin domains that largely exceed the promoter region. How is this achieved? First, histone acetylation, which is necessary for proper transcription initiation and is present at promoters of active genes (for review see ref.61), could be propagated in a similar fashion as it was describe for H3K9me3 spreading in yeast.62 Thus, the initial level of histone acetylation is transmitted throughout the entire gene and modulates alternative splicing of exons located many thousands of nucleotides downstream of the promoter. Alternatively, promoters and the complexes which form on promoters, might directly contact downstream regions by DNA looping. Recently, it was shown that promoters contact exons, preferentially alternative exons, in a tissue specific manner and this promoter-exon association correlates with alternative splicing changes.63 In this view, histone acetyltransferases bound to the promoter could directly modify histones downstream within the gene.
A mutation or deletion of the first CRE site resulting in decreased histone acetylation had no effect on the overall expression of the model mini-gene reporter. This result suggests that promoter elements, other than the first CRE site, control formation of the transcription initiation complex and transcription efficiency. This finding supports previous results that different features of the promoter, other than its strength, are important for promoter dependent splicing regulation.46 We have previously shown that increased histone acetylation of alternative exons locally enhanced Pol II elongation rate, which in turn affected alternative exon inclusion.42 Here we propose that the elongation rate of Pol II in the vicinity of alternative exons is modulated through promoter controlled histone acetylation. The total amount of RNA transcribed from the gene would not be significantly altered due to the fact that the RNA amount depends mainly on the number of transcribing polymerases and not their speed. Thus, different elements in the promoter could independently control transcription efficiency and alternative splicing.
The CRE site is also found in the endogenous FN1 gene. It was previously shown using a mini-gene reporter that the mutation of the FN1 CRE site, together with the mutation of the CCAAT site, largely promoted inclusion of another FN1 alternative exon – EDA (ED I).46 These data are consistent with our results and show that the mutation of the CRE site in different promoters affects alternative splicing the same way. In addition, we showed that alternative splicing of three other genes, all of which contain CRE sites in their promoters, is sensitive to p300 knockdown.
More than half of all human protein-coding genes contain alternative promoters64 and more than 95% of multi-exon genes are subject to alternative splicing.65 Our data suggest promoters and enhancers contain elements that can control alternative splicing independently of transcription regulation and that this regulation involves chromatin acetylation. In contrast to histone methylation, histone acetylation is more dynamic and provides a regulatory system that could rapidly react to changes in the cells environment.
Materials and Methods
Cell culture and treatment
HeLa and SRSRF1-GFP (HeLa, BAC) cell lines were cultured in DMEM supplemented with 10% fetal calf serum, penicillin and streptomycin (Invitrogen) and treated with 5 mM sodium butyrate for 12 (splicing assays) or 6 (nChIP assay) hours.
Antibodies
Polyclonal antibody for acetylated histone H4 was purchase from Upstate. Anti-H3, anti-GAPDH and anti-p300 (used for ChIP) antibodies were purchase from Abcam. The monoclonal m104 antibody, which recognizes phosphorylated SR proteins, was a gift from K. Neugebauer (Max Planck Institute of Molecular Cell Biology and Genetics, Germany). Nonspecific mouse IgG, anti-p300 (used for western blotting) and anti-CBP antibodies were purchased from Sigma. The anti-GFP antibody was purchase from Santa Cruz Biotechnology.
Plasmids and transfections
The FN1 cassette containing the EDB exon, together with its neighboring introns and exons (~3kb) were amplified from human genomic DNA, analyzed by sequencing and cloned into the BamHI site in the pcDNA3 plasmid (Invitrogen) (wt). The deletion mutants (Δ all, Δ 1, Δ 3–4, Δ 2–4, Δ 1–3) were prepared by PCR with specific primers. The mut1 plasmid was constructed by PCR site-specific mutagenesis. The sequence of the FN1 promoter was amplified from genomic DNA (403bp) and cloned into the BglII and HindIII restriction sites of pcDNA3 plasmid. The FN1 cassette was then inserted into the BamHI site. The Tet responsive promoter was removed from from the pTet-ON plasmid66 with BglII and HindIII restriction enzymes and inserted into the pcDNA3 plasmid into the same restriction sites. The FN1 cassette was then inserted into the BamHI site in pcDNA3. All promoter changes were confirmed by DNA sequencing. Plasmids were transiently transfected with Lipofectamine® LTX (Invitrogen) according to the manufacturer’s protocol and incubated for 24 h. Pre-annealed siRNA duplexes were obtained from Ambion: p300 #1: 5` GGACUACCCUAUCAAGUAAtt 3`, p300 #2: 5` GCCUGGUUAUAUAACCGCAtt 3`, p300 #3 5` CCACUACUGGAAUUCGGAAtt 3`, CBP #1: 5` GAAUCUUUCCCAUAUCGAAtt 3`, CBP #2: 5` CAGCUAUCAGAAUAGGUAUtt 3`.The negative control # 5 siRNA from Ambion was used as a negative control siRNAs (f.c.: 20 nM or 70nM) were transfected with Oligofectamine (Invitrogen) according to the manufacture’s protocol. Cells were incubated for 48 h. For co-transfection experiments, cells were transfected with siRNAs and after 24 h transfected with reporters and incubated additional 24 h.
RNA isolation and RT-PCR/RT-qPCR
Total RNA was isolated with TRIzol (Invitrogen). Reverse transcription was performed using SuperScript III (Invitrogen) and cDNA amplified by Taq polymerase (Fermentas). Primers used for RT-PCR and qPCR are detailed in the primer list. For RT-qPCR reactions, SyberGreen mix (Invitrogen) was used. The ratio of mRNA with EDB exon included/excluded was calculated from relative Ct values of primers EDBinclud and EDBexclud according to R = 2(CtEDBinclud – CtEDBexclud). The ratio of endogenous mRNA with EDB exon was calculated from relative Ct values of primers UP EDB (FN1 exon 24) and EDB (FN1 exon 25) according to R = 2(CtEDB – CtUP). Pol II elongation rate was calculated from relative Ct values of primer pairs A (upstream) and B (downstream) within particular intron according to pre-mRNA ratio distal/proximal = 2(CtA – CtB).
Native ChIP assay
We used native ChIP to increase effectiveness of histone immunoprecipitation. Hela cells transfected with splicing reporters for 24 h were washed by PBS and resuspended in 0.3 M sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 0.2% NP-40, 15 mM TRIS-HCl, pH 7.7, 0.5 mM DTT and complete protease inhibitor cocktail (Calbiochem). Nuclei were released by passaging through a 22 G needle and loaded onto a sucrose gradient (1.2 M sucrose, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 15 mM TRIS-HCl, pH 7.7, 0.5 mM DTT, protease inhibitors) and centrifuged for 20 min at 2000 g, 4 °C. Pellets were resuspended in MNase digestion buffer (0.32 M sucrose, 1 mM CaCl2, 4 mM MgCl2,15 mM TRIS-HCl pH 7.7 and protease inhibitors) and digestions were performed for 3 min at 37 °C (1U MNase/30ug chromatin). Reactions were stopped with the addition of EDTA (final concentration 10 mM) and centrifuged. The supernatant was taken and the pellet resuspended in 0.2 mM EDTA, 1 mM Tris/HCl, pH 7.7, incubated for 1 h at 4 °C, centrifuged again and both supernatants were mixed. ~100 µg of chromatin was diluted in nChIP buffer (50 mM NaCl, 5 mM EDTA, 50 mM Tris/HCl, pH 7.7) and incubated overnight at 4 °C with the appropriate antibody (5 ug nonspecific IgG, 5 ug anti-acetyl H4 or 5 ug anti-H3). The beads were washed once with nChIP buffer, then twice in the same buffer with increasing salt concentration (75 mM NaCl, 125 mM NaCl, 175 mM NaCl). Complexes were eluted with 1% SDS for 15 min at room temperature, treated with 20 µg proteinase K for 30 min at 45 °C and DNA was recovered with the QIAGEN PCR Purification Kit, quantified by qPCR and signals were compared with the input: 2Ct(input) – Ct(spec). The signal of acetylated-H4 was normalized to the H3 signal.
ChIP assays
We used classical ChIP for non-histone proteins. It is necessary to cross-linked bound proteins to chromatin to immunoprecipitate them, because they would not retained on the DNA during nuclease digestion of native chromatin.67 Hela cells transfected with reporters were washed with PBS, crosslinked with 1% formaldehyde/PBS for 10 min at room temperature and the reaction was stopped with the addition of glycine (final conc. 125 mM). Cells were resuspended in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM TRIS-HCl, pH 8.0, 5 mM EDTA, 0.5 mM PMSF, complete protease inhibitor cocktail (Calbiochem), 50 mM NaF and 0.2 mM sodium orthovanadate) and sonicated to generate ~500 nt chromatin fragments. The same total amount of protein (2 mg) was used for immunoprecipitation. Immunoprecipitation with the appropriate antibodies (2 µg anti-p300 or 2 µg IgG) was performed at 4 °C overnight. Subsequently, the beads were rinsed twice with RIPA, four times with 100 mM TRIS-HCl, pH 8.5, 500 mM LiCl, 1% Nonidet P-40, 1% deoxycholic acid, twice again with RIPA and twice with TE. Protein-DNA complexes were eluted with 1% SDS for 10 min at 65 °C, uncrosslinked in the presence of 200 mM NaCl for 5 h at 65 °C and treated with 20 µg proteinase K for 30 min at 45 °C. DNA was recovered with the QIAGEN PCR Purification Kit and amplified by quantitative real-time PCR on the LightCycler 480 System (Roche Applied Science). Data sets were normalized to ChIP input values.
Western blotting
Western blotting was performed as previously described.68 Proteins were extracted with TRIzol, after RNA isolation, according to manufacturer's protocol. Primers
Primers for RT-PCR/qPCR
FN1 ex24/ex26 5′- TGGAGTACAA TGTCAGTGTT-3′
5′- CTGGACCAAT GTTGGTGAAT C-3′
EDB includ. 5′- TGGACCAATG TTGGTGAATC-3′
5′- AGCCGGGCAT TGACTATGAT-3′
EDB exclude. 5′- GAGGAACAGC TGGGATGATG-3′
5′- TCACTATAGG GAGACCCAAG C -3′
FN1 ex24 5′- GGAAGAAGTG GTCCATGCTG-3′
5′- GGGACACTTT CCTTGTCATC C-3′
FN1 EDB 5′- AGGTGCCCCA ACTCACTGAC C-3′
5′- TGCCGCAACT ACTGTGATGC GGTA-3′
Amp 5′- TTGCCGGGAA GCTAGAGTAA-3′
5′- GCGGCCAACT TACTTCTGAC-3′
reporterRT 5′- ATTTAGGTGA CACTATAG-3′
FN1 ex26 5′- AGTTGGTTAA ATCAATGGAT G-3′
OCEL1 5′- CCGAGGCCTC AAAACCAG -3′
5′- TCATCTCAAA CTCCCTCCAA A -3′
UBE2H 5′- TGAAGGCGGA GTATGGAAAG -3′
5′- GCCAATAACT GAGGCAGGAA -3′
EXO1 5′- TGGGGATACA GGGATTGCTA -3′
5′- TTCCCCTCAC GAAGAAGTTG -3′
Primers for qPCR (nChIP,ChIP, Pol II elongation rate)
Reporter promoter 5′- CACCAAAATC AACGGGACTT-3′
5′- AGCCAGTAAG CAGTGGGTTC-3′
Reporter down 5′- TAGTTGCCAG CCATCTGTTG-3′
5′- GCGATGCAAT TTCCTCATTT-3′
FN1 promoter 5′- TTGATGACCG CAAAGGAAAC -3′
5′- TCGCAGCGAA CAAAAGAGAT -3′
FN1 exon 1 5′- CCGTCTCAAC ATGCTTAGGG -3′
5′- ATTTGCTGAG CCTGCCTCTT -3′
FN1 exon 7 5′- ATTAGGATCT GGCCCCTTCA -3′
5′- TGTGACACAG TGGCCATAGG -3′
FN1 intron 14 - exon 15 5′- AAAATGATGT TGGCGACGAG -3′
5′- CGTCTCTCCT GTCACGGTGT -3′
FN1 exon 24 5′- GGAAGAAGTG GTCCATGCTG -3′
5′- GGGACACTTT CCTTGTCATC C -3′
FN1 EDB 5′- AGGTGCCCCA ACTCACTGAC C -3′
5′- TGCCGCAACT ACTGTGATGC GGTA -3′
FN1 intron 25 5′- GGGTAGAGTG GATGGGCATT -3′
5′- CATGCTTGTC CCCAGACTGT -3′
FN1 exon 38 5′- CACCCAATTC CTTGCTGGTA -3′
5′- GGACCACTTC TCTGGGAGGA -3′
FN1 exon 42 5′- ACCAGTGCCA CTCTGACAGG -3′
5′- TTCCCGAACC TTATGCCTCT -3′
intergenic region 5′- GGCTAATCCT CTATGGGAGT CTGTC -3′
Chromosome 10 5′- CCAGGTGCTC AAGGTCAACA TC -3′
contig AL392045
FN1 exon 24 – intron 24, A 5′- CACTGTCAAG GATGACAAGG AA -3′
5′- CCCCACTCTT ATTGGAAGTG TC -3′
FN1 intron 24 – EDB, B 5′- TTTTTCCCTC TATTTTCCTT TTG -3′
5′- GTTATATCAA CAAAGCTTAG GTCAGTG -3′
FN1 intron 25, A 5′- GGGTAGAGTG GATGGGCATT -3′
5′- CATGCTTGTC CCCAGACTGT -3′
FN1 intron 25 – exon 26, B 5′- CCTTTCCAGC TACTTCGTTA GC -3′
5′- AATGTTGGTG AATCGCAGGT -3′
CRE 5′- GCCAGATATA CGCGTTGACA -3′
5′- GGAAAGTCCC TATTGGCGTT A -3′
Reporters:
Δ1–4 5′- Pho- ATGGGAGTTT GTTTTGGC -3′
5′- Pho- GAACTCCATA TATGGGCT -3′
Δ 1–3 5′- Pho- ATGACGGTAA ATGGCCCG -3′
5′- Pho- GAACTCCATA TATGGGCT -3′
Δ 1 5′- Pho- ATAATGACGT ATGTTCCC -3′
5′- Pho- GAACTCCATA TATGGGCT -3′
Δ 3–4 5′- Pho- ATAGGGGGCG TACTTGGC -3′
5′- Pho- ATGGGAGTTT GTTTTGGC -3′
Δ 2–4 5′- Pho-ATGGAAAGTC CCTATTGG -3′
5′- Pho- ATGGGAGTTT GTTTTGGC -3′
Mut1 5′- CCAACGACCC CCGCCCATTG GCTTCAATAA TGACGTATGT TCC -3′
5′- GGAACATACG TCATTATTGA AGCCAATGGG CGGGGGTCGT TGG -3′
FN1p 5′- GGAAGATCTC TCAAACACTA CCACCACCC -3′
5′- CCCAAGCTTG AGCCGGGGCT TATATG -3′
Supplementary Material
Acknowledgments
We thank Kim Kotovic for English proofreading and Karla Neugebauer for providing us with the SRSF1-GFP BAC cell line and the m104 antibody. This work was supported by the Academy of Sciences of the Czech Republic (RVO68378050) and the Czech Science Foundation (P305/12/G034). E.D. was supported by the grant 274111 from the Charles University Grant Agency.
References
- 1.Croft L, Schandorff S, Clark F, Burrage K, Arctander P, Mattick JS. ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nat Genet. 2000;24:340–1. doi: 10.1038/74153. [DOI] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–5. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 5.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 6.Brody Y, Neufeld N, Bieberstein N, Causse SZ, Böhnlein EM, Neugebauer KM, Darzacq X, Shav-Tal Y. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 2011;9:e1000573. doi: 10.1371/journal.pbio.1000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh J, Padgett RA. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol. 2009;16:1128–33. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huranová M, Ivani I, Benda A, Poser I, Brody Y, Hof M, Shav-Tal Y, Neugebauer KM, Stanek D. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J Cell Biol. 2010;191:75–86. doi: 10.1083/jcb.201004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt U, Basyuk E, Robert MC, Yoshida M, Villemin JP, Auboeuf D, Aitken S, Bertrand E. Real-time imaging of cotranscriptional splicing reveals a kinetic model that reduces noise: implications for alternative splicing regulation. J Cell Biol. 2011;193:819–29. doi: 10.1083/jcb.201009012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin RM, Rino J, Carvalho C, Kirchhausen T, Carmo-Fonseca M. Live-cell visualization of pre-mRNA splicing with single-molecule sensitivity. Cell Rep. 2013;4:1144–55. doi: 10.1016/j.celrep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacadie SA, Rosbash M. Cotranscriptional spliceosome assembly dynamics and the role of U1 snRNA:5’ss base pairing in yeast. Mol Cell. 2005;19:65–75. doi: 10.1016/j.molcel.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol. 2006;13:815–22. doi: 10.1038/nsmb1135. [DOI] [PubMed] [Google Scholar]
- 14.Sapra AK, Ankö ML, Grishina I, Lorenz M, Pabis M, Poser I, Rollins J, Weiland EM, Neugebauer KM. SR protein family members display diverse activities in the formation of nascent and mature mRNPs in vivo. Mol Cell. 2009;34:179–90. doi: 10.1016/j.molcel.2009.02.031. [DOI] [PubMed] [Google Scholar]
- 15.Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Lührmann R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun. 2012;3:994. doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- 16.Das R, Dufu K, Romney B, Feldt M, Elenko M, Reed R. Functional coupling of RNAP II transcription to spliceosome assembly. Genes Dev. 2006;20:1100–9. doi: 10.1101/gad.1397406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gromak N, Talotti G, Proudfoot NJ, Pagani F. Modulating alternative splicing by cotranscriptional cleavage of nascent intronic RNA. RNA. 2008;14:359–66. doi: 10.1261/rna.615508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kornblihtt AR. Coupling transcription and alternative splicing. Adv Exp Med Biol. 2007;623:175–89. doi: 10.1007/978-0-387-77374-2_11. [DOI] [PubMed] [Google Scholar]
- 19.Perales R, Bentley D. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell. 2009;36:178–91. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neugebauer KM. On the importance of being co-transcriptional. J Cell Sci. 2002;115:3865–71. doi: 10.1242/jcs.00073. [DOI] [PubMed] [Google Scholar]
- 21.Bird G, Zorio DA, Bentley DL. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotranscriptional pre-mRNA splicing and 3′-end formation. Mol Cell Biol. 2004;24:8963–9. doi: 10.1128/MCB.24.20.8963-8969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dower K, Rosbash M. T7 RNA polymerase-directed transcripts are processed in yeast and link 3′ end formation to mRNA nuclear export. RNA. 2002;8:686–97. doi: 10.1017/S1355838202024068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCracken S, Rosonina E, Fong N, Sikes M, Beyer A, O’Hare K, Shuman S, Bentley D. Role of RNA polymerase II carboxy-terminal domain in coordinating transcription with RNA processing. Cold Spring Harb Symp Quant Biol. 1998;63:301–9. doi: 10.1101/sqb.1998.63.301. [DOI] [PubMed] [Google Scholar]
- 24.Sisodia SS, Sollner-Webb B, Cleveland DW. Specificity of RNA maturation pathways: RNAs transcribed by RNA polymerase III are not substrates for splicing or polyadenylation. Mol Cell Biol. 1987;7:3602–12. doi: 10.1128/mcb.7.10.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–32. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Eperon LP, Graham IR, Griffiths AD, Eperon IC. Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase? Cell. 1988;54:393–401. doi: 10.1016/0092-8674(88)90202-4. [DOI] [PubMed] [Google Scholar]
- 27.Kadener S, Fededa JP, Rosbash M, Kornblihtt AR. Regulation of alternative splicing by a transcriptional enhancer through RNA pol II elongation. Proc Natl Acad Sci U S A. 2002;99:8185–90. doi: 10.1073/pnas.122246099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nogués G, Muñoz MJ, Kornblihtt AR. Influence of polymerase II processivity on alternative splicing depends on splice site strength. J Biol Chem. 2003;278:52166–71. doi: 10.1074/jbc.M309156200. [DOI] [PubMed] [Google Scholar]
- 29.Roberts GC, Gooding C, Mak HY, Proudfoot NJ, Smith CW. Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res. 1998;26:5568–72. doi: 10.1093/nar/26.24.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–69. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carrillo Oesterreich F, Bieberstein N, Neugebauer KM. Pause locally, splice globally. Trends Cell Biol. 2011;21:328–35. doi: 10.1016/j.tcb.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Hnilicová J, Staněk D. Where splicing joins chromatin. Nucleus. 2011;2:182–8. doi: 10.4161/nucl.2.3.15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornblihtt AR, Schor IE, Allo M, Blencowe BJ. When chromatin meets splicing. Nat Struct Mol Biol. 2009;16:902–3. doi: 10.1038/nsmb0909-902. [DOI] [PubMed] [Google Scholar]
- 34.Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14:153–65. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 35.Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saint-André V, Batsché E, Rachez C, Muchardt C. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol. 2011;18:337–44. doi: 10.1038/nsmb.1995. [DOI] [PubMed] [Google Scholar]
- 38.Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–76. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011;18:977–83. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 40.Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A. 2011;108:13564–9. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bieberstein NI, Carrillo Oesterreich F, Straube K, Neugebauer KM. First exon length controls active chromatin signatures and transcription. Cell Rep. 2012;2:62–8. doi: 10.1016/j.celrep.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 42.Hnilicová J, Hozeifi S, Dušková E, Icha J, Tománková T, Staněk D. Histone deacetylase activity modulates alternative splicing. PLoS One. 2011;6:e16727. doi: 10.1371/journal.pone.0016727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou HL, Hinman MN, Barron VA, Geng C, Zhou G, Luo G, Siegel RE, Lou H. Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc Natl Acad Sci U S A. 2011;108:E627–35. doi: 10.1073/pnas.1103344108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schor IE, Rascovan N, Pelisch F, Alló M, Kornblihtt AR. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci U S A. 2009;106:4325–30. doi: 10.1073/pnas.0810666106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cramer P, Cáceres JF, Cazalla D, Kadener S, Muro AF, Baralle FE, Kornblihtt AR. Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell. 1999;4:251–8. doi: 10.1016/S1097-2765(00)80372-X. [DOI] [PubMed] [Google Scholar]
- 46.Cramer P, Pesce CG, Baralle FE, Kornblihtt AR. Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci U S A. 1997;94:11456–60. doi: 10.1073/pnas.94.21.11456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pagani F, Stuani C, Zuccato E, Kornblihtt AR, Baralle FE. Promoter architecture modulates CFTR exon 9 skipping. J Biol Chem. 2003;278:1511–7. doi: 10.1074/jbc.M209676200. [DOI] [PubMed] [Google Scholar]
- 48.Auboeuf D, Hönig A, Berget SM, O’Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002;298:416–9. doi: 10.1126/science.1073734. [DOI] [PubMed] [Google Scholar]
- 49.Kadener S, Cramer P, Nogués G, Cazalla D, de la Mata M, Fededa JP, Werbajh SE, Srebrow A, Kornblihtt AR. Antagonistic effects of T-Ag and VP16 reveal a role for RNA pol II elongation on alternative splicing. EMBO J. 2001;20:5759–68. doi: 10.1093/emboj/20.20.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Mol Cell. 2000;6:307–16. doi: 10.1016/S1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 51.Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. J Biol Chem. 2002;277:43110–4. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]
- 52.Hnilicová J, Hozeifi S, Stejskalová E, Dušková E, Poser I, Humpolíčková J, Hof M, Staněk D. The C-terminal domain of Brd2 is important for chromatin interaction and regulation of transcription and alternative splicing. Mol Biol Cell. 2013;24:3557–68. doi: 10.1091/mbc.E13-06-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 54.Du K, Peng Y, Greenbaum LE, Haber BA, Taub R. HRS/SRp40-mediated inclusion of the fibronectin EIIIB exon, a possible cause of increased EIIIB expression in proliferating liver. Mol Cell Biol. 1997;17:4096–104. doi: 10.1128/mcb.17.7.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muro AF, Bernath VA, Kornblihtt AR. Interaction of the -170 cyclic AMP response element with the adjacent CCAAT box in the human fibronectin gene promoter. J Biol Chem. 1992;267:12767–74. [PubMed] [Google Scholar]
- 56.Alonso CR, Pesce CG, Kornblihtt AR. The CCAAT-binding proteins CP1 and NF-I cooperate with ATF-2 in the transcription of the fibronectin gene. J Biol Chem. 1996;271:22271–9. doi: 10.1074/jbc.271.36.22271. [DOI] [PubMed] [Google Scholar]
- 57.Nejepinska J, Malik R, Moravec M, Svoboda P. Deep sequencing reveals complex spurious transcription from transiently transfected plasmids. PLoS One. 2012;7:e43283. doi: 10.1371/journal.pone.0043283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kornblihtt AR. Promoter usage and alternative splicing. Curr Opin Cell Biol. 2005;17:262–8. doi: 10.1016/j.ceb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Perales R, Erickson B, Zhang L, Kim H, Valiquett E, Bentley D. Gene promoters dictate histone occupancy within genes. EMBO J. 2013;32:2645–56. doi: 10.1038/emboj.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 62.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 63.Mercer TR, Edwards SL, Clark MB, Neph SJ, Wang H, Stergachis AB, John S, Sandstrom R, Li G, Sandhu KS, et al. DNase I-hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet. 2013;45:852–9. doi: 10.1038/ng.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xin D, Hu L, Kong X. Alternative promoters influence alternative splicing at the genomic level. PLoS One. 2008;3:e2377. doi: 10.1371/journal.pone.0002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shav-Tal Y, Darzacq X, Shenoy SM, Fusco D, Janicki SM, Spector DL, Singer RH. Dynamics of single mRNPs in nuclei of living cells. Science. 2004;304:1797–800. doi: 10.1126/science.1099754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31:76–82. doi: 10.1016/S1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- 68.Huranová M, Hnilicová J, Fleischer B, Cvacková Z, Stanek D. A mutation linked to retinitis pigmentosa in HPRP31 causes protein instability and impairs its interactions with spliceosomal snRNPs. Hum Mol Genet. 2009;18:2014–23. doi: 10.1093/hmg/ddp125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.