

Background: We previously showed that plasmin binding to streptokinase is a three-step mechanism with a slow off-rate.
Results: Using rapid kinetics and equilibrium binding, we defined the unknown mechanism of plasminogen binding to streptokinase.
Conclusion: Encounter complex formation and conformational tightening are weakened in the three-step binding mechanism.
Significance: The results define the molecular basis for plasminogen displacement by plasmin in complexes with streptokinase.
Keywords: Bacterial Pathogenesis, Fibrinolysis, Fluorescence, Kinetics, Plasmin, Streptokinase
Abstract
Rapid kinetics demonstrate a three-step pathway of streptokinase (SK) binding to plasminogen (Pg), the zymogen of plasmin (Pm). Formation of a fluorescently silent encounter complex is followed by two conformational tightening steps reported by fluorescence quenches. Forward reactions were defined by time courses of biphasic quenching during complex formation between SK or its COOH-terminal Lys414 deletion mutant (SKΔK414) and active site-labeled [Lys]Pg ([5-(acetamido)fluorescein]-d-Phe-Phe-Arg-[Lys]Pg ([5F]FFR-[Lys]Pg)) and by the SK dependences of the quench rates. Active site-blocked Pm rapidly displaced [5F]FFR-[Lys]Pg from the complex. The encounter and final SK·[5F]FFR-[Lys]Pg complexes were weakened similarly by SK Lys414 deletion and blocking of lysine-binding sites (LBSs) on Pg kringles with 6-aminohexanoic acid or benzamidine. Forward and reverse rates for both tightening steps were unaffected by 6-aminohexanoic acid, whereas benzamidine released constraints on the first conformational tightening. This indicated that binding of SK Lys414 to Pg kringle 4 plays a role in recognition of Pg by SK. The substantially lower affinity of the final SK·Pg complex compared with SK·Pm is characterized by a ∼25-fold weaker encounter complex and ∼40-fold faster off-rates for the second conformational step. The results suggest that effective Pg encounter requires SK Lys414 engagement and significant non-LBS interactions with the protease domain, whereas Pm binding additionally requires contributions of other lysines. This difference may be responsible for the lower affinity of the SK·Pg complex and the expression of a weaker “pro”-exosite for binding of a second Pg in the substrate mode compared with SK·Pm.
Introduction
The serine proteinase plasmin (Pm)2 is primarily known for its role in dissolving fibrin thrombi (1). It also causes cell surface remodeling, signaling, and cancer progression (2). Proteolytic activation of the zymogen plasminogen (Pg) by tissue plasminogen activator and urokinase-type plasminogen activator differs from conformational activation by the non-enzymatic streptococcal pathogenicity factor streptokinase (SK). We studied SK from Streptococcus dysgalactiae subsp. equisimilis because of its 90% homology with phylogenetic cluster 1SKs from the human host-specific, virulent Streptococcus pyogenes (3). S. dysgalactiae subsp. equisimilis, which is generally opportunistic in horses, also causes severe human infections such as bacteremia, pneumonia, endocarditis, arthritis, and streptococcal toxic shock syndrome (4, 5).
The Pg activation mechanism by SK is unique (6–8). Stoichiometric binding of SK to Pg and Pm forms catalytically active SK·Pg* and SK·Pm complexes that bind Pg as a substrate in SK·Pg*·Pg and SK·Pm·Pg assemblies and cleave Arg561-Val562 in the Pg protease domain to form Pm (6, 8–14). Conformational activation of Pg in the catalytic SK·Pg* complex by the molecular sexuality mechanism involves insertion of the NH2-terminal Ile1-Ala2 residues of SK into the binding cleft of the Pg protease domain (9, 11, 12, 15–17). Ile1 binds Pg Asp194 (chymotrypsinogen numbering), causing expression of the substrate-binding site and formation of the oxyanion hole (15, 16, 18, 19). The mechanism is also valid for conformational prothrombin activation by staphylocoagulase and von Willebrand factor-binding protein from Staphylococcus aureus (20, 21). This mechanism allows group A and C streptococci to hijack Pg in the human fibrinolytic system by quorum sensing-induced secretion of SK. This results in localized plasmin generation for dissolution of host fibrin barriers and facilitated bacterial spreading (22–25).
In our unified model, the conformationally activated SK·Pg* complex binds Pg as a substrate and cleaves it to Pm. This is the trigger step in a self-limiting mechanism (6–8). After 1 SK eq of Pm is formed, it displaces Pg from the SK·Pg* complex to form the tight SK·Pm catalytic complex (with dissociation constant (KD) of 12 pm (26, 27)) that cleaves the remaining free Pg to Pm in a second catalytic cycle, the bullet cycle (6). In the SK·Pm complex, the three SK β-grasp domains rearrange from a beads-on-a-string conformation (28) to a crater surrounding the Pm active site (19). This forms a novel exosite for substrate Pg binding (19, 26).
[Glu]Pg, the circulating form of Pg, has an NH2-terminal plasminogen, apple, nematode (PAN) module, five kringles (K1–K5) with lysine-binding sites (LBSs), and a COOH-terminal serine protease domain (29–31). K1, K2, K4, and K5 bind lysine analogs and small aromatic anionic and cationic ligands (32). K1, K4, and K5 also bind COOH-terminal lysines on fibrin and other proteins (33–38). In the compact [Glu]Pg, the NH2-terminal PAN module occupies the LBS on K5, and in this spiral α-form, [Glu]Pg activation is inefficient (27, 39–43). Cleavage of the 77-residue PAN module by Pm converts [Glu]Pg to [Lys]Pg with a partially extended β-conformation that exposes kringle LBSs and is readily activated (6, 44–47). Occupying these LBSs with the lysine analog 6-aminohexanoic acid (6-AHA) fully extends [Lys]Pg to the γ-form (39, 43). SK binds weakly to [Glu]Pg in the absence and presence of 6-AHA, whereas SK binding to [Lys]Pg is tighter due in part to the interaction of the COOH-terminal Lys414 residue of SK with exposed LBSs on kringle domains in [Lys]Pg (48). This interaction is weakened ∼13–20-fold by blocking the LBSs with 6-AHA (27, 48). 6-AHA binds isolated kringles K1, K4, and K5 (30, 32, 49, 50), whereas benzamidine binds K1, K2, and K5 (32, 51, 52) and only partially extends [Glu]Pg and [Lys]Pg to the β-conformation (39, 43). K4 and K5 of [Glu]Pg were shown to bind 6-AHA cooperatively (53), which may be of importance in interpreting differences in binding of SK lysines to Pg and Pm. In this study, we used 6-AHA and benzamidine to study differential effects of LBS occupation on the formation of a stabilized SK·Pg complex.
Allosteric linkage between the protease active site and its exosite(s) allows investigating equilibrium binding of ligands to serine proteases labeled at their active sites with fluorescent probes (54–56). Active site labeling of the conformationally activated zymogens plasminogen and prothrombin (27, 57) provides the advantage of studying ligand binding uncoupled from catalytic activity. Introducing the fluorescent label 5-fluorescein ([5F]) and a tripeptide chloromethyl ketone in the Pm active site (FFR-Pm) does not affect the affinity for SK, whereas labeled [Glu]Pg and [Lys]Pg analogs bind SK with ∼5-fold lower affinity than the native proteins (26, 48, 58). We compared the binding kinetics of labeled Pg and Pm that have their active sites similarly locked in a substrate-binding conformation by the tripeptide chloromethyl ketone.
Here we explore for the first time the steps on the pathway of SK binding to Pg and identify critical differences with Pm binding (59) that are the basis for the ∼4,000-fold lower affinity of [5F]FFR-[Lys]Pg for SK (27). Stopped-flow kinetics of SK binding to labeled [Lys]Pg and [Glu]Pg defined the forward reactions of complex stabilization. Reverse reactions were studied by competitive displacement of labeled [Lys]Pg by active site-blocked FFR-Pm in the complex with SK. Forward and reverse reactions were biexponential, and overall off-rates were fast, requiring stopped-flow monitoring. Parameters from numerical integration of full forward and reverse time traces were consistent with those from the SK dependences of the forward rates of these conformational changes. This approach allowed a comparison of the elementary reaction steps in the sequence of SK·Pg* and SK·Pm formation. The data support a three-step mechanism of encounter complex formation followed by two tightening conformational steps as shown previously for SK·Pm (59) but with dramatic decreases in affinity of the encounter complex and ∼10–40-fold increases in off-rates for both conformational steps. Based on selective blocking of LBSs on Pg and Pm and binding experiments with SK lacking Lys414, we propose that the SK·Pg* complex is stabilized by SK Lys414 binding to LBSs on Pg and non-LBS interactions of SK within the Pg catalytic domain. These interactions are also present in the SK·Pm complex; however, additional contributions of SK lysines other than Lys414 may be partially responsible for the substantially tighter SK-Pm interaction.
EXPERIMENTAL PROCEDURES
Protein Purification and Characterization
[Glu]Pg carbohydrate form 2 was purified from human plasma and activated to [Lys]Pg and [Lys]Pm (Pm) as described (8, 26, 27, 60, 61). Pm was purified by affinity chromatography on soybean trypsin inhibitor-agarose and dialyzed against 5 mm HEPES, 0.3 m NaCl, 10 mm 6-AHA, 1 mg/ml PEG 8000 at pH 7.0 and 4 °C. The active Pm concentration (∼90%) was determined by active site titration with fluorescein mono-p-guanidinobenzoate (62). Pm (10–15 μm) was covalently inactivated with a 5-fold molar excess of d-Phe-Phe-Arg-CH2Cl (FFR-CH2Cl) in 0.1 m HEPES, 0.3 m NaCl, 1 mm EDTA, 10 mm 6-AHA, 1 mg/ml PEG 8000, pH 7.0 buffer at 25 °C for 30–60 min until hydrolysis of d-Val-Leu-Lys-p-nitroanilide was undetectable. Excess inhibitor was removed by dialysis against >250 volumes of 50 mm HEPES, 0.3 m NaCl, 1 mm EDTA, pH 7.0 at 4 °C. Native SK (nSK; Diapharma) was purified from outdated therapeutic SK from the S. dysgalactiae subsp. equisimilis strain H46A (26, 27). Recombinant wild-type SK (WT-SK) and the SKΔK414 and SKΔ(R253–L260)ΔK414-His6 mutants were prepared as published (48, 63). Proteins were quick frozen in 2-propanol/dry ice and stored at −80 °C. Protein concentrations were determined by absorbance at 280 nm using the following absorption coefficients ((mg/ml)−1 cm−1) and molecular weights: [Glu]Pg, 1.69 and 92,000; [Lys]Pg, 1.69 and 84,000; Pm, 1.9 and 84,000 (47, 61, 64); SK and SKΔK414, 0.81 and 47,000 (65, 66); SKΔ(R253–L260)ΔK414-His6, 0.78 and 49,213 (63).
Active Site Labeling of Pg
[Glu]Pg and [Lys]Pg were labeled at the active site as described previously (27, 58, 63). The SKΔ(R253–L260)ΔK414-His6 mutant activates [Lys]Pg conformationally, but the complex does not readily cleave Pg to Pm, and the use of this SK construct for Pg labeling significantly increased the yield of labeled Pg and reduced the preparation time (63). Labeled Pg concentration and probe incorporation (∼90%) were determined from the probe and protein absorbances in 6 m guanidine as described (48, 54, 55). Proteins were homogeneous by SDS gel electrophoresis.
Stopped-flow Kinetics of nSK, WT-SK, and SKΔK414 Binding to [5F]FFR-Pg
Complete progress curves of SK binding to labeled Pg were captured with an Applied Photophysics SX-18MV stopped-flow spectrofluorometer in single mixing mode with excitation at 500 nm and an emission cut-on filter (Melles-Griot) with 50% transmission at 515 nm. Changes in fluorescence intensity were measured for all the reactions, and for the interaction of native SK with [5F]FFR-[Lys]Pg in the absence of lysine analogs, changes in fluorescence anisotropy were also monitored. The reaction volume was 200 μl, the path length was 2 mm, and experiments were performed at 25 °C. Binding was studied under pseudo-first-order conditions (SK ≥5-fold over labeled Pg) in 50 mm HEPES, 0.125 m NaCl, 1 mm EDTA, 1 mg/ml PEG 8000, 1 mg/ml bovine serum albumin, 1 μm FFR-CH2Cl, pH 7.4 in the absence and presence of 50 mm 6-AHA or in 50 mm HEPES, 0.075 m NaCl, 1 mm EDTA, 1 mg/ml PEG 8000, 1 mg/ml bovine serum albumin, 1 μm FFR-CH2Cl, pH 7.4 containing 50 mm benzamidine to maintain constant ionic strength. Binding of nSK and SKΔK414 (0.050–18 μm) to [5F]FFR-[Lys]Pg (10–20 nm) was studied in all three buffer systems. Binding of WT-SK (0.050–10 μm) to [5F]FFR-[Lys]Pg (20 nm) in the absence of effectors was included as a control. Binding of native SK (0.1–6 μm) to [5F]FFR-[Glu]Pg (13 and 20 nm) was studied in the absence and presence of 50 mm 6-AHA. Averaged time traces (1,000 data points/trace and 10 traces for each SK concentration) of the decrease in fluorescence intensity or increase in anisotropy ranged from 0.4 to 50 s, depending on the SK concentration and the presence of effector. Averaged time traces of blank titrations containing buffer and SK only and buffer and [5F]FFR-Pg only were obtained to measure background light scattering and initial probe fluorescence, respectively, and to permit transformation of raw data into the fractional change in initial fluorescence ((Fobs − Fo)/Fo = ΔF/Fo) or anisotropy ((robs − ro)/ro = Δr/ro). Subtracting background scattering was critical as the signal-to-noise ratio for reactions with [5F]FFR-Pg (∼25% quench and ∼7.5% maximal scattering) was up to 5-fold smaller than that for reactions with [5F]FFR-Pm (∼50% quench and ∼6% maximal scattering) (59). Experiments were limited to labeled Pg concentrations up to 20 nm due to the lower solubility of the SK·Pg complex compared with that of SK·Pm, which resulted in a substantial increase in background scattering at Pg concentrations above ∼40 nm. None of our previously published studies of SK binding and kinetics have used [Lys]Pg and [Glu]Pg concentrations exceeding 20 and 30 nm, respectively. Averaged time traces were analyzed using Equation 1.
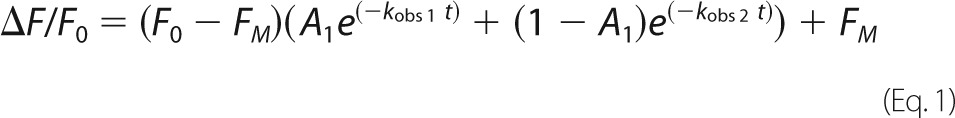 |
where Fo is the starting fluorescence, FM is the final fluorescence, A1 is the fractional amplitude of the fast exponential component, (1 − A1) is the fractional amplitude of the slow exponential component, and kobs 1 and kobs 2 are the observed first-order rate constants for the fast and the slow conformational changes. The rate constants were analyzed as a function of the total SK concentration ([SK]o) using Equation 2.
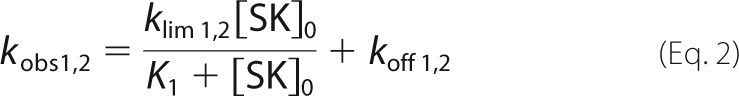 |
where K1 is the dissociation constant for the SK·Pg encounter complex and klim 1,2 and koff 1,2 are the limiting rates and the reverse rate constants for each conformational step, respectively.
Competitive Dissociation of [5F]FFR-[Lys]Pg from its Complex with nSK, WT-SK, and SKΔK414 by FFR-Pm
In stopped-flow experiments, [5F]FFR-[Lys]Pg and SK or SKΔK414 were preincubated in the dark for 5 min at 25 °C and loaded in one syringe. FFR-Pm was loaded in the second syringe, and time traces of fluorescence increase for the reverse reactions were recorded until displacement was >90% complete, ranging from 15 to 200 s. Final concentrations in the cell at the dead time of mixing were: [5F]FFR-[Lys]Pg, 10–20 nm; SK or SKΔK414, 59–2,000 nm; and FFR-Pm, 100–2,000 nm. Background scattering was subtracted, and Fo values of free [5F]FFR-[Lys]Pg were established, i.e. the signal at 100% displacement. Fluorescence quenches of the preformed SK·[5F]FFR-[Lys]Pg complexes in the dead time (4 ms) of the mixing step with FFR-Pm were compared for consistency with the values for forward reactions of SK and [5F]FFR-[Lys]Pg under identical conditions. Unlike the SK reactions with labeled Pm, the reactions with Pg were not stoichiometric due to weaker SK binding, and a range of SK and FFR-Pm concentrations was used to obtain time traces at various degrees of SK saturation with labeled [Lys]Pg and FFR-Pm. The faster and much larger displacement signal for FFR-Pm compared with FFR-Pg and the vastly lower scattering background of the SK·FFR-Pm complex were major reasons for performing displacement experiments with FFR-Pm rather than with FFR-Pg. The time traces were fit by a double exponential function (analogous to Equation 1) to obtain the observed first-order rate constants kdisp 1 and kdisp 2 for the fast and slow displacement processes.
Equilibrium Binding of [5F]FFR-Pg to SK and SKΔK414 in the Presence of Benzamidine
[5F]FFR-Pg (10 nm) was titrated with SK or SKΔK414 at 25 °C in 50 mm HEPES, 0.075 m NaCl, 1 mm EDTA, 1 mg/ml PEG 8000 buffer, pH 7.4 containing 50 mm benzamidine, 1 mg/ml BSA, and 1 μm FFR-CH2Cl. Fluorescence titrations were performed with a Photon Technology International, Inc. fluorometer at excitation and emission wavelengths of 500 and 516 nm, respectively, with 2/8-nm excitation/emission band passes. Fluorescence changes were measured after equilibration for 5–10 min. Measurements were corrected for background (≤10%) by subtraction of blanks lacking [5F]FFR-Pg. Data were analyzed by the quadratic equation for binding of a single ligand (55). This analysis gave the dissociation constant (KD) for binding of SK or SKΔK414 to [5F]FFR-Pg and the maximum fluorescence intensity change (ΔFmax/Fo) with a stoichiometric factor (n) of 1 for binding of SK or SKΔK414 to labeled Pg.
Two-exponential time traces of forward and reverse reactions, SK dependences of kobs 1,2, and equilibrium binding of SK and SKΔK414 to [5F]FFR-Pg in benzamidine buffer were analyzed by nonlinear least square fitting with SCIENTIST Software (MicroMath). All reported estimates of error represent ±2 S.D.
Numerical Integration Analysis of the Forward and Reverse Reactions
Arrays of progress curves for SK·[5F]FFR-Pg formation and displacement of labeled Pg from the complex were analyzed globally with the numerical integration program KinTek Explorer 3.0 (67–69) for each set of reactants, concentration ranges, and buffer conditions. Five arrays were performed in the absence of lysine analogs: fluorescence amplitude changes of [5F]FFR-[Lys]Pg binding to nSK, WT-SK, and SKΔK414; fluorescence amplitude changes of [5F]FFR-[Glu]Pg binding to nSK; and anisotropy changes of [5F]FFR-[Lys]Pg binding to nSK. Three arrays were performed in 6-AHA: fluorescence amplitude changes of [5F]FFR-[Lys]Pg binding to nSK and SKΔK414 and [5F]FFR-[Glu]Pg binding to nSK. Two arrays were performed in benzamidine: fluorescence amplitude changes of [5F]FFR-[Lys]Pg binding to nSK and SKΔK414.
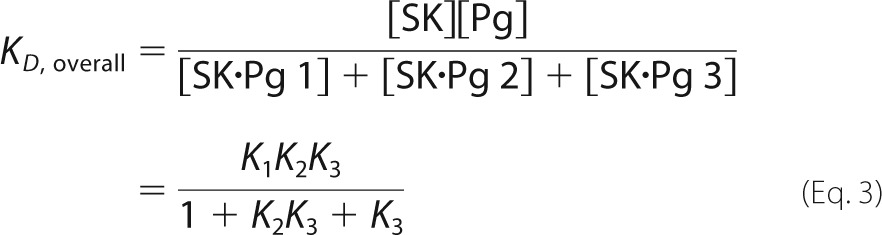 |
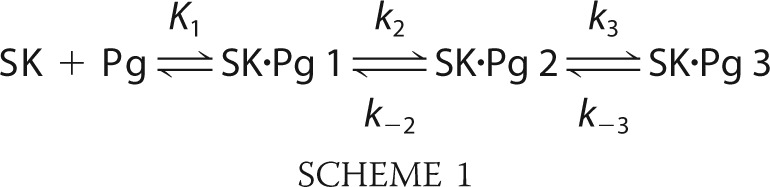 |
The mechanism included Scheme 1 for three-step SK·[5F]FFR-Pg binding and Scheme 2 for competitive three-step SK·FFR-Pm binding. The dissociation constants K1 and K4 for formation of the SK·Pg 1 and SK·Pm 1 encounter complexes represent the ratios k−1/k1 and k−4/k4 where k1 and k4 are the second-order association rate constants and k−1 and k−4 are the first-order rate constants for dissociation of the encounter complex. K1, k2, k−2, k3, and k−3 in this mechanism are equivalent to K1, klim 1, koff 1, klim 2, and koff 2, respectively, in Equation 2. The three-step mechanism for SK·Pm stabilization was validated in a previous study (59).
Time traces of fluorescence quenches were transformed to increases by plotting ΔF/Fo expressed as functions of the formation and stabilization of SK·Pg 1, SK·Pg 2, and SK·Pg 3 complexes using positive amplitude factors as KinTek Explorer does not accept negative parameters. The set of fluorescence anisotropy increases was analyzed without transformation. The fractional change in fluorescence intensity or anisotropy was expressed as ΔF/Fo = f2 × ([SK·Pg 2]/[Pg]o) + f3 × ([SK·Pg 3]/[Pg]o) where [Pg]o is the total [5F]FFR-Pg concentration, which is equal to the sum of Pgfree, SK·Pg 1, SK·Pg 2, and SK·Pg 3; [SK·Pg 2] and [SK·Pg 3] are the concentrations of these complexes at time t; and f2 and f3 the respective fractional amplitude factors for these complexes. The SK·Pg 1 complex does not contribute to fluorescence change. This expression allowed simultaneous analysis of time traces with different [5F]FFR-Pg concentrations.
Fitting Strategy
The on-rate constant k1 for formation of the encounter complex was initially constrained at 1 × 108 m−1 s−1 as determined experimentally for SK binding to unlabeled [Lys]Pg (7), and the assumption was made that similar on-rates would apply for reactions of SK with [5F]FFR-[Lys]Pg and [5F]FFR-[Glu]Pg and of SKΔK414 with [5F]FFR-[Lys]Pg in all of our experimental buffers. Upon refinement of the other parameters, fitting k1 yielded values that were close to 1 × 108 m−1 s−1 under all these conditions, justifying our choice of this value as an initial estimate. The parameters K1, k2, k−2, k3, and k−3 were initially constrained to K1, klim 1, koff 1, klim 2, and koff 2 obtained from the SK dependences (Table 1, superscript b), and refinement of these initial estimates ultimately provided the final fits (superscripts a and aa).
TABLE 1.
Kinetic and equilibrium binding parameters for the formation of SK·Pg and SKΔK414·Pg complexes
Kinetic constants obtained from simultaneous numerical integration of the forward and reverse reactionsa, forward reactions measured as anisotropy changesaa, and SK dependences of the fast and slow phases of the forward reactionsb are listed for reaction Scheme 1 in the absence of kringle ligands (no effector) and in the presence of saturating 6-AHA or benzamidine. KD, overall was calculated from the individual kinetic parametersc and measured by fluorescence titrationd. For analysis of near-linear SK dependences K1 was fixed to the value obtained by numerical analysisfixed. Amplitudes of change in fluorescence intensity (ΔFmax/Fo) were from numerical analysis (f2 and f3 for SK·Pg 2 and SK·Pg 3, respectively) and equilibrium binding (overall value). Reported errors are 2 × S.D. and were calculated by error propagation for compound parameters.
| K1 (encounter) | k2 (≈klim 1) | k−2 (≈koff 1) | k3 (≈klim 2) | k−3 (≈koff 2) | KD, overall | ΔFmax/Fo | |
|---|---|---|---|---|---|---|---|
| μm | s−1 | s−1 | s−1 | s−1 | μm | % | |
| [5F]FFR-[Lys]Pg, no effector | |||||||
| nSK | 2.8 ± 0.3a | 34 ± 2a | 3.5 ± 0.4a | 0.34 ± 0.04a | 0.15 ± 0.01a | 0.086 ± 0.018a,c | −30 ± 9a, −25 ± 6a |
| 1.8 ± 0.2aa | 33 ± 2aa | 1.8 ± 0.1aa | 0.26 ± 0.06aa | 0.12 ± 0.02aa | 0.030 ± 0.018aa,c | ||
| 3.4 ± 1.0b | 33 ± 3b | 4.0 ± 1.0b | 0.90 ± 0.30b | 0.25 ± 0.10b | 0.044 ± 0.009d | −28 ± 1d | |
| WT-SK | 0.94 ± 0.01a | 27 ± 1a | 3.5 ± 0.2a | 0.44 ± 0.03a | 0.22 ± 0.01a | 0.039 ± 0.005a,c | −26 ± 8a, −24 ± 4a |
| 2.1 ± 1.2b | 30 ± 6b | 4.5 ± 2.0b | 3.60 ± 1.60b | 0.24 ± 0.16b | 0.028 ± 0.006d | −23 ± 1d | |
| SKΔK414 | 20.3 ± 0.2a | 31 ± 1a | 2.5 ± 0.1a | 0.17 ± 0.01a | 0.20 ± 0.01a | 0.84 ± 0.09a,c | −24 ± 4a, −30 ± 5a |
| 19.0 ± 4.0b | 25 ± 4b | 2.3 ± 0.6b | 0.10 ± 0.08b | 0.30 ± 0.06b | 0.60 ± 0.20d | −24 ± 2d | |
| 6-AHA | |||||||
| nSK | 10.5 ± 0.7a | 28 ± 3a | 3.7 ± 0.2a | 0.10 ± 0.02a | 0.23 ± 0.01a | 0.89 ± 0.20a,c | −22 ± 8a, −37 ± 8a |
| 7.0 ± 5.0b | 25 ± 8b | 2.0 ± 1.0b | 2.00 ± 2.00b | 0.19 ± 0.09b | 0.56 ± 0.09d | −23 ± 1d | |
| SKΔK414 | 16.3 ± 0.6a | 31 ± 2a | 2.7 ± 0.1a | 0.19 ± 0.02a | 0.16 ± 0.01a | 0.64 ± 0.10a,c | −23 ± 6a, −29 ± 3a |
| 14.0 ± 2.0b | 31 ± 3b | 1.0 ± 0.6b | 0.15 ± 0.18b | 0.23 ± 0.02b | 0.75 ± 0.35d | −24 ± 3d | |
| Benzamidine | |||||||
| nSK | 11.4 ± 0.3a | 156 ± 3a | 3.2 ± 0.1a | 0.07 ± 0.01a | 0.15 ± 0.02a | 0.16 ± 0.04a,c | −18 ± 7a, −17 ± 8a |
| 12.0 ± 2.0b | 150 ± 15b | 2.0 ± 1.0b | 0.20 ± 0.02d | −30 ± 1d | |||
| SKΔK414 | 16.0 ± 0.3a | 69 ± 4a | 4.8 ± 0.2a | 0.26 ± 0.02a | 0.24 ± 0.02a | 0.52 ± 0.06a,c | −27 ± 7a, −23 ± 3a |
| 16.0 ± 10.0b | 70 ± 30b | 4.0 ± 2.0b | 1.30 ± 0.40b | 0.27 ± 0.10b | 0.80 ± 0.10d | −38 ± 1d | |
| [5F]FFR-[Glu]Pg, no effector | |||||||
| nSK | 18.1 ± 0.2a | 42 ± 1a | 2.8 ± 0.1a | 0.10 ± 0.01a | 0.19 ± 0.01a | 0.76 ± 0.20a,c | −31 ± 3a, −25 ± 4a |
| 18.1b,fixed | 46 ± 5b | 2.0 ± 0.5b | 1.00 ± 0.20b | 0.17 ± 0.02b | 0.58 ± 0.08d | −32 ± 1d | |
| 6-AHA | |||||||
| nSK | 15.4 ± 0.2a | 30 ± 1a | 3.2 ± 0.1a | 0.35 ± 0.01a | 0.19 ± 0.01a | 0.56 ± 0.08a,c | −18 ± 8a, −16 ± 7a |
| 15.4b,fixed | 36 ± 5b | 1.8 ± 0.6b | 2.94 ± 0.21b | 0.12 ± 0.03b | 0.60 ± 0.07d | −23 ± 1d | |
Analysis of [5F]FFR-[Lys]Pg displacement required known concentrations of free Pg and the intermediates SK·Pg 1, SK·Pg 2, and SK·Pg 3 present at the start of the reaction with FFR-Pm as there was substantial partitioning among these species at equilibration of the SK·[5F]FFR-[Lys]Pg complex. They were calculated iteratively using the starting concentrations of SK and [5F]FFR-[Lys]Pg used to form the complex, the forward and reverse rate constants, and the known dissociation constant for the competitive, unlabeled SK·FFR-Pm complex. The sum of the calculated free Pg, SK·Pg 1, SK·Pg 2, and SK·Pg 3 concentrations was in agreement with the total Pg concentration, indicating that mass balance was conserved during the fits. Complexes of SK with labeled and unlabeled Pm have indistinguishable affinities in the absence of lysine analogs (26, 48) and in 6-AHA (26, 27), suggesting that the binding parameters for SK are very similar for labeled and unlabeled FFR-Pm. This allowed fixing K4, k5, k−5, k6, and k−6 to our previously determined values for [5F]FFR-Pm binding in each buffer system (59). Displacement of [5F]FFR-[Lys]Pg binding to SK and SKΔK414 in benzamidine was analyzed with fitted KD values of 227 ± 11 and 200 ± 20 pm for FFR-Pm binding, respectively, in agreement with the previously determined 130 and 250 pm (59).
The large scattering background introduced variable uncertainty in the amplitude factors f2 and f3 for [SK·Pg 2] and [SK·Pg 3] at increasing SK concentrations, resulting in non-random residuals when imposing global f2 and f3 fits on the complete data sets. Initial estimates of the rate constants obtained by global fitting of f2 and f3 were fixed, and individual f2 and f3 amplitude factors were assigned as fitted parameters for time traces at each SK concentration. This largely eliminated the non-random deviations. Subsequent fixing of all the individual amplitude parameters provided further refinement of the fitted rate constants with only subtle differences from the original estimates.
The overall KD values for the final, stabilized complexes were calculated from Equation 3 using the rate constants obtained by numerical analysis and compared with KD, overall obtained independently from equilibrium binding.
 |
RESULTS
Stopped-flow Kinetics of SK Binding to [5F]FFR-[Lys]Pg
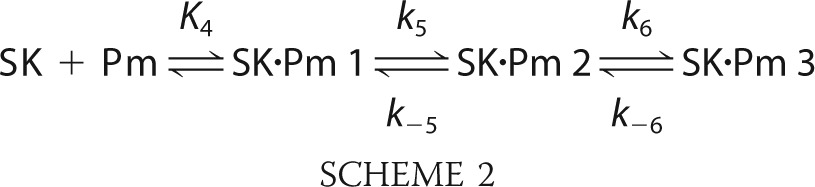
Time traces of fractional quenches of fluorescence intensity (ΔF/Fo) and increases of anisotropy (Δr/ro) following rapid mixing of [5F]FFR-Pg with excess SK in the absence of lysine analogs were distinctly biexponential with first-order rate constants kobs 1 and kobs 2 fitted by Equation 1. Time traces started at zero ΔF/Fo with F at 4 ms ≈ Fo of a control reaction with only [5F]FFR-Pg, indicating no significant fluorescence change associated with encounter complex formation. Representative changes in fluorescence intensity and anisotropy of [5F]FFR-[Lys]Pg binding to nSK are shown in Fig. 1. Colored lines represent global fits of forward and reverse reactions by numerical analysis. The first-order rate constants kobs 1 and kobs 2 for the fast and slow fluorescence changes obtained from the individual biexponential fits increased hyperbolically with increasing SK concentration. Fig. 2 shows the nSK and WT-SK dependences in the absence of effectors. The hyperbolic dependences of kobs 1 and the much smaller kobs 2, respectively, indicated saturation of the encounter complex and the subsequent conformational intermediate (Fig. 2, inset). The parameters K1, klim 1, klim 2, koff 1, and koff 2 obtained by fitting the binding rate constants by Equation 2 are given in Table 1 (superscript b). The reverse rate constants koff 1 and koff 2 for the two conformational steps given by the extrapolated intercepts of kobs 1 and kobs 2 at zero SK were 4.0 ± 1.0 and 0.25 ± 0.10 s−1. The dissociation constants K1 for the encounter complexes of [5F]FFR-[Lys]Pg with nSK and WT-SK were 3.4 ± 1.0 and 2.1 ± 1.2 μm.
FIGURE 1.
Stopped-flow fluorescence changes of SK binding to [5F]FFR-[Lys]Pg. A and B, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and nSK versus time are shown in the absence of lysine analogs at 20 nm [5F]FFR-[Lys]Pg and 0.42, 0.84, 1.58, 3.00, and 5.00 μm nSK (A) and at 10 nm [5F]FFR-[Lys]Pg and 8.2, 11.7, and 17.6 μm nSK (B). C and D, fractional fluorescence anisotropy changes (Δr/ro) are shown for 20 nm [5F]FFR-[Lys]Pg and 0.075, 0.15, 0.20, and 0.52 μm nSK (C) and for 4.7, 7.0, and 14 μm nSK (D). Green and blue solid lines represent the fits from numerical integration with the parameters given in Table 1 as described under “Experimental Procedures.”
FIGURE 2.

SK concentration dependence of the kinetics of [5F]FFR-[Lys]Pg binding in the absence of lysine analogs. Dependences of kobs 1 and kobs 2 (●, nSK, fluorescence intensity; ○, nSK, fluorescence anisotropy; ▴, WT-SK, fluorescence intensity) on [SK]o are shown for binding to 10–20 nm [5F]FFR-[Lys]Pg. The inset shows the kobs 2 dependence on an enlarged scale. Solid and dashed lines represent the least square fits by Equation 2 with the parameters given in Table 1 for the reactions with nSK and WT-SK, respectively.
Stopped-flow Kinetics of SKΔK414 Binding to [5F]FFR-[Lys]Pg
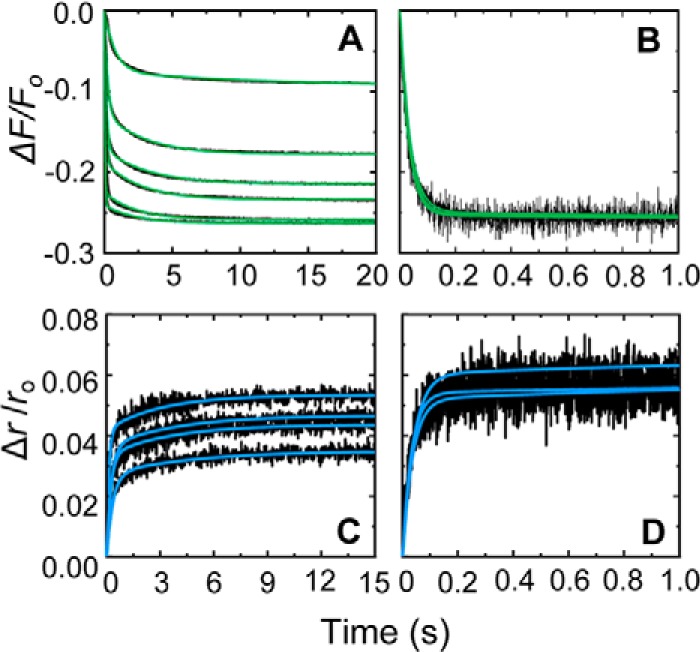
Progress curves of SKΔK414 binding to [5F]FFR-[Lys]Pg and the SKΔK414 dependence of kobs 1 and kobs 2 are shown in Fig. 3 with colored lines representing the global fits of the forward and reverse reactions by numerical analysis. The SKΔK414 mutant bound ∼6-fold more weakly than nSK with a K1 of 19 ± 4 μm (Table 1, superscript b), suggesting that the COOH-terminal SK Lys414 increases the efficiency of initial docking of SK with Pg by interacting with a kringle.
FIGURE 3.
Kinetics of SKΔK414 binding to [5F]FFR-[Lys]Pg in the absence of lysine analogs. A, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and SKΔK414 versus time are shown in the absence of lysine analogs at 20 nm [5F]FFR-[Lys]Pg and 0.1, 0.25, 0.5, 1.0, 2.5, 5.0, and 10 μm SKΔK414. Green solid lines represent the fits from numerical integration as described under “Experimental Procedures.” B, dependences of kobs 1 and kobs 2 (● and ○) on the total SKΔK414 concentration ([SKΔK414]o) are shown for binding to 20 nm [5F]FFR-[Lys]Pg. The inset shows the kobs 2 dependence on an enlarged scale. Solid lines represent the fits by Equation 2 with the parameters given in Table 1. Experiments were performed and analyzed as described under “Experimental Procedures.”
Effects of 6-AHA on the Kinetics of SK and SKΔK414 Binding to [5F]FFR-[Lys]Pg
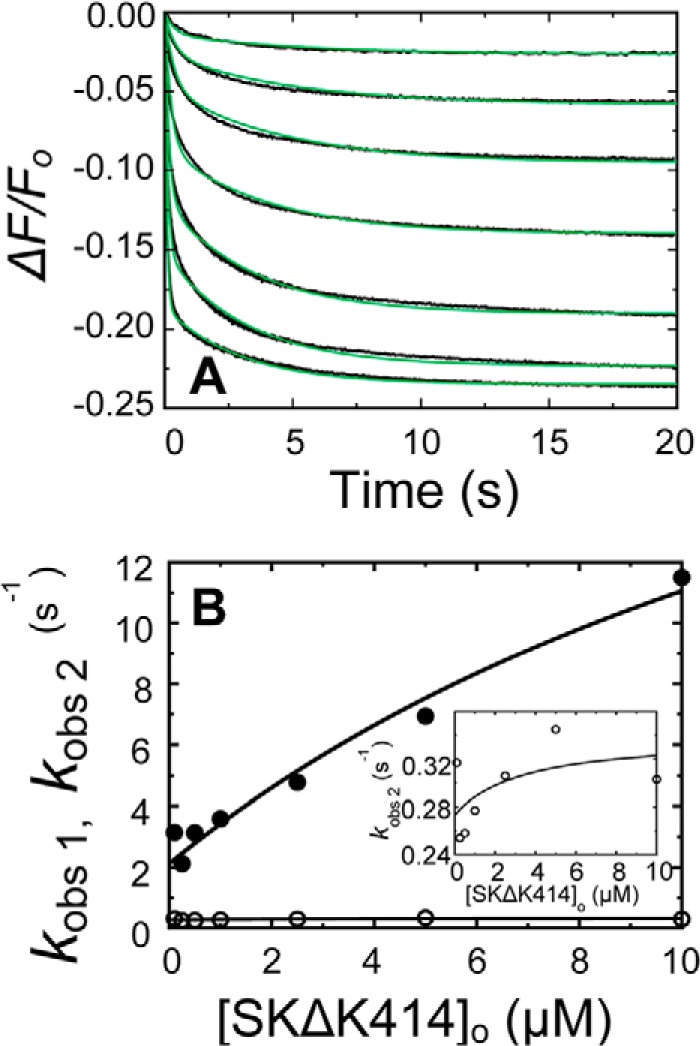
Time traces of the forward reactions and the SK dependences of kobs1 and kobs2 in 50 mm 6-AHA are shown in Fig. 4. Blocking the LBSs on kringles K1, K4, and K5 with 6-AHA decreased the affinity of the encounter complex to a K1 value of 7 μm (Table 1, superscript b), which is comparable with that of SKΔK414 binding in the absence of 6-AHA. The SK and SKΔK414 dependences of these weak binding interactions were not saturable, preventing accurate determination of K1; hence values ranging from 7 to 20 μm may be considered comparable. A weak encounter complex with a K1 value of 14 μm was also observed for SKΔK414 binding in 6-AHA (Fig. 5). The limiting rate constants klim 1 and klim 2 and the off-rates koff 1 and koff 2 determining the conformational changes following encounter complex formation were similar for SK and SKΔK414 binding to [5F]FFR-[Lys]Pg in the absence and presence of 6-AHA. Total amplitudes of the time traces fit by Equation 1 reflected overall maximal fluorescence changes (ΔFmax/Fo) for SK and SKΔK414 binding to [5F]FFR-Pg in agreement with equilibrium binding results in the absence and presence of 6-AHA (27, 48).
FIGURE 4.
Kinetics of SK binding to [5F]FFR-[Lys]Pg in 50 mm 6-AHA. A and B, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and nSK versus time in 50 mm 6-AHA are shown for 20 nm [5F]FFR-[Lys]Pg and 0.10, 0.26, 0.53, 1.53, and 3.06 μm nSK (A) and at 10 nm [5F]FFR-[Lys]Pg and 2.93, 4.40, and 8.21 μm nSK (B). Green solid lines represent the fits from numerical integration as described under “Experimental Procedures.” C, dependences of kobs 1 (●) and kobs 2 (○) on the total nSK concentration ([SK]o) are shown for binding to 10–20 nm [5F]FFR-[Lys]Pg. The inset shows the kobs 2 dependence on an enlarged scale. Solid lines represent the least-squares fits by Equation 2 with the parameters given in Table 1. Experiments were performed and analyzed as described under “Experimental Procedures.”
FIGURE 5.
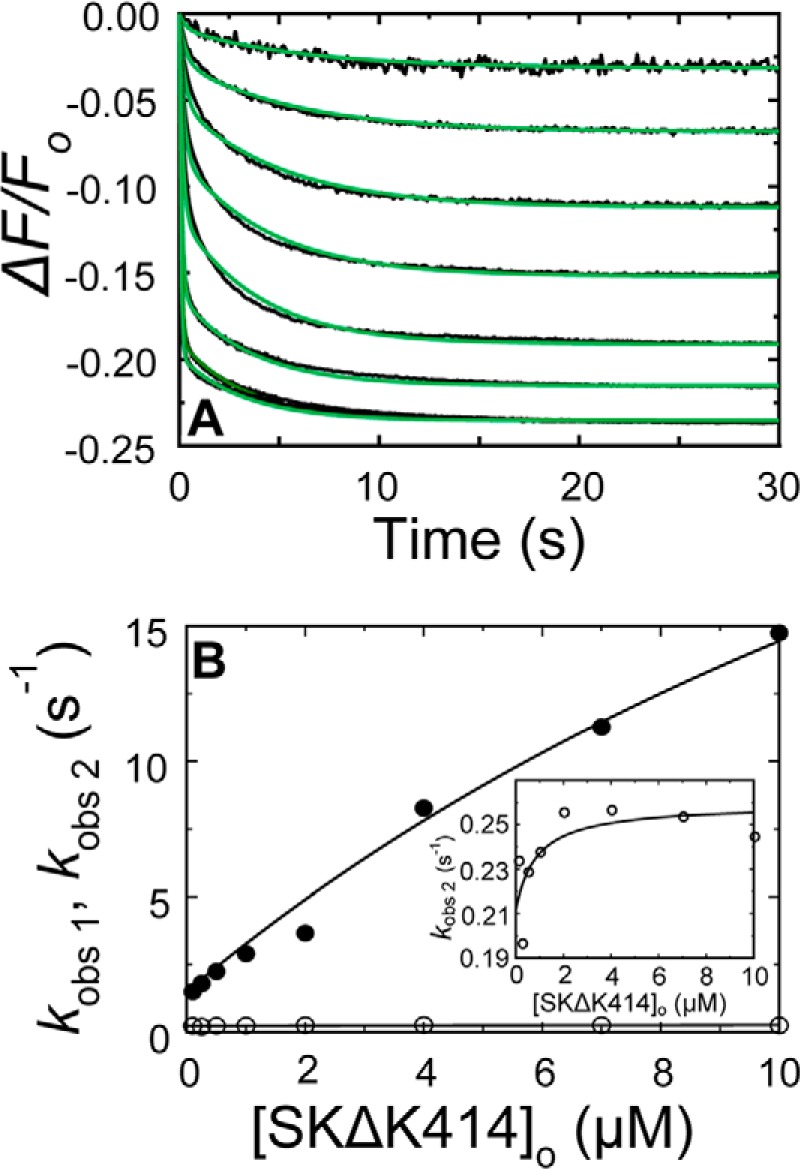
Kinetics of SKΔK414 binding to [5F]FFR-[Lys]Pg in 50 mm 6-AHA. A, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and SKΔK414 versus time in 50 mm 6-AHA are shown for 20 nm [5F]FFR-[Lys]Pg and 0.1, 0.25, 0.5, 1, 2, 4, 7, and 10 μm SKΔK414. Green solid lines represent the fits from numerical integration as described under “Experimental Procedures.” B, dependences of kobs 1 (●) and kobs 2 (○) on the total SKΔK414 concentration ([SKΔK414]o) are shown for binding to 20 nm [5F]FFR-[Lys]Pg. The inset shows the kobs 2 dependence on an enlarged scale. Solid lines represent the least square fits by Equation 2 with the parameters given in Table 1. Experiments were performed and analyzed as described under “Experimental Procedures.”
Effects of Benzamidine on the Kinetics of SK and SKΔK414 Binding to [5F]FFR-[Lys]Pg
Blocking kringles K1, K2, and K5 with 50 mm benzamidine weakened the K1 of the [5F]FFR-[Lys]Pg encounter complex with SK to 12 μm (Table 1, superscript b). Time traces of the forward reactions of SK binding were not resolvable into two phases and appeared as single exponential curves, whereas SKΔK414 binding was clearly biphasic. Progress curves for binding of SK and SKΔK414 and their concentration dependences of kobs 1 and kobs 2 in benzamidine are shown in Fig. 6. The limiting rate klim 1 in benzamidine was ∼5-fold faster for SK binding and ∼2.3-fold faster for SKΔK414 binding compared with the values in 6-AHA and in the absence of lysine analogs (Table 1, superscript b). The ∼30% lower maximal fluorescence changes than those for equilibrium binding in the presence of benzamidine described below (Table 1) may be due to the scattering properties of Pg complexes in benzamidine being differentially affected by the optical cell geometry and path length of the Photon Technology International, Inc. fluorometer and the stopped-flow instrument.
FIGURE 6.
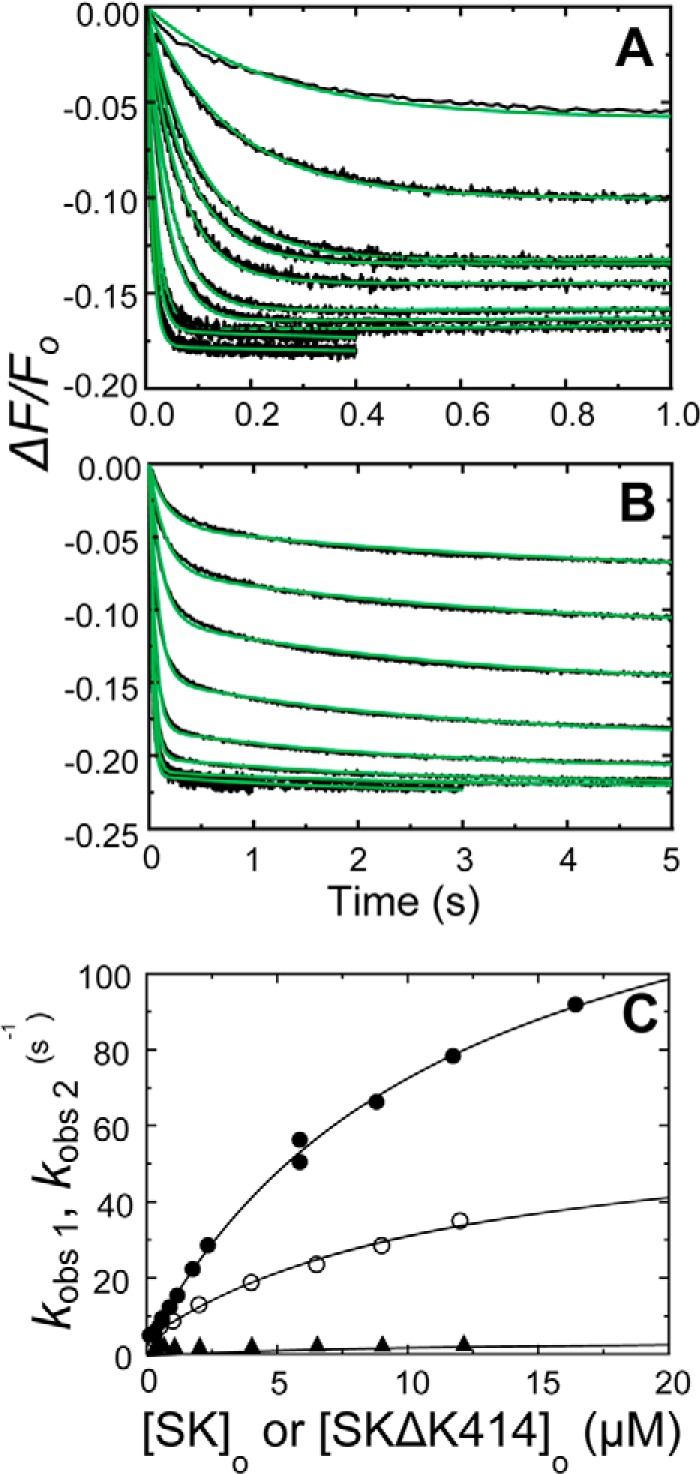
Kinetics of SK and SKΔK414 binding to [5F]FFR-[Lys]Pg in 50 mm benzamidine. A, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and nSK versus time in 50 mm benzamidine are shown for 10 or 15 nm [5F]FFR-[Lys]Pg and 0.12, 0.29, 0.59, 0.88, 1.17, 1.76, 2.34, 5.86, 8.8, 11.73, and 16.42 μm nSK. B. the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Lys]Pg and SKΔK414 versus time in 50 mm benzamidine are shown for 20 nm [5F]FFR-[Lys]Pg and 0.25, 1, 2, 4, 6.5, 9, and 12 μm SKΔK414. Green solid lines represent the fits from numerical integration as described under “Experimental Procedures.” C, dependences of kobs1 (●) on the total nSK concentration ([SK]o) and kobs 1 and kobs 2 (○ and ▴) on the total SKΔK414 concentration ([SKΔK414]o) are shown for binding to 10–20 nm [5F]FFR-[Lys]Pg. Solid lines represent the least square fits by Equation 2 with the parameters given in Table 1. Experiments were performed and analyzed as described under “Experimental Procedures.”
Stopped-flow Kinetics of SK Binding to [5F]FFR-[Glu]Pg
Biexponential binding of SK to [5F]FFR-[Glu]Pg was not saturable, and the k2/K1 ratios in the absence and presence of 6-AHA were indistinguishable and similar to those for [5F]FFR-[Lys]Pg binding to SKΔK414 in the absence of kringle ligands and to nSK and SKΔK414 binding in 6-AHA (Fig. 7). Fitting of these near linear dependences was performed using fixed, lower limit K1 values that were reasonably resolvable by numerical integration (see below). The limiting rate constants klim 1 and klim 2 and the off-rates koff 1 and koff 2 were similar to those for SK and SKΔK414 binding to [5F]FFR-[Lys]Pg in the absence of lysine analogs and in 6-AHA (Table 1, superscript b).
FIGURE 7.
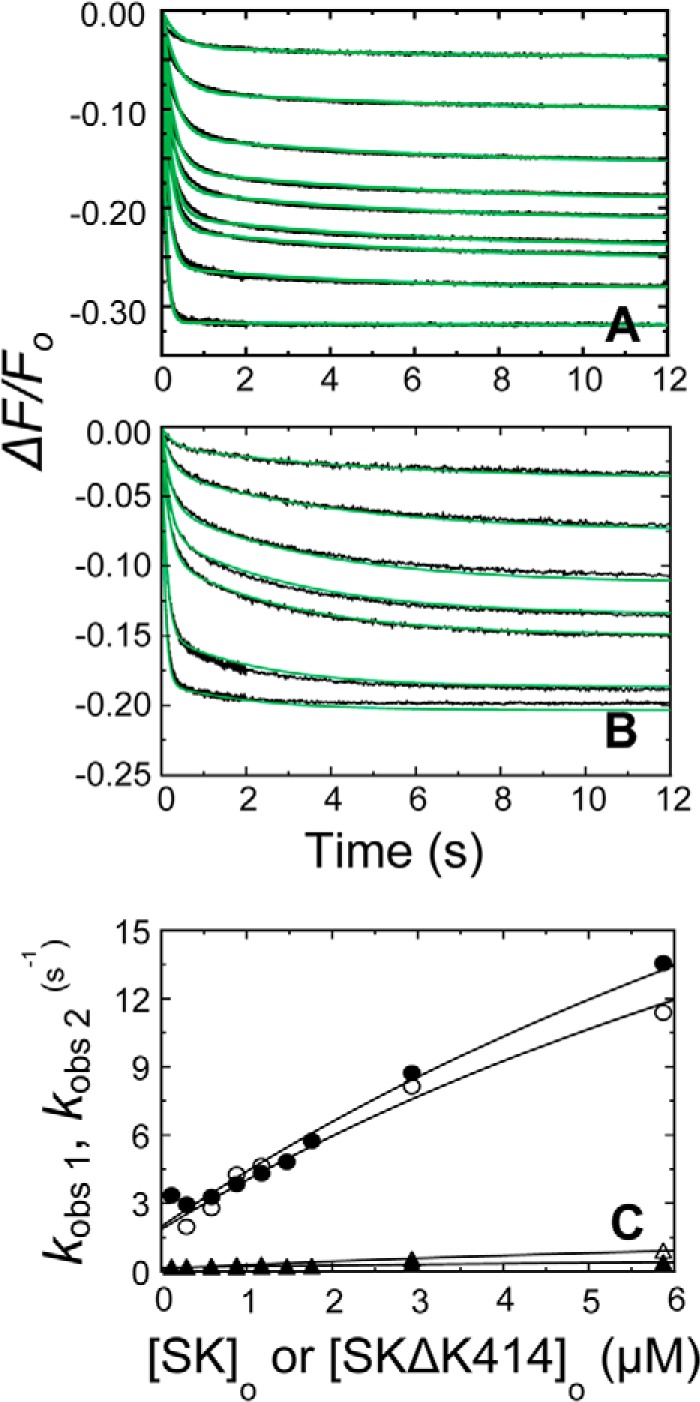
Kinetics of SK binding in the [5F]FFR-[Glu]Pg complex in the absence of lysine analogs and at saturating 6-AHA. A, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Glu]Pg and nSK versus time in the absence of lysine analogs are shown for 13 or 20 nm [5F]FFR-[Glu]Pg and 0.12, 0.29, 0.59, 0.88, 1.17, 1.47, 1,76, 2.93, and 5.87 μm nSK. B, the fractional fluorescence intensity changes (ΔF/Fo) following rapid mixing of [5F]FFR-[Glu]Pg and nSK versus time in 50 mm 6-AHA are shown for 13 or 20 nm [5F]FFR-[Glu]Pg and 0.12, 0.29, 0.59, 0.88, 1.17, 2.93, and 5.87 μm nSK. Green solid lines represent the fits from numerical integration as described under “Experimental Procedures.” C, dependences of kobs 1 (● and ○) and kobs 2 (▴ and ▵) on the total nSK concentration ([SK]o) are shown for binding to 10–20 nm [5F]FFR-[Glu]Pg in the absence of lysine analogs (filled symbols) and in 50 mm 6-AHA (open symbols). Solid lines represent the least square fits by Equation 2 with the parameters given in Table 1. Experiments were performed and analyzed as described under “Experimental Procedures.”
Competitive Displacement of [5F]FFR-[Lys]Pg from Its Complex with nSK, WT-SK, and SKΔK414 by FFR-Pm
Mixing a ∼5–100-fold excess of unlabeled FFR-Pm with the preformed complexes of SK and SKΔK414 with [5F]FFR-[Lys]Pg at varying degrees of saturation caused a rapid, biexponential increase of fluorescence, approaching the initial fluorescence intensity. Analysis of the time traces by Equation 1 yielded perfect fits with random residuals (not shown) and gave kdisp 1 and kdisp 2 values for the fast and slow exponential phases of the displacement reactions. Representative averaged traces for SK and SKΔK414 displacement by FFR-Pm are shown in Figs. 8 and 9 with the colored lines representing global fits of forward and reverse reactions by numerical analysis. These processes represent rapid reversal of [5F]FFR-[Lys]Pg binding to SK and SKΔK414 and parallel formation of non-fluorescent complexes with FFR-Pm. Saturation of Pg required high SK and SKΔK414 concentrations and consequently high FFR-Pm to bind free SK and SKΔK414 and to displace labeled Pg from the complexes. Therefore we expressed kdisp 1 and kdisp 2 as dependences of free rather than total FFR-Pm calculated by numerical integration. The rates were independent of free FFR-Pm, consistent with extremely tight binding of SK and SKΔK414 to plasmin (59), and were similar for [Lys]Pg and [Glu]Pg in the absence and presence of lysine analogs. The off-rate for the fast process, kdisp 1, was 0.90 ± 0.60 s−1, which is modestly lower than the averaged koff 1 of 2.60 ± 1.2 s−1 determined from the SK dependences of the forward reactions and the equivalent averaged k−2 of 3.2 ± 1.6 s−1 from numerical analysis (see below). The kdisp 2 off-rate for the slow phase was 0.13 ± 0.09 s−1, which is similar to the averaged koff 2 of 0.22 ± 0.12 s−1 from the SK dependences and the equivalent averaged k−3 of 0.19 ± 0.08 s−1 from numerical analysis.
FIGURE 8.
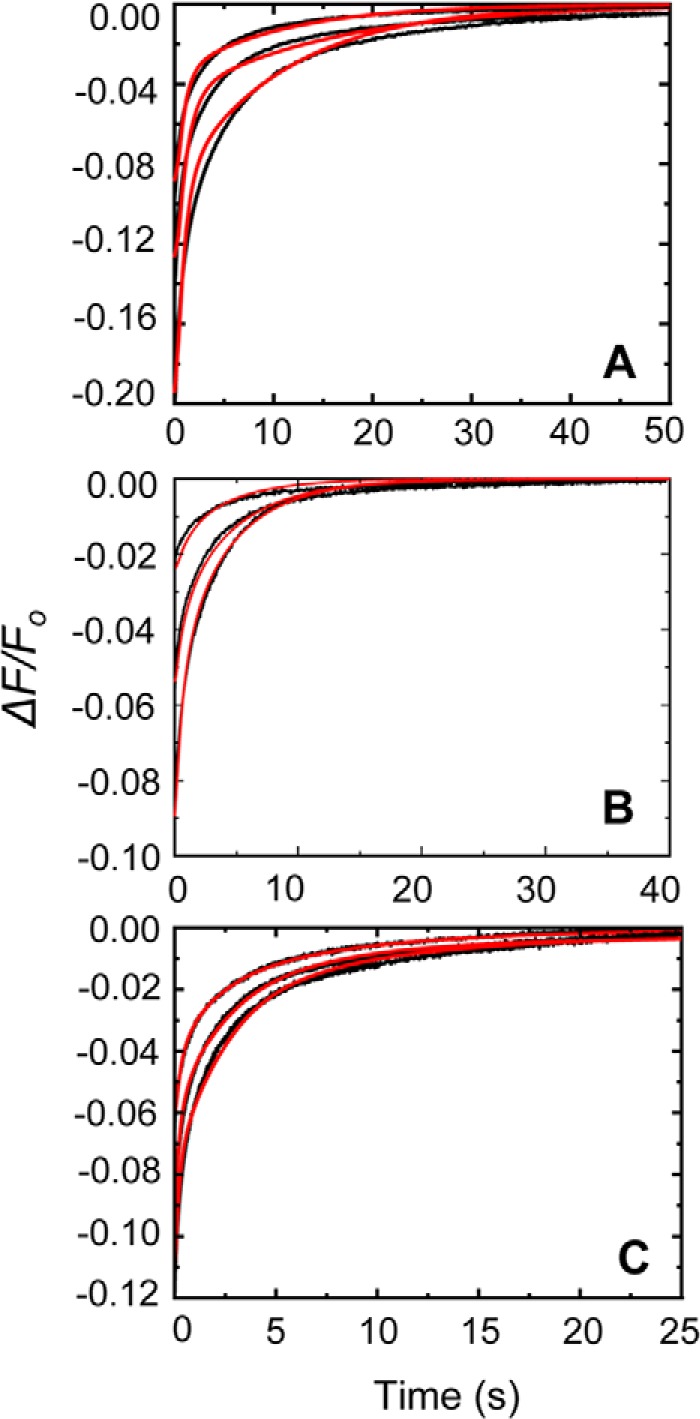
Competitive dissociation of [5F]FFR-[Lys]Pg from its complex with nSK by FFR-Pm. A, displacement of [5F]FFR-[Lys]Pg from its stabilized complex with nSK by FFR-Pm in the absence of lysine analogs is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.06 μm nSK, and 0.1 μm FFR-Pm (top); 0.12 μm nSK and 0.2 μm FFR-Pm (middle); and 0.23 μm nSK and 0.4 μm FFR-Pm (bottom). B, displacement of [5F]FFR-[Lys]Pg from its stabilized complex with nSK by FFR-Pm in 50 mm 6-AHA is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.1 μm nSK, and 0.2 μm FFR-Pm (top); 0.25 μm nSK and 0.3 μm FFR-Pm (middle); and 0.5 μm nSK and 0.6 μm FFR-Pm (bottom). C, displacement of [5F]FFR-[Lys]Pg from its stabilized complex by FFR-Pm in 50 mm benzamidine is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.1 μm nSK, and 0.2 μm FFR-Pm (top); 0.25 μm nSK and 0.3 μm FFR-Pm (middle); and 0.5 μm nSK and 0.6 μm FFR-Pm (bottom). Red solid lines represent the fits from numerical integration as described under “Experimental Procedures.”
FIGURE 9.
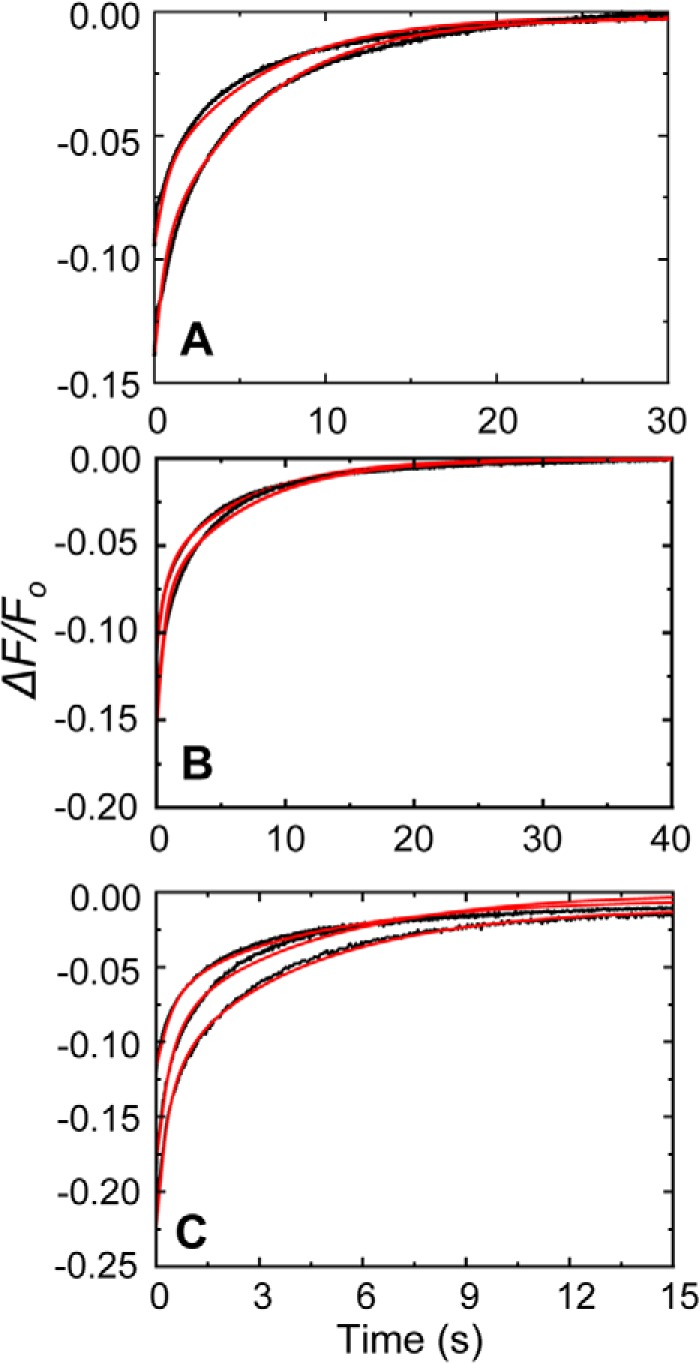
Competitive dissociation of [5F]FFR-[Lys]Pg from its complex with SKΔK414 by FFR-Pm. A. Displacement of [5F]FFR-[Lys]Pg from its stabilized complex with SKΔK414 by FFR-Pm in the absence of lysine analogs is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.5 μm SKΔK414, and 0.5 μm FFR-Pm (top) and 1.0 μm SKΔK414 and 1.0 μm FFR-Pm (bottom). B, displacement of [5F]FFR-[Lys]Pg from its stabilized complex with SKΔK414 by FFR-Pm in 50 mm 6-AHA is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.5 μm SKΔK414, and 0.7 μm FFR-Pm (top) and 1 μm SKΔK414 and 1.4 μm FFR-Pm (bottom). C, displacement of [5F]FFR-[Lys]Pg from its stabilized complex with SKΔK414 by FFR-Pm in 50 mm benzamidine is shown at dead time mixing concentrations of 20 nm [5F]FFR-[Lys]Pg, 0.5 μm SKΔK414, and 0.5 μm FFR-Pm (top); 1 μm SKΔK414 and 1.2 μm FFR-Pm (middle); and 2 μm SKΔK414 and 2 μm FFR-Pm (bottom). Red solid lines represent the fits from numerical integration as described under “Experimental Procedures.”
Equilibrium Binding of [5F]FFR-[Lys]Pg to SK and SKΔK414 in the Presence of 50 mm Benzamidine
We determined the affinity and fluorescence change for SK and SKΔK414 equilibrium binding to [5F]FFR-[Lys]Pg in 50 mm benzamidine to characterize the effect of this K1, K2, and K5 ligand on the overall equilibrium binding constant KD, overall and to compare this affinity with KD, overall calculated from the forward and reverse constants obtained by the binding kinetics (Fig. 10). Analysis of the titrations indicated that SK bound with a KD of 200 ± 20 nm and ΔFmax/Fo of −30 ± 1%. The affinity was ∼5-fold weaker than in the absence of kringle ligands but still ∼3-fold tighter than in 6-AHA. SKΔK414 bound labeled [Lys]Pg with a ΔFmax/Fo of −38 ± 1% and KD of 800 ± 100 nm. This affinity was similar to that of SK in 6-AHA, SKΔK414 with and without 6-AHA (48), and SK binding to [5F]FFR-[Glu]Pg with and without 6-AHA (27, 48).
FIGURE 10.

Equilibrium binding of SK and SKΔK414 to [5F]FFR-[Lys]Pg in the presence of benzamidine. The fractional change in fluorescence (ΔF/Fo) of 20 nm [5F]FFR-[Lys]Pg in buffer containing 50 mm benzamidine is plotted as a function of the total nSK (●) or SKΔK414 (○) concentration ([SK]o or [SKΔK414]o). Solid lines represent least square fits of the quadratic equation for binding of a single ligand with the parameters listed in the text and Table 1. Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Numerical Integration Analysis of the Forward and Reverse Reactions
Fitted values for K1, the rate constants for both conformational steps, and the fluorescence amplitudes were in good agreement with those obtained from two-exponential analysis and equilibrium binding and are given in Table 1 (superscript a, fluorescence intensity, and superscript aa, fluorescence anisotropy). The results indicated that formation of fluorescently silent SK·Pg 1 occurs in the dead time of the reaction and that subsequent partitioning occurs between SK·Pg 2 and SK·Pg 3.
The SK·[5F]FFR-[Lys]Pg encounter complex was weakened ∼10–20-fold by blocking LBSs on the Pg kringles and by loss of Lys414. The rate constant k2 for the first conformational step ranged from 25 to 45 s−1 in the absence and presence of 6-AHA but increased substantially in benzamidine, suggesting a decrease in conformational restraint.
The rate constants k−2 and k−3 for the reverse reactions were equivalent to koff 1 and koff 2 from hyperbolic fitting of the SK dependences of the forward reaction rates and to kdisp 1 and kdisp 2 for the biexponential appearance of free [5F]FFR-[Lys]Pg in competitive displacement by FFR-Pm. The analytical solution of the overall koff value for a three-step reaction is only straightforward under conditions of single exponential kinetics (70); however, the agreement of koff 2 and kdisp 2 with k−3 from numerical analysis suggests that dissociation is limited by k−3. The off-rates were unaffected by lysine analogs.
In the absence of effectors, KD, overall for SK binding to [5F]FFR-[Lys]Pg ranged from 30 ± 18 to 86 ± 18 nm in agreement with the results from equilibrium binding (27, 48). Deletion of SK Lys414 or blocking the LBSs with 6-AHA caused an increase of KD, overall to 0.5–0.9 μm, which is identical to that for [Glu]Pg binding. In benzamidine, KD, overall for binding of intact SK to labeled [Lys]Pg was 0.2 μm, possibly reflecting the contribution of the large forward rate for the first tightening step.
Within global data sets, the errors in the amplitude factors f2 for the fast conformational step and f3 for the slow step were ∼30 and ∼18%, respectively (2 × S.D.). Numerical integration fits for the forward and reverse reactions are shown as colored lines in the figures.
DISCUSSION
The present study demonstrates a minimal three-step sequential mechanism for binding of SK to [5F]FFR-Pg, consisting of an encounter complex with affinity in the low micromolar range followed by at least two resolvable conformational steps (Fig. 11), which increase the affinity of the stabilized complex to ∼30–86 nm. The first conformational step is the main tightening event, whereas the second step does not confer additional tightening. However, deletion of this second step in the mechanism resulted in single exponential curves that did not fit the data.
FIGURE 11.
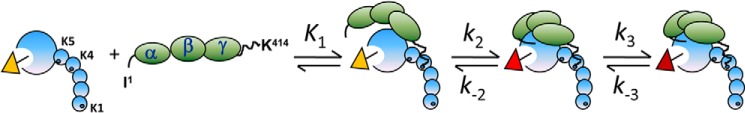
The three-step mechanism of SK·[5F]FFR-[Lys]Pg catalytic complex formation. [5F]FFR-[Lys]Pg is shown as blue circles in a hypothetical partially extended β-conformation. The five kringle domains are small circles with the LBSs of K1, K4, and K5 as tiny black dimples. The zymogen catalytic domain is the larger blue circle with the activated catalytic site in white locked into its conformation by the fluorescein probe (ocher triangle) covalently attached to the peptide chloromethyl ketone that has alkylated His57 (black stem). SK is shown by three green ovals representing the three β-grasp domains marked α, β, and γ. The NH2 terminus of SK is indicated by I,1 and the COOH-terminal Lys414 is at the end of a long disordered segment (squiggle) of the γ-domain. During formation of the initial SK·Pg encounter complex (governed by K1), Lys414 engages the LBS of K4, whereas the domains of SK are thought to not be fully engaged, and this does not produce a change in fluorescence. The first, tightening, conformational change governed by k2 and k−2 with the largest decrease in fluorescence (red triangle) is shown hypothetically to involve insertion of SK Ile1 into the NH2-terminal binding cleft forming the Asp194 salt bridge and settling of the SK domains into a more ordered arrangement. The last conformational step controlled by k3 and k−3 completes the arrangement of SK domains accompanied by a smaller fluorescence decrease (maroon triangle).
We previously discovered that a three-step mechanism also governs SK binding to labeled plasmin but with ∼4,000-fold tighter KD, overall values of 7–12 pm (59). The conformational changes caused a ∼9,000-fold tightening of the Pm encounter complex but only a ∼50-fold increase in affinity for Pg. We show here that substantial decreases in affinity of the encounter complex and the second conformational event are mainly responsible for the weaker SK binding to Pg in the stabilized complex.
The results suggest that the SK interactions with LBSs on [Lys]Pg are mainly limited to SK Lys414 binding to K4, whereas plasmin binding involves another SK lysine interacting with K5 in addition to Lys414 binding to K4. It is noteworthy that non-LBS interactions with the protease domain are significant sources of binding energy in both plasminogen and plasmin binding (71). Until now, SK binding to Pg had only been studied by equilibrium binding, and although the published KD values report the affinities of the final complexes, they do not provide information on the intermediates in this multistep mechanism.
The results support the following sequential steps on the pathway to a stabilized complex with labeled Pg: SK Lys414 binding to a Pg kringle during formation of a weak, fluorescently silent encounter complex and two conformational steps of SK reorganization from a flexible to a more organized form during binding to the Pg protease domain accompanied by expression of a pro-exosite for binding of a second Pg molecule in the substrate mode. This reorganization is reported by biphasic fluorescence changes of the probe in the active site on the protease domain of Pg. Two striking differences between SK binding to labeled Pm and [Lys]Pg were immediately obvious: a ∼40-fold weaker binding of SK in the encounter complex illustrated by higher SK concentrations required for saturation of the rates of fluorescence change and the requirement of stopped flow to study Pg displacement from the complex by FFR-Pm evidenced by the large increase in the off-rate constants k−2 and k−3. Whereas displacement from the SK·Pm complex required several hours of incubation with excess FFR-Pm, the complexes with [Lys]Pg were easily reversed in a matter of seconds.
Binding to Pg also involves insertion of the NH2 terminus of SK in the activation pocket of the Pg catalytic domain; however, adding a conformational step to the mechanism did not improve the fits. Stopped-flow fluorescence of SK binding to labeled Pg may not allow identifying the timing of the NH2-terminal insertion or whether NH2-terminal insertion contributes to the affinity of the Pg complex, and further studies are required to resolve this complex event.
Binding of SK Lys414 to a kringle facilitates formation of the encounter complexes with both [Lys]Pg and Pm as a similar 6–8-fold reduction in their affinity was observed when Lys414 was deleted. K1 of the encounter complex with labeled [Lys]Pg increased from ∼3 to ∼19 μm upon deleting SK Lys414. Saturation of the LBSs did not decrease the affinity of SK and SKΔK414 any further, indicating no other SK lysine-LBS interactions, and the 10–19 μm affinity range of LBS-blocked SK and SKΔK414 complexes likely represents the contribution of non-LBS binding to the Pg catalytic domain (Table 1). K1 of the encounter complex with Pm increases from ∼0.08 to ∼0.67 μm upon SK Lys414 deletion (59); however, the SKΔK414·Pm encounter complex still exhibits substantial affinity, reflecting the sum of the LBS interactions with other lysine residues and non-LBS interactions with the protease domain. Kringle K5 harbors an LBS that preferentially interacts with ligands not carrying a free carboxylate function, such as alkylamines (51, 72, 73), and K5 on Pm may bind a non-COOH-terminal SK lysine. Saturation of Pm with 6-AHA disengages Lys414 and other lysines, and as expected, this affinity is not weakened further by SK Lys414 deletion. The remaining encounter affinity of ∼5–8 μm likely represents the non-LBS interactions with the Pm catalytic domain (59). Multiple LBS interactions in the tighter encounter complex with Pm may be made possible by an increased flexibility of two-chain Pm compared with single chain Pg. This flexibility might also allow more intimate contacts during stabilization of the SK·Pm complex.
6-AHA binds kringles K1, K4, and K5, and the Pg binding results likely eliminate the involvement of K2 and K3 in SK Lys414 binding. Similarly, the weak SK binding to [Glu]Pg eliminates K1 as a candidate for Lys414 interaction as this is the only kringle in [Glu]Pg exposed for fibrin binding (74). Kringle K4 is not accessible in [Glu]Pg due to steric hindrance by the Pg NH2-terminal PAN module, which binds K5 (75, 76). The identical encounter complex affinity of SK for [Glu]Pg in the absence and presence of 6-AHA indicated that LBS interactions do not play a role in [Glu]Pg binding.
Benzamidine blocks kringles K1, K2, and K5 and leaves kringle K4 available for lysine binding. The affinity of the SK·Pg encounter complex in 6-AHA and benzamidine was similar, suggesting that Lys414 binding to K4 in Pg does not increase the affinity when K5 is blocked. However, the SK·Pm encounter complex was ∼2-fold tighter in benzamidine than that in 6-AHA and was further weakened by deletion of Lys414 (59). Further studies are required to clarify these different effects on Pg and Pm binding.
The 42-residue COOH-terminal sequence is not resolved in the SK·μPm crystal structure (19). Lys414 at the end of this disordered, mobile sequence may guide the pathway by initial interaction with the LBS on K4; however, this does not contribute much to the free energy of binding of the encounter complex. Calculating changes in free energy of association for SK and SKΔK414 binding to Pg and Pm from ΔG0 = RT ln(KD) using averaged K1 values from Table 1 and our previous study (59) shows that the non-LBS interactions contribute ∼83 and ∼73% of the binding energy in Pg and Pm encounter complex formation, respectively. SK Lys414 contributes ∼17 and ∼13%. Binding of (an)other SK lysine residue(s) to Pm contributes ∼14%. Although LBS interactions are important for efficient docking of SK, it appears that non-LBS interactions are the major source of the binding energy both for Pg and for Pm encounter. Previous equilibrium binding studies with α-domain-truncated SK showed that the LBS-independent interactions with the Pg/Pm protease domain largely reside in the SK α-domain, whereas the β- and γ-domains participate in LBS-dependent interactions with Pg/Pm kringles (71). It is likely that Pm-binding lysines other than the C-terminal Lys414 reside in the SK β- and γ-domains.
SK·Pg and SK·Pm differ in their rate constants for the two conformational steps. The k2 values for the SK·Pm complex were ∼10 s−1 for intact and Lys414-deleted SK and ∼37 s−1 in 6-AHA (59); the latter is comparable with k2 for all the SK and SKΔK414 interactions with Pg in this study except those in benzamidine. For SK·Pm, the conformational restraint reflected by a low k2 may be due to binding of a non-COOH-terminal SK lysine to K5, which also makes the SK·Pm encounter complex tighter. This restraint is absent in [Lys]Pg, suggesting no binding contribution from SK lysines other than Lys414. Benzamidine enhanced k2 by 5- and 6-fold in SK binding to Pg and Pm, respectively (59). This enhancement was weaker with SKΔK414, suggesting that Lys414 binding to K4 may release conformational restraints. The large k2 value may contribute to a ∼4-fold tighter KD, overall for SK·Pg in benzamidine compared with that for SK·Pg in 6-AHA and SKΔK414·Pg in all buffer systems. 6-AHA causes transition of [Glu]Pg from the compact α-form to the fully extended γ-form and of [Lys]Pg from the partially extended β- to the γ-form, whereas benzamidine keeps [Lys]Pg in the β-form (43). This suggests that the release of constraints on k2 does not depend on α → β → γ conformational changes in Pg. The k−2, k3, and k−3 values were similar in all data sets, indicating that these steps are LBS-independent.
In summary, we demonstrate here for the first time that the three-step kinetic model for the pathway of Pg binding to SK is substantially different from that of Pm binding with the main differences being a weaker encounter complex and increased off-rates for the conformational steps. Whereas cooperative Lys414 and other lysine interactions with K4 and K5 and non-LBS interactions with the protease domain contribute to formation of the SK·Pm complex, the SK·Pg* complex assembly appears to be driven by non-LBS interactions and by SK Lys414 binding to Pg kringle K4. Consistent with the experimentally determined Km of ≥2 μm for substrate Pg binding to the SK·Pg* complex and of 270 nm for Pg binding to the SK·Pm complex (7) and in the absence of a crystal structure of the SK·Pg* complex, we hypothesize that the weaker interaction with Pg in the catalytic complex results in expression of a pro-exosite that binds substrate Pg with lower affinity than the corresponding exosite on the SK·Pm complex.
Differences in affinity of the SK·Pg* and SK·Pm complexes may be important in their partitioning on bacterial surface proteins and in binding to host fibrin(ogen). Group A streptococcal M-like surface proteins bind Pg with high affinity, and the M1 subset lacking Pm-binding motifs binds fibrinogen (Fbg). This allows indirect activation by way of SK·Pg*·Fbg ternary complex formation (7, 77), which proceeds by Fbg binding to the SK·Pg* complex rather than SK recruitment on the Pg·Fbg complex (7). Group A Streptococcus SK exhibits significant polymorphism (78–80), and considerable differences exist among SK allelic variants in their efficiency of activating Pg and their recruitment in complexes with Fbg, Fbg fragment D, fibrin, and the plasminogen-binding group A streptococcal M protein (81). [Glu]Pg binds fibrin(ogen) through a K1 interaction, whereas K1 and K4 of [Lys]Pg and Pm are involved in fibrin(ogen) binding (74), and these differences are expected to influence localization of SK·Pg* and SK·Pm complexes. Future stopped-flow studies will identify how fibrin(ogen) and streptococcal surface proteins affect the pathways of SK·Pg* and SK·Pm formation and will be instrumental in characterizing these pathways in complexes with allelic SK variants.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL056181 from the NHLBI (to P. E. B.).
- Pm
- plasmin
- SK
- streptokinase
- SKΔK414
- SK lacking the COOH-terminal Lys414 residue
- nSK
- native streptokinase
- Pg
- plasminogen
- [Glu]Pg
- intact native plasminogen
- [Lys]Pg
- native Pg lacking the N-terminal 77 residues
- [Lys]Pg*
- the conformationally activated form of Pg
- FFR-CH2Cl
- d-Phe-Phe-Arg-CH2Cl
- FFR-Pm
- Pm inhibited with d-Phe-Phe-Arg-CH2Cl
- [5F]FFR-[Lys]Pg
- [5-(acetamido)fluorescein]-d-Phe-Phe-Arg-[Lys]Pg
- [5F]FFR-[Glu]Pg
- [5-(acetamido)-fluorescein]-d-Phe-Phe-Arg-[Glu]Pg
- 6-AHA
- 6-aminohexanoic acid
- Pg*
- nonproteolytically activated form of the plasminogen zymogen
- LBS
- lysine-binding site
- PAN
- plasminogen, apple, nematode
- K
- kringle
- [5F]
- 5-fluorescein
- Fbg
- fibrinogen
- [SK]o
- total SK concentration.
REFERENCES
- 1. Collen D., Lijnen H. R. (1995) Molecular basis of fibrinolysis, as relevant for thrombolytic therapy. Thromb. Haemost. 74, 167–171 [PubMed] [Google Scholar]
- 2. Deryugina E. I., Quigley J. P. (2012) Cell surface remodeling by plasmin: a new function for an old enzyme. J. Biomed. Biotechnol. 2012, 564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carapetis J. R., Steer A. C., Mulholland E. K., Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 4. Preziuso S., Laus F., Tejeda A. R., Valente C., Cuteri V. (2010) Detection of Streptococcus dysgalactiae subsp. equisimilis in equine nasopharyngeal swabs by PCR. J. Vet. Sci. 11, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopardo H. A., Vidal P., Sparo M., Jeric P., Centron D., Facklam R. R., Paganini H., Pagniez N. G., Lovgren M., Beall B. (2005) Six-month multicenter study on invasive infections due to Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis in Argentina. J. Clin. Microbiol. 43, 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boxrud P. D., Bock P. E. (2004) Coupling of conformational and proteolytic activation in the kinetic mechanism of plasminogen activation by streptokinase. J. Biol. Chem. 279, 36642–36649 [DOI] [PubMed] [Google Scholar]
- 7. Nolan M., Bouldin S. D., Bock P. E. (2013) Full time course kinetics of the streptokinase-plasminogen activation pathway. J. Biol. Chem. 288, 29482–29493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boxrud P. D., Verhamme I. M., Bock P. E. (2004) Resolution of conformational activation in the kinetic mechanism of plasminogen activation by streptokinase. J. Biol. Chem. 279, 36633–36641 [DOI] [PubMed] [Google Scholar]
- 9. McClintock D. K., Bell P. H. (1971) The mechanism of activation of human plasminogen by streptokinase. Biochem. Biophys. Res. Commun. 43, 694–702 [DOI] [PubMed] [Google Scholar]
- 10. Wohl R. C., Summaria L., Arzadon L., Robbins K. C. (1978) Steady state kinetics of activation of human and bovine plasminogens by streptokinase and its equimolar complexes with various activated forms of human plasminogen. J. Biol. Chem. 253, 1402–1407 [PubMed] [Google Scholar]
- 11. Reddy K. N., Markus G. (1972) Mechanism of activation of human plasminogen by streptokinase. Presence of active center in streptokinase-plasminogen complex. J. Biol. Chem. 247, 1683–1691 [PubMed] [Google Scholar]
- 12. Schick L. A., Castellino F. J. (1974) Direct evidence for the generation of an active site in the plasminogen moiety of the streptokinase-human plasminogen activator complex. Biochem. Biophys. Res. Commun. 57, 47–54 [DOI] [PubMed] [Google Scholar]
- 13. Bajaj A. P., Castellino F. J. (1977) Activation of human plasminogen by equimolar levels of streptokinase. J. Biol. Chem. 252, 492–498 [PubMed] [Google Scholar]
- 14. Davidson D. J., Higgins D. L., Castellino F. J. (1990) Plasminogen activator activities of equimolar complexes of streptokinase with variant recombinant plasminogens. Biochemistry 29, 3585–3590 [DOI] [PubMed] [Google Scholar]
- 15. Boxrud P. D., Verhamme I. M., Fay W. P., Bock P. E. (2001) Streptokinase triggers conformational activation of plasminogen through specific interactions of the amino-terminal sequence and stabilizes the active zymogen conformation. J. Biol. Chem. 276, 26084–26089 [DOI] [PubMed] [Google Scholar]
- 16. Wang S., Reed G. L., Hedstrom L. (1999) Deletion of Ile1 changes the mechanism of streptokinase: evidence for the molecular sexuality hypothesis. Biochemistry 38, 5232–5240 [DOI] [PubMed] [Google Scholar]
- 17. Wang S., Reed G. L., Hedstrom L. (2000) Zymogen activation in the streptokinase-plasminogen complex. Ile1 is required for the formation of a functional active site. Eur. J. Biochem. 267, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 18. Bode W., Huber R. (1976) Induction of the bovine trypsinogen-trypsin transition by peptides sequentially similar to the N-terminus of trypsin. FEBS Lett. 68, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Lin X., Loy J. A., Tang J., Zhang X. C. (1998) Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science 281, 1662–1665 [DOI] [PubMed] [Google Scholar]
- 20. Friedrich R., Panizzi P., Fuentes-Prior P., Richter K., Verhamme I., Anderson P. J., Kawabata S., Huber R., Bode W., Bock P. E. (2003) Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature 425, 535–539 [DOI] [PubMed] [Google Scholar]
- 21. Kroh H. K., Bock P. E. (2012) Effect of zymogen domains and active site occupation on activation of prothrombin by von Willebrand factor-binding protein. J. Biol. Chem. 287, 39149–39157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malke H., Steiner K. (2004) Control of streptokinase gene expression in group A & C streptococci by two-component regulators. Indian J. Med. Res. 119, (suppl.) 48–56 [PubMed] [Google Scholar]
- 23. Walker M. J., McArthur J. D., McKay F., Ranson M. (2005) Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 13, 308–313 [DOI] [PubMed] [Google Scholar]
- 24. Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 25. Khil J., Im M., Heath A., Ringdahl U., Mundada L., Cary Engleberg N., Fay W. P. (2003) Plasminogen enhances virulence of group A streptococci by streptokinase-dependent and streptokinase-independent mechanisms. J. Infect. Dis. 188, 497–505 [DOI] [PubMed] [Google Scholar]
- 26. Boxrud P. D., Fay W. P., Bock P. E. (2000) Streptokinase binds to human plasmin with high affinity, perturbs the plasmin active site, and induces expression of a substrate recognition exosite for plasminogen. J. Biol. Chem. 275, 14579–14589 [DOI] [PubMed] [Google Scholar]
- 27. Boxrud P. D., Bock P. E. (2000) Streptokinase binds preferentially to the extended conformation of plasminogen through lysine binding site and catalytic domain interactions. Biochemistry 39, 13974–13981 [DOI] [PubMed] [Google Scholar]
- 28. Damaschun G., Damaschun H., Gast K., Gerlach D., Misselwitz R., Welfle H., Zirwer D. (1992) Streptokinase is a flexible multi-domain protein. Eur. Biophys. J. 20, 355–361 [DOI] [PubMed] [Google Scholar]
- 29. Henkin J., Marcotte P., Yang H. C. (1991) The plasminogen-plasmin system. Prog. Cardiovasc. Dis. 34, 135–164 [DOI] [PubMed] [Google Scholar]
- 30. Ponting C. P., Marshall J. M., Cederholm-Williams S. A. (1992) Plasminogen: a structural review. Blood Coagul. Fibrinolysis 3, 605–614 [PubMed] [Google Scholar]
- 31. Law R. H., Caradoc-Davies T., Cowieson N., Horvath A. J., Quek A. J., Encarnacao J. A., Steer D., Cowan A., Zhang Q., Lu B. G., Pike R. N., Smith A. I., Coughlin P. B., Whisstock J. C. (2012) The x-ray crystal structure of full-length human plasminogen. Cell Rep. 1, 185–190 [DOI] [PubMed] [Google Scholar]
- 32. Marti D. N., Hu C. K., An S. S., von Haller P., Schaller J., Llinás M. (1997) Ligand preferences of kringle 2 and homologous domains of human plasminogen: canvassing weak, intermediate, and high-affinity binding sites by 1H-NMR. Biochemistry 36, 11591–11604 [DOI] [PubMed] [Google Scholar]
- 33. Castellino F. J., McCance S. G. (1997) The kringle domains of human plasminogen. Ciba Found. Symp. 212, 46–60; discussion 60–65 [DOI] [PubMed] [Google Scholar]
- 34. Christensen U. (1985) C-terminal lysine residues of fibrinogen fragments essential for binding to plasminogen. FEBS Lett. 182, 43–46 [DOI] [PubMed] [Google Scholar]
- 35. Lucas M. A., Fretto L. J., McKee P. A. (1983) The binding of human plasminogen to fibrin and fibrinogen. J. Biol. Chem. 258, 4249–4256 [PubMed] [Google Scholar]
- 36. Wiman B., Lijnen H. R., Collen D. (1979) On the specific interaction between the lysine-binding sites in plasmin and complementary sites in α2-antiplasmin and in fibrinogen. Biochim. Biophys. Acta 579, 142–154 [DOI] [PubMed] [Google Scholar]
- 37. Bok R. A., Mangel W. F. (1985) Quantitative characterization of the binding of plasminogen to intact fibrin clots, lysine-Sepharose, and fibrin cleaved by plasmin. Biochemistry 24, 3279–3286 [DOI] [PubMed] [Google Scholar]
- 38. Clemmensen I., Petersen L. C., Kluft C. (1986) Purification and characterization of a novel, oligomeric, plasminogen kringle 4 binding protein from human plasma: tetranectin. Eur. J. Biochem. 156, 327–333 [DOI] [PubMed] [Google Scholar]
- 39. Marshall J. M., Brown A. J., Ponting C. P. (1994) Conformational studies of human plasminogen and plasminogen fragments: evidence for a novel third conformation of plasminogen. Biochemistry 33, 3599–3606 [DOI] [PubMed] [Google Scholar]
- 40. Mangel W. F., Lin B. H., Ramakrishnan V. (1990) Characterization of an extremely large, ligand-induced conformational change in plasminogen. Science 248, 69–73 [DOI] [PubMed] [Google Scholar]
- 41. Ponting C. P., Holland S. K., Cederholm-Williams S. A., Marshall J. M., Brown A. J., Spraggon G., Blake C. C. (1992) The compact domain conformation of human Glu-plasminogen in solution. Biochim. Biophys. Acta 1159, 155–161 [DOI] [PubMed] [Google Scholar]
- 42. Weisel J. W., Nagaswami C., Korsholm B., Petersen L. C., Suenson E. (1994) Interactions of plasminogen with polymerizing fibrin and its derivatives, monitored with a photoaffinity cross-linker and electron microscopy. J. Mol. Biol. 235, 1117–1135 [DOI] [PubMed] [Google Scholar]
- 43. Cockell C. S., Marshall J. M., Dawson K. M., Cederholm-Williams S. A., Ponting C. P. (1998) Evidence that the conformation of unliganded human plasminogen is maintained via an intramolecular interaction between the lysine-binding site of kringle 5 and the N-terminal peptide. Biochem. J. 333, 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoylaerts M., Rijken D. C., Lijnen H. R., Collen D. (1982) Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J. Biol. Chem. 257, 2912–2919 [PubMed] [Google Scholar]
- 45. Wohl R. C., Summaria L., Robbins K. C. (1980) Kinetics of activation of human plasminogen by different activator species at pH 7.4 and 37 °C. J. Biol. Chem. 255, 2005–2013 [PubMed] [Google Scholar]
- 46. Violand B. N., Byrne R., Castellino F. J. (1978) The effect of α-,ω-amino acids on human plasminogen structure and activation. J. Biol. Chem. 253, 5395–5401 [PubMed] [Google Scholar]
- 47. Violand B. N., Castellino F. J. (1976) Mechanism of the urokinase-catalyzed activation of human plasminogen. J. Biol. Chem. 251, 3906–3912 [PubMed] [Google Scholar]
- 48. Panizzi P., Boxrud P. D., Verhamme I. M., Bock P. E. (2006) Binding of the COOH-terminal lysine residue of streptokinase to plasmin(ogen) kringles enhances formation of the streptokinase·plasmin(ogen) catalytic complexes. J. Biol. Chem. 281, 26774–26778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lerch P. G., Rickli E. E., Lergier W., Gillessen D. (1980) Localization of individual lysine-binding regions in human plasminogen and investigations on their complex-forming properties. Eur. J. Biochem. 107, 7–13 [DOI] [PubMed] [Google Scholar]
- 50. Markus G., DePasquale J. L., Wissler F. C. (1978) Quantitative determination of the binding of ϵ-aminocaproic acid to native plasminogen. J. Biol. Chem. 253, 727–732 [PubMed] [Google Scholar]
- 51. Thewes T., Constantine K., Byeon I. J., Llinás M. (1990) Ligand interactions with the kringle 5 domain of plasminogen. A study by 1H NMR spectroscopy. J. Biol. Chem. 265, 3906–3915 [PubMed] [Google Scholar]
- 52. Váradi A., Patthy L. (1981) Kringle 5 of human plasminogen carries a benzamidine-binding site. Biochem. Biophys. Res. Commun. 103, 97–102 [DOI] [PubMed] [Google Scholar]
- 53. Christensen U., Mølgaard L. (1992) Positive co-operative binding at two weak lysine-binding sites governs the Glu-plasminogen conformational change. Biochem. J. 285, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bock P. E. (1992) Active-site-selective labeling of blood coagulation proteinases with fluorescence probes by the use of thioester peptide chloromethyl ketones. I. Specificity of thrombin labeling. J. Biol. Chem. 267, 14963–14973 [PubMed] [Google Scholar]
- 55. Bock P. E. (1992) Active-site-selective labeling of blood coagulation proteinases with fluorescence probes by the use of thioester peptide chloromethyl ketones. II. Properties of thrombin derivatives as reporters of prothrombin fragment 2 binding and specificity of the labeling approach for other proteinases. J. Biol. Chem. 267, 14974–14981 [PubMed] [Google Scholar]
- 56. Verhamme I. M., Olson S. T., Tollefsen D. M., Bock P. E. (2002) Binding of exosite ligands to human thrombin. Re-evaluation of allosteric linkage between thrombin exosites I and II. J. Biol. Chem. 277, 6788–6798 [DOI] [PubMed] [Google Scholar]
- 57. Panizzi P., Friedrich R., Fuentes-Prior P., Kroh H. K., Briggs J., Tans G., Bode W., Bock P. E. (2006) Novel fluorescent prothrombin analogs as probes of staphylocoagulase-prothrombin interactions. J. Biol. Chem. 281, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bock P. E., Day D. E., Verhamme I. M., Bernardo M. M., Olson S. T., Shore J. D. (1996) Analogs of human plasminogen that are labeled with fluorescence probes at the catalytic site of the zymogen. Preparation, characterization, and interaction with streptokinase. J. Biol. Chem. 271, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 59. Verhamme I. M., Bock P. E. (2008) Rapid-reaction kinetic characterization of the pathway of streptokinase-plasmin catalytic complex formation. J. Biol. Chem. 283, 26137–26147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deutsch D. G., Mertz E. T. (1970) Plasminogen: purification from human plasma by affinity chromatography. Science 170, 1095–1096 [DOI] [PubMed] [Google Scholar]
- 61. Castellino F. J., Powell J. R. (1981) Human plasminogen. Methods Enzymol. 80, 365–378 [DOI] [PubMed] [Google Scholar]
- 62. Bock P. E., Craig P. A., Olson S. T., Singh P. (1989) Isolation of human blood coagulation a-factor Xa by soybean trypsin inhibitor-Sepharose chromatography and its active-site titration with fluorescein mono-p-guanidinobenzoate. Arch. Biochem. Biophys. 273, 375–388 [DOI] [PubMed] [Google Scholar]
- 63. Laha M., Panizzi P., Nahrendorf M., Bock P. E. (2011) Engineering streptokinase for generation of active site-labeled plasminogen analogs. Anal. Biochem. 415, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sjöholm I. (1973) Studies on the conformational changes of plasminogen induced during activation to plasmin and by 6-aminohexanoic acid. Eur. J. Biochem. 39, 471–479 [DOI] [PubMed] [Google Scholar]
- 65. Taylor F. B., Jr., Botts J. (1968) Purification and characterization of streptokinase with studies of streptokinase activation of plasminogen. Biochemistry 7, 232–242 [DOI] [PubMed] [Google Scholar]
- 66. Jackson K. W., Tang J. (1982) Complete amino acid sequence of streptokinase and its homology with serine proteases. Biochemistry 21, 6620–6625 [DOI] [PubMed] [Google Scholar]
- 67. Johnson K. A. (2009) Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol. 467, 601–626 [DOI] [PubMed] [Google Scholar]
- 68. Johnson K. A., Simpson Z. B., Blom T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 69. Johnson K. A., Simpson Z. B., Blom T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 70. Tsodikov O. V., Record M. T., Jr. (1999) General method of analysis of kinetic equations for multistep reversible mechanisms in the single-exponential regime: application to kinetics of open complex formation between Eσ70 RNA polymerase and λP(R) promoter DNA. Biophys. J. 76, 1320–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bean R. R., Verhamme I. M., Bock P. E. (2005) Role of the streptokinase α-domain in the interactions of streptokinase with plasminogen and plasmin. J. Biol. Chem. 280, 7504–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chang Y., Mochalkin I., McCance S. G., Cheng B., Tulinsky A., Castellino F. J. (1998) Structure and ligand binding determinants of the recombinant kringle 5 domain of human plasminogen. Biochemistry 37, 3258–3271 [DOI] [PubMed] [Google Scholar]
- 73. Christensen U. (1984) The AH-site of plasminogen and two C-terminal fragments. A weak lysine-binding site preferring ligands not carrying a free carboxylate function. Biochem. J. 223, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ho-Tin-Noé B., Rojas G., Vranckx R., Lijnen H. R., Anglés-Cano E. (2005) Functional hierarchy of plasminogen kringles 1 and 4 in fibrinolysis and plasmin-induced cell detachment and apoptosis. FEBS J. 272, 3387–3400 [DOI] [PubMed] [Google Scholar]
- 75. Váli Z., Patthy L. (1982) Location of the intermediate and high affinity ω-aminocarboxylic acid-binding sites in human plasminogen. J. Biol. Chem. 257, 2104–2110 [PubMed] [Google Scholar]
- 76. McCance S. G., Castellino F. J. (1995) Contributions of individual kringle domains toward maintenance of the chloride-induced tight conformation of human glutamic acid-1 plasminogen. Biochemistry 34, 9581–9586 [DOI] [PubMed] [Google Scholar]
- 77. Sanderson-Smith M. L., Walker M. J., Ranson M. (2006) The maintenance of high affinity plasminogen binding by group A streptococcal plasminogen-binding M-like protein is mediated by arginine and histidine residues within the a1 and a2 repeat domains. J. Biol. Chem. 281, 25965–25971 [DOI] [PubMed] [Google Scholar]
- 78. Cook S. M., Skora A., Gillen C. M., Walker M. J., McArthur J. D. (2012) Streptokinase variants from Streptococcus pyogenes isolates display altered plasminogen activation characteristics: implications for pathogenesis. Mol. Microbiol. 86, 1052–1062 [DOI] [PubMed] [Google Scholar]
- 79. Kalia A., Bessen D. E. (2004) Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J. Bacteriol. 186, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang Y., Liang Z., Hsueh H. T., Ploplis V. A., Castellino F. J. (2012) Characterization of streptokinases from group A streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M protein and cluster 2b streptokinase. J. Biol. Chem. 287, 42093–42103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cook S. M., Skora A., Walker M. J., Sanderson-Smith M. L., McArthur J. D. (2014) Site-restricted plasminogen activation mediated by group A streptococcal streptokinase variants. Biochem. J. 458, 23–31 [DOI] [PubMed] [Google Scholar]