

Abstract
The development of efficient methods for accessing fluorinated functional groups is desirable. Herein, we report a two-step method that utilizes catalytic Cu for the decarboxylative trifluoromethylation of propargyl bromodifluoroacetates. This protocol affords a mixture of propargyl trifluoromethanes and trifluoromethyl allenes.
Keywords: copper, catalysis, trifluoromethylation, decarboxylation, alkynes
The development of methods that enable the incorporation of the trifluoromethyl group into organic compounds can impact agricultural chemistry,1 chemical biology,2 material sciences,3 and medicinal chemistry.2 Among the numerous approaches for trifluoromethylation,4 copper(0) and copper salts are frequently employed to both generate and harness reactive CF3 complexes. In recent years, improved methods have enabled the generation of copper–trifluoromethyl (Cu–CF3) species from common starting materials, including R3Si–CF3,5 trifluoromethane (CHF3),6 halodifluoroacetates,7 and S-(trifluoromethyl)diarylsulfonium salts.8 In these reactions, Cu–CF3 complexes typically display excellent functional group compatibility, and can be used in the presence of hard electrophiles, such as aldehydes and ketones.6 Further, these species tolerate high temperatures7b–f and the presence of protic solvents, including water.9
Given these benefits, the reaction of Cu–CF3 species10 with activated electrophiles can provide trifluoromethanes under mild conditions. A range of allyl, benzyl, propargyl and aromatic electrophiles react with Cu–CF3 complexes to provide trifluoromethane-containing products (Scheme 1).5–8 While the use of stoichiometric Cu enables a variety of important transformations,4 the principles of green chemistry encourage the development of trifluoromethylation reactions that only utilize catalytic quantities of copper.11
Scheme 1.

Generation of Cu–CF3 from various reagents enables the synthesis of trifluoromethane-containing products
The conversion of propargyl electrophiles (bromides, chlorides, mesylates, and trifluoroacetates) into trifluoromethanes represents one such transformation. Several methods that utilize stoichiometric quantities of Cu–CF3 have been recently reported (Scheme 2, eq 1–3).12 Depending upon the nature of the substrate and the Cu–CF3 species, two classes of products were obtained: propargyl trifluoromethanes, and trifluoromethyl allenes. Most commonly, primary propargyl electrophiles yielded propargyl trifluoromethanes (Scheme 2, eq 1), whereas secondary substrates provided trifluoromethyl allenes (Scheme 2, eq 2).12 Propargyl trifluoromethanes were also accessed from secondary propargyl chlorides; however, the reaction proceeded via the initial formation of trifluoromethyl allene, followed by a rearrangement that afforded a propargyl trifluoromethane (Scheme 2, eq 3).12b In addition to these copper-mediated reactions, an alternate copper-catalyzed trifluoromethylation employed copper(I) thiophenes-2-carboxylate (CuTC) and trimethyl(trifluoromethyl)silane (TMS–CF3) with potassium fluoride (KF) as an activator (Scheme 2, eq 4 and 5).13 The regioselectivity of this transformation was dictated by the substrate, with primary propargyl chlorides providing propargyl trifluoromethanes (Scheme 2, eq 4), and secondary propargyl chlorides affording trifluoromethyl allenes (Scheme 2, eq 5).13
Scheme 2.

Methods for the conversion of propargyl electrophiles into trifluoromethanes
In contrast, alternate electrophiles for nucleophilic substitution include propargyl halodifluoroacetates, which undergo decarboxylative trifluoromethylation upon treatment with stoichiometric copper(I) iodide (CuI).14 However, only a single example of this transformation exists, which converts propargyl chlorodifluoroacetate to trifluoromethyl allene (Scheme 2, eq 6).14 While this strategy utilized decarboxylation as an effective method to generate reactive fluorinated species, the use of stoichiometric copper(I) iodide encourages the development of a catalytic process.
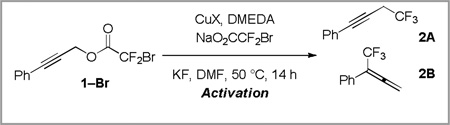
In order to establish whether this strategy could be expanded more generally to substituted propargyl substrates, we subjected 3-phenylpropynyl chlorodifluoroacetate (1–Cl) to the previously reported conditions utilizing stoichiometric copper(I) iodide.14 Interestingly, this reaction provided a 1.7:1 mixture of propargyl (2A) and allenyl (2B) products (Scheme 3). With the goal of developing a catalytic variant of the reaction, subjecting 1–Cl to similar conditions with 10 mol % of copper(I) iodide provided a low yield of trifluoromethylated product (Scheme 3).
Scheme 3.

Decarboxylative trifluoromethylation provides a mixture of products
Given the poor reactivity of chlorodifluoroacetates compared to bromodifluoroacetates,7a, 14 the reaction of 3-phenylpropynl bromodifluoroacetate (1–Br) was explored. Promotion of the reaction with stoichiometric copper provided 57% of the trifluoromethylated product with 2.6:1 regioselectivity (Table 1, entry 1). In contrast, catalytic turnover was realized using just 10 mol % copper(I) iodide, providing the trifluoromethylated product in 65% yield (Table 1, entry 2). Based on previous work in our laboratory,7a we hypothesized that the addition of N,N’-dimethylethylenediamine (DMEDA), and the use of an activation procedure might improve the yield of product. While the use of DMEDA alone was detrimental to the reaction (Table 1, entry 3), possibly because of the uncatalyzed reaction of the amine with the substrate, the employment of DMEDA, sodium bromo(difluoro)acetate (NaO2CCF2Br) and an activation procedure7a provided 75% of trifluoromethane-containing product and a 2.7 : 1 ratio of 2A:2B (Table 1, entry 4). Heating copper(I) iodide, DMEDA, KF, and NaO2CCF2Br in N,N-dimethylformamide at 50 °C for 10 minutes prior to the addition of substrate may facilitate the formation of an active (DMEDA)Cu–CF3 species (Scheme 4) and circumvent an induction period during which the substrate could be destroyed via non-productive pathways.
Table 1.
Catalytic Decarboxylative Trifluoromethylation Improved by DMEDA and an Activation Procedurea
 | ||||
|---|---|---|---|---|
| Entry | CuX (mol %) |
DMEDA (mol %) |
Activationb | % Yieldc (A : B)d |
| 1e | I (100) | 0 | – | 57 (2.6 : 1) |
| 2 | I (10) | 0 | – | 65 (3.3 : 1) |
| 3 | I (10) | 10 | – | 51 (3.6 : 1) |
| 4 | I (10) | 10 | √ | 75 (2.7 : 1) |
| 5 | TC (10) | 10 | √ | 52 (2.7 : 1) |
| 6f,g | TC (5) | 0 | – | <5 (N.D.) |
| 7f,h | TC (5) | 0 | – | 0 (–) |
Reactions were performed with 1-Br (0.20 mmol), and KF (0.40 mmol) in DMF (0.20 mL).
Activation involved heating CuI, DMEDA, NaO2CCF2Br, and KF in DMF for 10 min prior to injection of 1-Br.
Combined yield of 2A and 2B as determined by 19F NMR analysis, using α,α,α-trifluorotoluene as an internal standard.
Determined by 19F NMR spectroscopic analysis. ND = not determined.
DMF (0.60 mL).
KF (0.30 mmol), THF (1.2 mL), 20 h.
TMSCF3 (0.30 mmol) was added to the reaction.
75% of 1-Br remained, as determined by 19F NMR spectroscopic analysis.
Scheme 4.

Activation provides access into the proposed catalytic cycle
Attempted optimization of several other parameters did not lead to an improvement in the yield or selectivity. A broad screen of N- and O-based ligands did not result in increased yields or selectivity for the formation of 2A. The regioselectivity of the reaction was not influenced dramatically by temperature, and isomerization was not observed upon prolonged heating. Incomplete conversion of starting material was observed at 8–10-hour time points; therefore, an extended reaction time of 14 h was selected for the general reaction conditions. In addition, various control reactions were conducted to probe the use of copper(I) thiophenes-2-carboxylate as a catalyst for the present reaction. This salt has been employed for the regioselective conversion of propargyl chlorides to propargyl trifluoromethanes.13 Treatment of 1-Br with copper(I) thiophenes-2-carboxylate provided a decreased yield (52%) and similar regiochemical outcome (2.7:1) (Table 1, entry 5). Further, subjection of 1-Br to the exact conditions that facilitated the conversion of propargyl chlorides to trifluoromethanes [TMSCF3 (1.5 equiv) and KF (1.5 equiv) in THF at 60 °C for 20 h] formed less than 5% of desired material, which demonstrates that there are inherent differences in the reactivity of propargyl chlorides and bromodifluoroacetates (Table 1, entry 6). When this reaction was conducted in the absence of TMSCF3, only 25% conversion of 1 occurred, which suggests that decarboxylation does not occur under these conditions (Table 1, entry 7). Based on the results in entries 5–7, we hypothesize that the selection of appropriate solvent is critical for the present reaction.
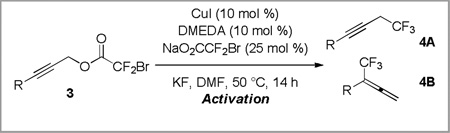
The copper(I) iodide/DMEDA-catalyzed trifluoromethylation of propargyl bromodifluoroacetates (3) tolerates many useful and important functional groups. Electron-donating aryl ethers provided trifluoromethane-containing products in moderate yield (Table 2, entries 1 and 2). A variety of carbonyl containing functional groups were compatible with the reaction conditions, including: esters, ketones, carbamates, and trifluoroacetamides (Table 2, entries 3–6). In addition, the successful reaction of the trifluoroacetamide provided the desired product, albeit in low yield, which provides additional evidence that Cu–CF3 species tolerate protic functional groups (Table 2, entry 6).9 The present trifluoromethylation reaction was conducted on an increased scale (7 mmol), and provided a typical yield according to 19F NMR spectroscopy (Table 2, entry 9). In addition to aromatic substrates, an aliphatic substrate afforded trifluoromethylated product in moderate yield, and displayed distinct regioselectivity compared to the aromatic substrates (Table 2, entry 10). Based on the similarity of propargyl bromodifluoroacetates and cinnamyl bromodifluoroacetates, and the identical catalyst systems employed for decarboxylative trifluoromethylation, it is anticipated that other functional groups, including aryl bromides and triflates, thiophenes, anilines, and phthalimides should be tolerated under the reaction conditions.7a While attempts were made to separate regioisomeric products, we were unable to achieve sufficient separation via standard silica gel chromatography to enable practical isolation of pure products.
Table 2.
Copper(I) Iodide/DMEDA-Catalyzed Reaction Tolerating Important Functional Groupsa
 | |||
|---|---|---|---|
| Entry | Product | Yield (%)b | A:Bc |
| 1 | 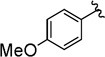 |
72 (77) | 4.0:1 (3.5:1) |
| 2 | 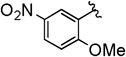 |
70 (78) | 2.1:1 (2.3:1) |
| 3 | 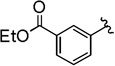 |
78 (83) | 2.3:1 (2.6:1) |
| 4 | 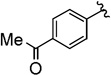 |
66 (70) | 2.6:1 (2.9:1) |
| 5 | 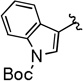 |
59 (62) | 3.0:1 (2.9:1) |
| 6 | 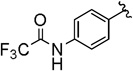 |
40 (44) | 6.3:1 (3.4:1) |
| 7 | 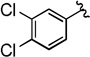 |
79 (84) | 2.2:1 (2.5:1) |
| 8 | 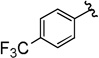 |
70 (73) | 1.7:1 (1.8:1) |
| 9d | 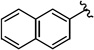 |
57 (77) | 3.8:1 (2.2:1) |
| 10 | 70 (69) | 1:2.1 (1:1.6) | |
Reactions were performed with 3 (0.20 mmol), CuI (0.020 mmol), DMEDA (0.020 mmol), NaO2CCF2Br (0.050 mmol), and KF (0.40 mmol) in DMF (0.20 mL) at 50 °C for 14 h following 10 min activation period.
Isolated yield of a purified mixture of regioisomers 4A and 4B; the figure in parentheses represents the combined yield of 4A and 4B as determined by 19F NMR spectroscopic analysis, using α,α,α-trifluorotoluene as an internal standard.
Ratio of regioisomers in the isolated material as determined by 1H NMR spectroscopic analysis; the ratio in parentheses represents the ratio of isomers in the crude reaction mixture as determined by 19F NMR spectroscopic analysis.
Reaction conducted on a 7 mmol scale.
Using the standard reaction conditions, a secondary propargyl substrate was less reactive than primary substrates, and provided 16% of trifluoromethylated product after 12 hours at 50 °C. However, under more forcing conditions (70 °C, 24 h), both propargyl trifluoromethane 6A and trifluoromethyl allene 6B were formed (Scheme 5). For the reactions of propargyl bromodifluoroacetates, both primary and secondary substrates provided similar regiochemical outcomes, and propargylic trifluoromethanes were observed as the major product (Scheme 5). In contrast, previous copper-catalyzed trifluoromethylation reactions of propargyl electrophiles displayed substrate-dependent regioselectivity, with primary electrophiles providing propargyl trifluoromethanes, and secondary electrophiles yielding trifluoromethyl allenes (Scheme 2, eq 4 and 5).13
Scheme 5.

Copper-catalyzed decarboxylative trifluoromethylation of secondary propargyl bromodifluoroacetates displays atypical reactivity. The ratio of products represents an average of multiple runs
The present copper/DMEDA-based catalyst system demonstrated unique reactivity compared to other copper-based catalyst systems. Several Cu–CF3 complexes commonly react with aryl iodides under mild reaction conditions to furnish trifluoromethylarenes.5, 15 In order to determine whether propargylic trifluoromethylation could be selectively achieved in the presence of aryl iodides, an exogenous aryl iodide was added to a standard decarboxylative trifluoromethylation reaction.16 The addition of one equivalent of aryl iodide had no effect on the yield or selectivity of the reaction (Scheme 6, eq 1). GC analysis of the reaction revealed that 92% of aryl iodide 7 remained unconsumed. In addition, less than 1% of trifluoromethylarene 8 was observed, which demonstrates the unique reactivity of this system. In order to confirm that substrates containing aryl iodides were compatible with the reaction conditions, 4-iodophenylpropynyl bromodifluoroacetate (9) was subjected to decarboxylative trifluoromethylation. As expected, a good combined yield (80%) of trifluoromethylated products 10A and 10B was obtained with typical regioselectivity (2.1:1, Scheme 6, eq 2). Again, only trace amounts of aromatic trifluoromethyl products 4A-8 and 4B-8 (see Table 2, entry 8) were observed.
Scheme 6.

Propargylic trifluoromethylation is accomplished selectively in the presence of aryl iodides
In conclusion, a two-step, copper-catalyzed protocol enables the conversion of propargyl bromodifluoroacetic esters into a mixture of propargyl trifluoromethanes and trifluoromethyl allenes. This decarboxylative strategy utilizes the combination of bromo(difluoro)acetate and potassium fluoride as an attractive trifluoromethylation reagent that produces carbon dioxide as a benign, easily separable byproduct. For the copper-catalyzed trifluoromethylation, the use of DMEDA as a ligand, and an activation procedure, helped establish the catalyst system. Ongoing work in our laboratory aims to develop more selective and efficient catalyst systems for the current trifluoromethylation reaction, as well as other related fluoroalkylation reactions.
Experimental Section
Unless otherwise noted, chemicals were purchased from commercial sources and used without further purification. Potassium fluoride (spray-dried) was ground to a fine powder with a mortar and pestle and dried in a vacuum oven (180 °C) for a minimum of 24 h prior to use. Dry solvents were used directly from a solvent purification system, in which solvent was dried by passage through two columns of activated alumina under argon, or purchased from commercial sources in Sure-Seal® bottles. All reactions were conducted under an atmosphere of dry N2 using oven-dried glassware. Trifluoromethylation reactions were performed in resealable 15 mL test tubes sealed with PTFE septa, and all other reactions were performed in round bottom flasks sealed with rubber septa. Reactions were monitored by thin-layer chromatography using Analtech UNIPLATETM Silica Gel HLF 250 micron glass plates precoated with 230–400 mesh silica impregnated with a fluorescent indicator (250 nm), visualizing with fluorescence quenching or p-anisaldehyde solution. Flash column chromatography was performed using a CombiFlash® RF–4x purification system. Silica gel was purchased from Sorbent Technologies (cat. # 30930M-25, 60 Å, 40–63 µm). Yields of products reported in the experimental section refer to the isolated yield of a single experiment. 19F NMR yields reported in tables were determined using α,α,α-trifluorotoluene (TFT) as an internal standard, and represent the average of at least two independent runs. Uncorrected melting points were measured on a Thomas Hoover Capillary Melting Point apparatus. Infrared spectra were recorded using a Shimadzu FTIR-8400S Fourier Transform Infrared Spectrometer. 1H NMR spectra were recorded on a Bruker 400 Avance spectrometer (400 MHz) or a Bruker 500 Avance spectrometer (500 MHz). 13C NMR spectra were recorded on a Bruker 500 Avance spectrometer (126 MHz). 19F NMR spectra were recorded on a Bruker 400 Avance spectrometer (376 MHz). Chemical shifts (δ) for protons are reported in parts per million (ppm) downfield from tetramethylsilane, and are referenced to the proton resonance of residual CHCl3 in the NMR solvent (δ = 7.27 ppm). Chemical shifts for carbon are reported in parts per million downfield from tetramethylsilane, and are referenced to the carbon resonances of the solvent peak (δ = 77.16 ppm). Chemical shifts for fluorine are reported in parts per millions, and are referenced to α,α,α-trifluorotoluene (δ = −63.72 ppm). Low-resolution mass spectra were recorded on a Shimatzu GCMS-QP2010 SE mass spectrometer. High-resolution mass spectra were recorded on a Waters LCT PremierTM mass spectrometer in the ESI mode.
Propargyl Alcohols; Representative Procedure
An oven-dried 25 mL round bottom flask was charged with CuI (76 mg, 0.40 mmol) and Pd(PPh3)2Cl2 (0.14 g, 0.20 mmol). The flask was sealed and then evacuated and backfilled with N2 three times. MeCN (0.010 L) and 4-iodoanisole (2.3 g, 0.010 mol) were injected, and the suspension was cooled to −10 °C. NEt3 (6.3 mL, 45 mmol) was added dropwise, and the mixture was stirred for 10 min. Propargyl alcohol (0.64 mL, 11 mmol) was injected dropwise, and then the reaction was allowed to warm to 23 °C. After 4 h, the solvent was removed in vacuo, and the crude mixture was dissolved in EtOAc (60 mL). The solution was passed through a pad of silica, which was washed with additional EtOAc (3×30 mL). Further chromatographic purification (hexanes–EtOAc 1:0→4:1) afforded 3-(4-methoxyphenyl)prop-2-yn-1-ol as a pale yellow solid (1.56 g, 96%).
Mp 69–70 °C (lit.17 74–75).
1H NMR (400 MHz, CDCl3): δ = 7.42–7.35 (m, 2 H), 6.89−6.81 (m, 2 H), 4.49 (d, J = 6.1 Hz, 2 H), 3.82 (s, 3 H), 1.64 (t, J = 6.1 Hz, 1 H).
Propargyl Bromodifluoroacetates; Representative Procedure
An oven-dried single-neck round-bottom flask (flask 1) was charged with bromodifluoroacetic acid (0.74 g, 4.2 mmol), and the system was attached to a bubbler. DCM (0.010 L) and DMF (0.070 mL, 0.90 mmol) were injected, and then oxalyl chloride (0.33 mL, 3.9 mmol) was added dropwise. In a separate oven-dried 2-neck round-bottom flask (flask 2), 3-(4-methoxyphenyl)prop-2-yn-1-ol (0.49 g, 3.0 mmol), NEt3 (0.84 mL, 6.0 mmol) and DCM (0.010 L) were combined, and the system was attached to a bubbler via a glass adapter. The solution was cooled to 0 °C, and then the solution in flask 1 was transferred to flask 2 via cannula. The reaction was allowed to warm to 23 °C and stirred for 3 h. The mixture was diluted with DCM (30 mL) and washed with 1 M HCl (25 mL), H2O (25 mL), and brine (25 mL). The organic solution was dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. Chromatographic purification (hexanes–EtOAc 19:1) afforded 3-(4-methoxyphenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate as a colorless oil (540 mg, 56%).
IR (film): 3010, 2839, 1780, 1606, 1510, 1290, 1249, 1172, 1120, 1031, 946, 833, 709, 603 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.47−7.38 (m, 2 H), 6.91−6.79 (m, 2 H), 5.16 (s, 2 H), 3.83 (s, 3 H).
13C NMR (126 MHz, CDCl3): δ = 160.4, 159.2 (t, J = 31.9 Hz), 133.7, 114.1, 113.5, 108.6 (t, J = 314.4 Hz), 88.9, 79.1, 56.8, 55.4.
19F NMR (376 MHz, CDCl3): δ = −62.30 (s, 2 F).
HRMS (EI): m/z [M]+ calcd for C12H9BrF2O3: 317.9703; found: 317.9700.
Trifluoromethane-Containing Compounds; General Procedure
KF (23 mg, 0.40 mmol) was added to a resealable 15 mL test tube and dried in a vacuum oven for a minimum of 24 h. The test tube was removed from the oven, sealed with a PTFE septum, and cooled under N2. CuI (3.8 mg, 0.020 mmol) and NaO2CCF2Br (9.8 mg, 0.050 mmol) were added, and the test tube was evacuated and backfilled with N2 three times. DMEDA (2.2 µL, 0.020 mmol) and DMF (0.20 mL) were injected into the test tube, which was placed into an oil bath at 50 °C. The mixture was heated for 10 min, during which bubbling was observed and the solution changed from teal/blue to yellow. Next, propargyl bromodifluoroacetate (0.20 mmol) was injected into the test tube, and heating was maintained for 14 h. The mixture was diluted with EtOAc (3 mL), and TFT (24.6 µL, 0.200 mmol) was added as an internal standard. An aliquot was removed, and a 19F NMR spectrum was obtained. The aliquot was recombined, and the reaction mixture was diluted further with EtOAc (15 mL). The organic solution was washed with aq NH4Cl solution (10 mL) and brine (10 mL), dried over anhydrous Na2SO4, filtered, and the solvent was removed in vacuo. Chromatographic purification afforded a mixture of propargyl trifluoromethane (A) and trifluoromethyl allene (B). The ratio or regioisomers was determined by 1H NMR spectroscopy (propargylic CH2/terminal CH2 of allene). Note: the following numbering system is used for compounds 4: 4A/B-x, where×is an integer referring to the specific entry in Table 2.
Compounds 4A/B-113,18
The general procedure was followed using 3-(4-methoxyphenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (64 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→49:1) afforded a mixture of regioisomers as a yellow oil (31 mg, 72%). Analysis of the 1H NMR spectrum revealed a 4.0:1 ratio of A/B.
1H NMR (400 MHz, CDCl3): δ = 7.43–7.34 (m, 4 H, A/B), 6.94–6.89 (m, 2 H, B), 6.88–6.82 (m, 2 H, A), 5.51 (q, J = 3.4 Hz, 2 H, B), 3.83 (s, 3 H, B), 3.82 (s, 3 H, A), 3.26 (q, J = 9.6 Hz, 2 H, A).
19F NMR (376 MHz, EtOAc): δ = −61.76 (t, J = 3.6 Hz, 3 F, B), −67.76 (t, J = 10.0 Hz, 3 F, A).
Compounds 4A/B-2
The general procedure was followed using 3-(2-methoxy-5-nitrophenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (73 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→3:1) afforded a mixture of regioisomers as a colorless solid (36 mg, 70%). Analysis of the 1H NMR spectrum revealed a 2.1:1 ratio of A/B.
Mp 76–81 °C.
IR (film): 3119, 3094, 2947, 2920, 2847, 1983, 1610, 1580, 1514, 1493, 1492, 1439, 1418, 1344, 1275, 1246, 1190, 1148, 1103, 1018, 968, 906, 891, 868, 833, 797, 750, 735, 694, 665, 638 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.31 (d, J = 2.8 Hz, 1 H, A), 8.28 (dd, J = 9.1, 2.8 Hz, 1 H, B), 8.24–8.20 (m, 2 H, A/B), 7.01 (d, J = 9.1 Hz, 1 H, B), 6.96 (d, J = 9.2 Hz, 1 H, A), 5.42 (q, J = 3.4 Hz, 2 H, B), 4.00 (s, 3 H, A), 3.96 (s, 3 H, B), 3.35 (q, J = 9.5 Hz, 2 H, A).
13C NMR (126 MHz, CDCl3): δ = 209.41 (q, J = 3.7 Hz, B), 164.97 (A), 162.43 (B), 141.23 (B), 141.09 (A), 129.59 (A), 126.67 (B), 126.51 (B), 126.21 (A), 124.10 (q, J = 277.0 Hz, A), 122.87 (q, J = 273.9 Hz, B), 119.90 (B), 112.56 (A), 110.98 (B), 110.48 (A), 95.74 (q, J = 37.2 Hz, B), 83.93 (q, J = 5.0 Hz, A), 82.19 (B), 78.61 (A), 56.79 (A), 56.58 (B), 27.15 (q, J = 34.9 Hz, A).
19F NMR (376 MHz, CDCl3): δ = −62.45 (t, J = 3.5 Hz, 3 F, B), −67.35 (t, J = 9.9 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C11H8F3NO3: 259.0; found: 259.0.
Compounds 4A/B-3
The general procedure was followed using ethyl 3-(3-(2-bromo-2,2-difluoroacetoxy)prop-1-yn-1-yl)benzoate (72 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→49:1) afforded a mixture of regioisomers as a pale green oil (0.040 g, 78%). Analysis of the 1H NMR spectrum revealed a 2.3:1 ratio of A/B.
IR (film): 3067, 2984, 2932, 2854, 1971, 1720, 1472, 1367, 1298, 1256, 1231, 1173, 1148, 1111, 1084, 1026, 908, 872, 754 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.16–8.09 (m, 2 H, A/B), 8.05–7.97 (m, 2 H, A/B), 7.66–7.60 (m, 2 H, A/B), 7.47 (t, J = 7.8 Hz, 1 H, B), 7.41 (t, J = 7.8 Hz, 1 H, A), 5.61 (q, J = 3.4 Hz, 2 H, B), 4.46–4.33 (m, 4 H, A/B), 3.30 (q, J = 9.5 Hz, 2H, A), 1.41 (t, J = 7.1 Hz, 6 H, A/B).
13C NMR (126 MHz, CDCl3): δ = 208.71 (q, J = 4.0 Hz, B), 166.23 (B), 165.92 (A), 136.02 (A), 133.06 (A), 131.27 (B), 131.26 (q, J = 1.3 Hz, B), 130.94 (A), 129.85 (A), 129.79 (B), 129.39 (B), 128.94 (B), 128.59 (A), 128.44 (B), 124.24 (q, J = 277.4 Hz, A), 123.28 (q, J = 273.9 Hz, B), 122.65 (A), 101.42 (q, J = 35.7 Hz, B), 84.23 (B), 83.56 (A), 78.59 (q, J = 5.1 Hz, A), 61.42 (A), 61.38 (B), 26.93 (q, J = 34.9 Hz, A), 14.46 (A/B).
19F NMR (376 MHz, CDCl3): δ = −61.59 (t, J = 3.6 Hz, 3 F, B), −67.51 (t, J = 10.0 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C13H11F3O2: 256.1; found: 256.1.
Compounds 4A/B-4
The general procedure was followed using 3-(4-acetylphenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (66 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→49:1) afforded a mixture of regioisomers as a yellow oil (0.030 g, 66%). Analysis of the 1H NMR spectrum revealed a 2.6:1 ratio of A/B.
IR (film): 3067, 2964, 2932, 2854, 1969, 1933, 1686, 1603, 1558, 1418, 1404, 1362, 1306, 1263, 1178, 1150, 1109, 1016, 957, 935, 906, 833, 717, 679, 628, 592 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.99–7.94 (m, 2 H, B), 7.94–7.89 (m, 2 H, A), 7.57–7.52 (m, 4 H, A/B), 5.64 (q, J = 3.3 Hz, 2 H, B), 3.32 (q, J = 9.5 Hz, 2 H, A), 2.62 (s, 3 H, B), 2.61 (s, 3 H, A).
13C NMR (126 MHz, CDCl3): δ = 209.23 (q, J = 3.9 Hz, B), 197.50 (B), 197.42 (A), 136.80 (A), 136.60 (B), 134.07 (B), 132.15 (A), 129.99 (A), 128.85 (B), 128.35 (A), 127.17 (q, J = 1.3 Hz, B), 127.08 (A), 124.13 (q, J = 277.4 Hz, A) 123.17 (q, J = 273.9 Hz, B), 101.72 (B), 84.45 (B), 83.74 (A), 80.99 (q, J = 5.1 Hz, A), 27.03 (q, J = 35.0 Hz, A), 26.81 (A), 26.78 (B).
19F NMR (376 MHz, EtOAc): δ = −63.30 to −63.53 (m, 3 F, B), −67.66 (t, J = 10.0 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C12H9F3O: 226.1; found: 226.1.
Compounds 4A/B-5
The general procedure was followed using tert-butyl 3-(3-(2-bromo-2,2-difluoroacetoxy)prop-1-yn-1-yl)-1H-indole-1-carboxylate (86 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→9:1) afforded a mixture of regioisomers as a viscous orange oil (38 mg, 59%). Analysis of the 1H NMR spectrum revealed a 3.0:1 ratio of A/B.
IR (film): 3159, 3055, 2980, 2932, 2851, 1740, 1558, 1475, 1454, 1420, 1375, 1357, 1308, 1279, 1234, 1256, 1234, 1111, 1049, 1032, 854, 831, 746, 729 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.19 (d, J = 8.5 Hz, 1 H, B), 8.15 (d, J = 8.2 Hz, 1 H, A), 7.87 (dt, J = 8.0, 1.0 Hz, 1 H, B), 7.79 (s, 1 H, A), 7.73 (s, 1 H, B), 7.68–7.60 (m, 1 H, A), 7.37 (td, J = 7.8, 1.4 Hz, 2 H, A/B), 7.31 (td, J = 7.5, 1.1 Hz, 1 H, A), 7.29–7.24 (m, 1 H, B), 5.69 (qd, J = 3.0, 0.9 Hz, 2 H, B), 3.37 (q, J = 9.6 Hz, 2 H, A), 1.70 (s, 9 H, B), 1.68 (s, 9 H, A).
13C NMR (126 MHz, CDCl3): δ = 208.94 (q, J = 3.7 Hz, B), 149.49 (B), 149.12 (A), 135.48 (B), 134.66 (A), 130.53 (A), 129.57 (A), 128.40 (B), 125.40 (A), 125.17 (B), 124.35 (q, J = 277.1 Hz, A), 124.21 (q, J = 1.3 Hz, B), 123.41 (A), 123.33 (q, J = 273.7 Hz, B), 123.07 (B), 120.07 (A), 119.93 (B), 115.46 (B), 115.40 (A), 107.96 (B), 102.44 (A), 95.70 (q, J = 36.1 Hz, B), 84.59 (A), 84.58 (A) 84.52 (B), 81.08 (q, J = 5.1 Hz, A), 76.64 (B) 28.30 (B), 28.28 (A), 27.20 (q, J = 34.8 Hz, A).
19F NMR (376 MHz, CDCl3): δ = −63.41 (t, J = 3.5 Hz, 3 F, B), −67.66 (t, J = 9.9 Hz, 3 F, A).
HRMS (ESI): m/z [2 M + Na]+ calcd for C34H32F6N2O4Na: 669.2164; found: 669.2179.
Compounds 4A/B-6
The general procedure was followed using 3-(4-(2,2,2-trifluoroacetamido)phenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (80 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–DCM 1:0→1:1) afforded a mixture of regioisomers as a colorless solid (24 mg, 40%). Analysis of the 1H NMR spectrum revealed a 6.3:1 ratio of A/B.
IR (film): 3300, 3202, 3136, 2964, 1705, 1607, 1547 1512, 1410, 1366, 1281, 1246, 1202, 1155, 1107, 959, 906, 839, 727, 704, 654 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.89 (s, 1 H, A), 7.83 (s, 1 H, B), 7.62–7.58 (m, 2 H, B), 7.58–7.54 (m, 2 H, A), 7.52–7.45 (m, 4 H, A/B), 5.60 (q, J = 3.4 Hz, 2 H, B), 3.29 (q, J = 9.5 Hz, 2 H, A).
13C NMR (126 MHz, CDCl3): δ = 208.65 (q, J = 4.4 Hz, B), 154.89 (q, J = 37.5 Hz, B),154.80 (q, J = 37.5 Hz, A), 135.38 (A), 135.00 (B), 133.10 (A), 128.16 (q, J = 1.5 Hz, B), 124.25 (q, J = 276.9 Hz, A), 123.29 (q, J = 275.6 Hz, B), 120.71 (A), 120.32 (B), 120.25 (A), 120.18 (B), 115.70 (q, J = 288.8, B), 115.69 (q, J = 288.8 Hz, A), 101.24 (q, J = 35.4 Hz, B), 84.25 (B), 83.51 (A), 78.48 (q, J = 5.0 Hz, A), 26.94 (q, J = 34.8 Hz, A).
19F NMR (376 MHz, CDCl3): δ = −61.55 (t, J = 3.0 Hz, 3 F, B), −67.35 (t, J = 9.5 Hz, 3 F, A), −76.69 (6 F, A/B).
MS (CI): m/z [M]+ calcd for C12H7F6NO: 295.0; found: 295.0.
Compounds 4A/B-7
The general procedure was followed using 3-(3,4-dichlorophenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (72 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes) afforded a mixture of regioisomers as a pale yellow oil (0.040 g, 79%). Analysis of the 1H NMR spectrum revealed a 2.2:1 ratio of A/B.
IR (film): 3074, 2928, 1973, 1533, 1475, 1466, 1364, 1352, 1281, 1254, 1178, 1151, 1130, 1111, 1034, 906, 881, 822 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.55 (d, J = 2.0 Hz, 1 H, A), 7.52 (d, J = 2.2 Hz, 1 H, B), 7.45 (d, J = 8.5 Hz, 1 H, B), 7.40 (d, J = 8.3 Hz, 1 H, A), 7.30–7.26 (m, 4 H, A/B), 5.63 (q, J = 3.3 Hz, 2 H, B), 3.28 (q, J = 9.5 Hz, 2 H, A).
13C NMR (126 MHz, CDCl3): δ = 208.60 (q, J = 3.9 Hz, B), 133.64 (A), 133.41 (A), 133.16 (B), 132.75 (A), 132.58 (B), 131.10 (A), 130.80 (B), 130.54 (A), 129.38 (B), 129.03 (q, J = 1.7 Hz, B), 126.30 (q, J = 1.7 Hz, B), 124.12 (q, J = 276.9 Hz, A), 123.02 (q, J = 273.1 Hz, B), 122.18 (A), 100.59 (q, J = 35.2 Hz, B), 84.69 (B), 82.28 (A), 79.80 (q, J = 5.1 Hz, A), 26.92 (q, J = 34.9 Hz, A).
19F NMR (376 MHz, EtOAc): δ = −61.59 (t, J = 3.6 Hz, 3 F, B), −67.51 (t, J = 10.0 Hz, 3 F, A).
HRMS (EI): m/z [M]+ calcd for C10H5Cl2F3: 251.9720; found: 251.9721.
Compounds 4A/B-8
The general procedure was followed using 3-(4-(trifluoromethyl)phenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (71 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→49:1) afforded a mixture of regioisomers as a colorless oil (35 mg, 70%). Analysis of the 1H NMR spectrum revealed a 1.7:1 ratio of A/B.
IR (film): 3063, 2934, 1971, 1927, 1618, 1406, 1366, 1329, 1281, 1267, 1151, 1130, 1105, 1068, 1018, 937, 906, 870, 843, 735, 723 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.67–7.53 (m, 8 H, A/B), 5.64 (q, J = 3.4 Hz, 2 H, B), 3.31 (q, J = 9.5 Hz, 2 H, A).
13C NMR (126 MHz, CDCl3): δ = 209.04 (q, J = 4.1 Hz, B), 133.10 (q, J = 1.6 Hz, B), 132.27 (A), 130.64 (q, J = 32.7 Hz, A), 130.36 (q, J = 32.7 Hz, B), 127.41 (q, J = 1.6 Hz, B), 126.06 (q, J = 1.7 Hz, A), 125.82 (q, J = 3.8 Hz, B), 125.42 (q, J = 3.9 Hz, A), 124.15 (q, J = 278.6 Hz, A), 124.02 (q, J = 272.2 Hz, B), 123.93 (q, J = 271.8 Hz, A), 123.13 (q, J = 274.8 Hz, B), 101.33 (q, J = 35.0 Hz, B), 84.47 (B), 83.21 (A), 80.23 (q, J = 5.1 Hz, A), 26.96 (q, J = 34.9 Hz, A).
19F NMR (376 MHz, EtOAc): δ = −61.50 (t, J = 3.5 Hz, 3 F, B), −63.79 (3 F, B), −63.93 (3 F, A), −67.42 (t, J = 9.9 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C11H6F6: 252.0; found: 252.0.
Compounds 4A/B-913, 18
The general procedure was followed using 3-(naphthalen-2-yl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (2.4 g, 7.0 mmol), CuI (130 mg, 0.70 mmol), DMEDA (75 µL, 0.70 mmol), NaO2CCF2Br (0.35 g, 1.8 mmol), KF (0.81 g, 14 mmol), with DMF (7.0 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→49:1) afforded a mixture of regioisomers as a pale yellow solid (0.93 g, 57%). Analysis of the 1H NMR spectrum revealed a 3.8:1 ratio of A/B.
Mp 42–45 °C.
1H NMR (400 MHz, CDCl3): δ = 8.00 (s, 1 H, A), 7.93 (s, 1 H, B), 7.88–7.77 (m, 6 H, A/B), 7.57–7.47 (m, 6 H, A/B), 5.63 (q, J = 3.3 Hz, 2 H, B), 3.34 (q, J = 9.6 Hz, 2 H, B).
19F NMR (376 MHz, CDCl3): δ = −61.30 (t, J = 3.6 Hz, 3 F, B), −67.56 (t, J = 10.1 Hz, 3 F, A).
Compounds 4A/B-10
The general procedure was followed using 5-phenylpent-2-yn-1-yl 2-bromo-2,2-difluoroacetate (63 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes) afforded a mixture of regioisomers as a tan oil (0.030 g, 70%). Analysis of the 1H NMR spectrum revealed a 1:2.1 ratio of A/B.
IR (film): 3088, 3065, 3030, 2932, 2862, 1985, 1954, 1605, 1497, 1454, 1366, 1333, 1281, 1261, 1200, 1157, 1115, 1055, 980, 908, 864, 744, 700 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.37–7.28 (m, 4 H, A/B), 7.26–7.19 (m, 6 H, A/B), 5.18 (sext, J = 3.6 Hz, 2 H, B), 3.01 (qt, J = 9.7, 2.4 Hz, 2 H, A), 2.90–2.73 (m, 4 H, A/B), 2.54–2.41 (m, 4 H, A/B).
13C NMR (126 MHz, CDCl3): δ = 206.73 (q, J = 4.1 Hz, B), 140.80 (B), 140.54 (A), 128.59 (A/B), 128.56 (B), 128.52 (A), 126.48 (A), 126.35 (B), 124.53 (q, J = 277.8 Hz, A), 123.94 (q, J = 274.5 Hz, B), 98.11 (q, J = 34.0 Hz, B), 84.28 (A), 82.54 (B), 69.21 (q, J = 5.1 Hz, A), 34.94 (A), 33.64 (B), 27.70 (B), 26.28 (q, J = 34.6 Hz, A), 20.99 (A).
19F NMR (376 MHz, CDCl3): δ = −61.60 (t, J = 3.7 Hz, 3 F, B), −67.54 (t, J = 10.0 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C12H11F3: 212.1; found: 212.1.
Compounds 6A/B
The general procedure was followed using 4-(3-nitrophenyl)but-3-yn-2-yl 2-bromo-2,2-difluoroacetate (70 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes–EtOAc 1:0→9:1) afforded a mixture of regioisomers as a yellow oil (0.020 g, 41%). Analysis of the 1H NMR spectrum revealed a 1.6:1 ratio of A/B (single run).
IR (film): 3090, 2961, 2926, 2856, 1963, 1535, 1481, 1441, 1352, 1327, 1248, 1178, 1155, 1119, 980, 964, 926, 901, 806, 739, 710, 687 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.41–8.37 (m, 1 H, A), 8.32–8.24 (m, 2 H, A/B), 8.20–8.14 (m, 1 H, B), 7.85–7.80 (m, 1 H, A), 7.79–7.73 (m, 1 H, B), 7.63–7.53 (m, 2 H, A/B), 6.08 (qt, J = 7.5, 3.1 Hz, 1 H, B), 4.43 (qq, J = 7.8, 2.5 Hz, 1 H, A), 1.97–1.93 (m, 6 H, A/B).
13C NMR (126 MHz, CDCl3): δ = 205.60 (q, J = 3.9 Hz, B), 148.66 (B), 148.47 (A), 135.54 (A), 134.62 (B), 132.72 (q, J = 1.8 Hz, B), 129.79 (B), 129.74 (A), 124.62 (A) 124.61 (B), 124.09 (q, J = 280.4 Hz, A), 124.00 (A), 123.07 (q, J = 274.8 Hz, B), 122.85 (A), 122.19 (q, J = 1.8 Hz, B), 100.12 (q, J = 35.3 Hz, B), 96.56 (B), 83.59 (A), 70.34 (q, J = 3.4 Hz, A), 43.30 (q, J = 31.8 Hz, A), 13.37 (B), 3.79 (A).
19F NMR (376 MHz, CDCl3): δ = −61.70 (s, 3 F, B), −71.74 (d, J = 7.5 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C11H8F3NO2: 243.1; found: 243.1.
Compounds 10A/B
The general procedure was followed using 3-(4-iodophenyl)prop-2-yn-1-yl 2-bromo-2,2-difluoroacetate (83 mg, 0.20 mmol), CuI (3.8 mg, 0.020 mmol), DMEDA (2.2 µL, 0.020 mmol), NaO2CCF2Br (9.8 mg, 0.050 mmol), KF (23 mg, 0.40 mmol), with DMF (0.20 mL) as solvent. Workup and chromatographic purification (hexanes) afforded a mixture of regioisomers as an amorphous tan solid (0.050 g, 80%). Analysis of the 1H NMR spectrum revealed a 2.0:1 ratio of A/B.
IR (film): 3065, 2978, 1961, 1541, 1485, 1391, 1366, 1319, 1279, 1263, 1254, 1173, 1148, 1111, 1061, 1007, 935, 906, 868, 820, 743, 665 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.73–7.69 (m, 2 H, B), 7.69–7.64 (m, 2 H, A), 7.21–7.15 (m, 4 H, A/B), 5.56 (q, J = 3.4 Hz, 2 H, B), 3.27 (q, J = 9.5 Hz, 2 H, A).
13C NMR (126 MHz, CDCl3): δ = 208.50 (q, J = 4.0 Hz, B), 138.00 (B), 137.66 (A), 133.47 (A/B), 128.87 (q, J = 1.5 Hz, B), 124.18 (q, J = 276.9 Hz, A), 123.19 (q, J = 273.6 Hz, B), 121.78 (A), 94.88 (A), 94.13 (B), 84.23 (A), 83.60 (B), 79.14 (q, J = 5.1 Hz, A), 26.99 (q, J = 34.9 Hz, A). Note: Terminal substituted carbon of allene B could not be distinguished from the baseline [expected to be a quartet (J ≈ 35 Hz) between δ 102–100].
19F NMR (376 MHz, EtOAc): δ = −61.60 (t, J = 3.7 Hz, 3 F, B), −67.54 (t, J = 10.0 Hz, 3 F, A).
MS (CI): m/z [M]+ calcd for C10H6F3I: 310.0; found: 310.0.
Supplementary Material
Acknowledgment
We thank the donors of the Herman Frasch Foundation for Chemical Research (701-HF12) and the American Chemical Society Petroleum Research Fund (52073-DNI1) for financial support, and the NIGMS Training Grant on Dynamic Aspects of Chemical Biology (T32 GM08545) for a graduate traineeship (B. R. A.). Further financial assistance from the University of Kansas Office of the Provost, Department of Medicinal Chemistry, and General Research Fund (2301795) is gratefully acknowledged. Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant # S10RR024664 and NSF Major Research Instrumentation Grant # 0320648.
Footnotes
Supporting Information for this article is available online at http://www.thiemeconnect.com/products/ejournals/journal/10.1055/s-00000084.
References
- 1.Theodoridis G. Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In: Tressaud A, editor. Fluorine and the Environment: Agrochemicals, Archaeology, Green Chemistry & Water. Amsterdam: Elsevier B.V.; 2006. [Google Scholar]
- 2.(a) Gouverneur V, Müller K, editors. Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications. London: Imperial College Press; 2012. [Google Scholar]; (b) Ojima I, editor. Fluorine in Medicinal Chemistry and Chemical Biology. West Sussex: Blackwell Publishing Ltd.; 2009. [Google Scholar]; (c) Bégué J-P, Bonnet-Delpon D, editors. Bioorganic and Medicinal Chemistry of Fluorine. Hoboken: John Wiley & Sons; 2008. [Google Scholar]
- 3.(a) Patil Y, Ameduri B. Prog. Polym. Sci. 2013;38:703. [Google Scholar]; (b) Dhara MG, Banerjee S. Prog. Polym. Sci. 2010;35:1022. [Google Scholar]
- 4.For some recent reviews, see: Liang T, Neumann CN, Ritter T. Angew. Chem. Int. Ed. 2013;52:8214. doi: 10.1002/anie.201206566. Liu H, Gu Z, Jiang X. Adv. Synth. Catal. 2013;355:617. Liu T, Shen Q. Eur. J. Org. Chem. 2012:6679. Studer A. Angew. Chem. Int. Ed. 2012;51:8950. doi: 10.1002/anie.201202624. Tomashenko OA, Grushin VV. Chem. Rev. 2011;111:4475. doi: 10.1021/cr1004293. Zheng Y, Ma J-A. Adv. Synth. Catal. 2010;352:2745.
- 5.(a) Morimoto H, Tsubogo T, Litvinas ND, Hartwig JF. Angew. Chem. Int. Ed. 2011;50:3793. doi: 10.1002/anie.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oishi M, Kondo H, Amii H. Chem. Commun. 2009:1909. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]; (c) Dubinina GG, Furutachi H, Vicic DA. J. Am. Chem. Soc. 2008;130:8600. doi: 10.1021/ja802946s. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lishchynskyi A, Novikov MA, Martin E, Escudero-Adán EC, Novák P, Grushin VV. J. Org. Chem. 2013;78:11126. doi: 10.1021/jo401423h. [DOI] [PubMed] [Google Scholar]; (b) Zanardi A, Novikox MA, Martin E, Benet-Buchholz J, Grushin VV. J. Am. Chem. Soc. 2011;133:20901. doi: 10.1021/ja2081026. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ambler BR, Altman RA. Org. Lett. 2013;15:5578. doi: 10.1021/ol402780k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen M, Buchwald SL. Angew. Chem. Int. Ed. 2013;52:11628. doi: 10.1002/anie.201306094. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schareina T, Wu X-F, Zapf A, Cotté A, Gotta M, Beller M. Top. Catal. 2012;55:426. [Google Scholar]; (d) Li Y, Chen T, Wang H, Zhang R, Jin K, Wang X, Duan C. Synlett. 2011;12:1713. [Google Scholar]; (e) McReynolds KA, Lewis RS, Ackerman LKG, Dubinina GG, Brennessel WW, Vicic DA. J. Fluorine Chem. 2010;131:1108. [Google Scholar]; (f) Langlois BR, Roques N. J. Fluorine Chem. 2007;128:1318. [Google Scholar]
- 8.(a) Dai J-J, Fang C, Xiao B, Yi J, Xu J, Liu Z-J, Fu Y. J. Am. Chem. Soc. 2013;135:8436. doi: 10.1021/ja404217t. [DOI] [PubMed] [Google Scholar]; (b) Lhu L, Liu S, Douglas JT, Altman RA. Chem.–Eur. J. 2013;19:12800. doi: 10.1002/chem.201302328. [DOI] [PubMed] [Google Scholar]; (c) Xu J, Fu Y, Luo D-F, Jiang Y-Y, Xiao B, Liu Z-J, Gong T-J, Liu L. J. Am Chem. Soc. 2011;133:15300. doi: 10.1021/ja206330m. [DOI] [PubMed] [Google Scholar]; (d) Kawai H, Furukawa T, Nomura Y, Tokunaga E, Shibata N. Org. Lett. 2011;13:3596. doi: 10.1021/ol201205t. [DOI] [PubMed] [Google Scholar]; (e) Zhang C-P, Wang Z-L, Chen Q-Y, Zhang C-T, Gu Y-C, Xiao J-C. Angew. Chem. Int. Ed. 2011;50:1896. doi: 10.1002/anie.201006823. [DOI] [PubMed] [Google Scholar]
- 9.Hu M, He Z, Gao B, Li L, Ni C, Hu J. J. Am. Chem. Soc. 2013;135:17302. doi: 10.1021/ja409941r. [DOI] [PubMed] [Google Scholar]
- 10.(a) Urata H, Fuchikami T. Tetrahedron Lett. 1991;32:91. [Google Scholar]; (b) Wiemers DM, Burton DJ. J. Am. Chem. Soc. 1986;108:832. [Google Scholar]; (c) Kobayashi Y, Yamamoto K, Kumadaki I. Tetrahedron Lett. 1979;42:4071. [Google Scholar]
- 11.Bryan MC, Dillon B, Hamann LG, Hughes GJ, Kopach ME, Peterson EA, Pourashraf M, Raheem I, Richardson P, Richter D, Sneddon HF. J. Med. Chem. 2013;56:6007. doi: 10.1021/jm400250p. [DOI] [PubMed] [Google Scholar]
- 12.(a) Kawai H, Furukawa T, Nomura Y, Tokunaga E, Shibata N. Org. Lett. 2011;13:3596. doi: 10.1021/ol201205t. [DOI] [PubMed] [Google Scholar]; (b) Zhao TSN, Szabó KJ. Org. Lett. 2012;14:3966. doi: 10.1021/ol3017287. [DOI] [PubMed] [Google Scholar]; (c) Jiang X, Qing F-L. Beilstein. J. Org. Chem. 2013;9:2862. doi: 10.3762/bjoc.9.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyake Y, Ota S-i, Shibata M, Nakajima K, Nishibayashi Y. Chem. Commun. 2013;49:7809. doi: 10.1039/c3cc44434a. [DOI] [PubMed] [Google Scholar]
- 14.Duan J-X, Chen Q-Y. J. Chem. Soc. Perkin Trans. 1. 1994:725. [Google Scholar]
- 15.(a) Mulder JA, Frutos RP, Patel ND, Qu B, Sun X, Tampone TG, Gao J, Sarvestani M, Eriksson MC, Haddad N, Shen S, Song JJ, Senanayake CH. Org. Process Res. Dev. 2013;17:940. [Google Scholar]; (b) Chen Q-Y, Wu S-W. J. Chem. Soc. Chem. Commun. 1989:705. [Google Scholar]
- 16.Collins KD, Glorius F. Nature Chem. 2013;5:597. doi: 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]
- 17.Paraskar AS, Sudalai A. Tetrahedron. 2006;62:5756. [Google Scholar]
- 18.Sam B, Montgomery TP, Krische MJ. Org. Lett. 2013;15:3790. doi: 10.1021/ol401771a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.