

Abstract
Background
Cardiomyocytes that differentiate from pluripotent stem cells (PSCs) provide a crucial cellular resource for cardiac regeneration. The mechanisms of mitochondrial metabolic and redox regulation for efficient cardiomyocyte differentiation are, however, still poorly understood. Here, we show that inhibition of the mitochondrial permeability transition pore (mPTP) by Cyclosporin A (CsA) promotes cardiomyocyte differentiation from PSCs.
Methods and Results
We induced cardiomyocyte differentiation from mouse and human PSCs and examined the effect of CsA on the differentiation process. The cardiomyogenic effect of CsA mainly resulted from mPTP inhibition rather than from calcineurin inhibition. The mPTP inhibitor NIM811, which does not have an inhibitory effect on calcineurin, promoted cardiomyocyte differentiation as much as CsA did, but calcineurin inhibitor FK506 only slightly increased cardiomyocyte differentiation. CsA‐treated cells showed an increase in mitochondrial calcium, mitochondrial membrane potential, oxygen consumption rate, ATP level, and expression of genes related to mitochondrial function. Furthermore, inhibition of mitochondrial oxidative metabolism reduced the cardiomyogenic effect of CsA while antioxidant treatment augmented the cardiomyogenic effect of CsA.
Conclusions
Our data show that mPTP inhibition by CsA alters mitochondrial oxidative metabolism and redox signaling, which leads to differentiation of functional cardiomyocytes from PSCs.
Keywords: metabolism, mitochondria, myocytes, redox, stem cells
Introduction
If not treated immediately, myocardial infarction can lead to permanent and irreversible loss of cardiomyocytes in the corresponding region of the heart. Substantial damage in the myocardium can manifest as reduced structural and functional capacity of the blood‐pumping organ, ultimately giving rise to congestive heart failure.1 Many researchers and clinicians are extensively investigating the feasibility of using putative cardiac stem cells and cardiomyocytes derived from adult and pluripotent stem cells (PSCs) for the functional recovery of damaged hearts.2–5 Thus far, replacing damaged cardiomyocytes with fresh and healthy cardiomyocytes by direct implantation is the predominate method among several potential approaches for cardiac regeneration.6–7 However, major hurdles remain that hinder cardiac stem cell therapy from becoming a clinically applicable strategy for restoring cardiac function.2,8–9 Among these major obstacles, obtaining a sufficient amount of purified cardiomyocytes from PSCs is of the greatest importance.
PSCs such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) have emerged as an attractive resource for generating de novo cardiomyocytes. Specifically, Flk1+ mesodermal precursor cells (MPCs), which can be obtained by differentiating PSCs, were previously identified as cardiovascular progenitors. Flk1+ MPCs can give rise to cardiomyocytes, endothelial cells (ECs), hematopoietic cells (HCs), and mural cells.10–14 Among several cardiomyogenic molecules, we have been interested in Cyclosporin A (CsA), which is best known as an immunosuppressive agent. Surprisingly, it has been reported recently that CsA induces cardiomyogenesis,15–16 but the exact mechanism of CsA's effects in cardiomyogenesis is unknown.
CsA inhibits the mitochondrial permeability transition pore (mPTP) via binding to peptidyl‐prolyl cis‐trans‐isomerase, also known as cyclophilin D, which is a matrix protein of mPTP. The mPTP is a nonselective pore that is permeable to any molecule of <1.5 kDa in size.17–18 The mPTPs open within the mitochondrial inner membrane under pathologic conditions such as mitochondrial calcium overload.18–19 Previous studies focused mainly on showing that pharmacologic inhibition of mPTP by CsA has a cardioprotective effect in an ischemia‐reperfusion injury model.17–18 However, the role of mPTP inhibition in stem cell differentiation and its downstream signaling has not been clearly elucidated.
Concomitant increase of myofibrils and mitochondria is a key process of cardiomyocyte differentiation from PSCs. Specifically, development of mitochondrial oxidative metabolic capacity in cardiomyocytes is essential to providing the energy necessary to sustain the beating function. Therefore, the connection between transcriptional and metabolic regulation is important for cardiomyocyte specification and differentiation, and a nucleus‐mitochondria interaction is required to translate signals that regulate cardiomyogenesis.20 Although previous studies reported that mitochondrial function and oxidative metabolism have some correlation with the differentiation of cardiomyocytes,20–25 the mechanism by which mitochondrial oxidative metabolism is regulated and the link between cardiomyogenesis and mitochondrial function are still poorly understood. Additionally, reactive oxygen species (ROS) modulate the differentiation of cardiomyocytes from ESCs,26–30 but the exact role of redox signaling in PSC‐derived cardiomyocyte differentiation remains controversial.31–32
In the present study, we demonstrate that CsA promotes differentiation of functional cardiomyocytes from PSC‐derived Flk1+ MPCs by inhibition of mPTP. This increase in differentiation appears to result from activation of mitochondrial oxidative metabolism. In addition, we found that antioxidant treatment augmented the cardiomyogenic effect of CsA. These data thus constitute novel evidence that activation of mitochondrial oxidative metabolism via inhibition of mPTP and subsequent changes in redox signaling pathways are key factors in PSC fate determination toward the cardiac lineage. Our findings suggest that modulation of mPTP and redox signaling could be a compelling target for cardiac stem cell therapy in the field of regenerative medicine.
Methods
Mouse and Human PSC and OP9 Cell Culture
EMG7 mouse ESCs, which have α‐myosin heavy chain (MHC) promoter‐driven enhanced green fluorescent protein (GFP) gene, E14Tg2a ESCs and OP9 cells were generated and maintained as described previously.11,33–34 Mouse iPSCs derived from FVB strain was obtained through a generous gift from Drs Hyun‐Jai Cho and Hyo‐Soo Kim (Seoul National University Hospital) and prepared as described previously.35 H9 human ESCs were purchased from the ATCC. We received approval from the institutional review board of KAIST to perform the experiments using human ESCs.
Induction of Mouse PSC‐Derived Flk1+ MPCs and Cardiomyocytes
For induction of Flk1+ MPCs, ESCs, and iPSCs were cultured without LIF (leukemia inhibitory factor) and plated on a 0.1% gelatin‐coated dish at cell density 1 to 1.5×103 cells/cm2 in the differentiation medium (alpha MEM, Invitrogen) with 10% fetal bovine serum (FBS) (Welgene), 2‐mercaptoethanol (Invitrogen), l‐glutamine (Invitrogen), and antibiotics (Invitrogen) for 4.5 days (96 to 112 hour) which was changed every 2 days. At day 4.5, differentiated ESCs and iPSCs were harvested with 0.25% trypsin‐EDTA and antigen recovery was performed in the differentiation medium for 30 minutes in an incubator. Then, cells were washed using phosphate buffered saline (PBS)/2% FBS and attached with biotin‐conjugated anti‐mouse Flk1+ antibody (clone AVAS12a1, eBioscience) and anti‐streptavidin MicroBeads (Miltenyi Biotec). Flk1+ MPCs were sorted by AutoMACS Pro Separator (Miltenyi Biotec). For induction of cardiomyocytes, sorted Flk1+ MPCs were plated onto the mitomycin‐C (MMC) (AG scientific) treated confluent OP9 cells at a density of 5 to 10×103 cells/cm2. For the induction of cardiomyocytes in a feeder free system, ESCs were plated on a 0.1% gelatin coated dish at cell density 3×103 cells/cm2 without OP9 cells. Cells were cultured in the differentiation medium and the medium was changed every 2 days. Embryoid bodies (EBs) were made from 2×103 ESCs using hanging drop method and incubated for 4.5 days. Then the EBs were harvested and plated on 0.1% gelatin‐coated dish, and cultured in the medium (DMEM, Welgene) containing 15% FBS (Welgene), NEAA (Invitrogen), 2‐mercaptoethanol (Invitrogen), and antibiotics (Invitrogen). The medium was changed every day or every other day.
Induction of Human ESC‐Derived Cardiomyocytes
Human ESC‐derived cardiomyocyte differentiation was induced as previously reported.36 Cells were harvested by incubation with Versene (Invitrogen) and plated onto Matrigel‐coated dishes at a density of 10.0×104 cells/cm2 in mouse embryonic fibroblast conditioned medium (MEF‐CM) with 4 ng/mL basic fibroblast growth factor (bFGF) for 2 to 3 days before induction. To induce cardiomyocyte differentiation, we replaced MEF‐CM with RPMI+B27 medium (RPMI1640, 2 mmol/L l‐glutamine, ×1 B27 supplement without insulin) with 100 ng/mL of activin A (R&D Systems) for 24 hours, followed by 10 ng/mL of human bone morphogenetic protein 4 (BMP4; R&D Systems) and 10 ng/mL human bFGF for 4 days. The culture medium was subsequently changed with RPMI+B27 supplemented with 100 ng/mL of Dickkopf‐related protein 1 (Dkk1, R&D Systems) for 2 days. At day 7, the culture medium was changed to RPMI+B27 with vascular endothelial growth factor (VEGF, R&D Systems). The medium was changed every 1 to 2 days. Beating cardiomyocytes were observed on days 8 to 9.
Culture of H9C2 Cardiac Cell Line
The rat cardiac cell line H9C2 was purchased from the ATCC. The cells were grown in DMEM (Welgene) supplemented with 10% FBS (Welgene) and antibiotics (Invitrogen). Before confluence, cells were split, plated at a lower density in culture dishes in 10% FBS culture medium and cultured for 1 to 2 days. Cells were then incubated in DMEM (welgene) supplemented with 1% FBS for myogenic differentiation.37 The culture medium was replaced every 2 days.
Reagents
CsA (a gift from Novartis Pharma, Korea), NIM811 (N‐methyl‐4‐isoleucine‐CsA, a gift from Novartis Pharma, Swiss Basel), FK506 (Sigma Aldrich), carbonyl cyanide‐p‐trifluoromethoxy phenylhydrazone (FCCP, Abcam), 6‐Hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylic acid (Trolox, Sigma Aldrich), IWP‐2, IWP‐4 (Stemgent) and SB431542 (Sellekchem) were dissolved in dimethyl sulfoxide (DMSO, Sigma Aldrich). N‐acetyl‐L‐cysteine (NAC, Sigma Aldrich) and tert‐butyl hydroperoxide (tBHP) were dissolved in distilled water. Reagents were treated at the time of medium change. DMSO was treated as a control vehicle.
Flow Cytometry Analysis
Differentiating Flk1+ MPCs on OP9 cells were harvested with 0.25% trypsin‐EDTA or dissociation buffer (Invitrogen). To analyze ECs and HCs, the cells were incubated for 20 minutes with the following antibodies: PE‐conjugated anti‐mouse CD31 (clone 390, eBioscience) and PE‐conjugated anti‐mouse CD45 (clone 30‐F11, eBioscience). To analyze cardiac Troponin T (cTnT)+ cardiomyocytes, the cells were permeabilized using Cytofix/Cytoperm solution (BD Biosciences) for 15 min. After permeabilization, the cells were incubated for 30 minutes with anti‐mouse cTnT (Clone 13‐11, Thermo Scientific) monoclonal antibody. After washing in 10% Perm/Wash buffer (BD Biosciences), the cells were incubated for 10 minutes with Cy5‐conjugated anti‐mouse IgG antibody (Invitrogen). The cells were washed with 10% Perm/Wash buffer (BD Biosciences) and then analyzed. In live cell analysis, dead cells were excluded using 7‐aminoactinomycin D (Invitrogen) and OP9 cells were excluded from Flk1+ MPCs by gating in flow cytometry. Analysis was performed by FACS Aria II (Beckton Dickinson). Data were analyzed using FlowJo Version 7.5.4 software (TreeStar).
Sorting of Cardiomyocytes Using Flow Cytometry
Cardiomyocytes were sorted using αMHC‐GFP. Differentiating Flk1+ MPCs on OP9 cells were harvested with 0.25% trypsin‐EDTA and cells were washed using PBS/10% FBS twice. αMHC‐GFP+ cardiomyocytes were sorted and plated onto 0.1% gelatin coated dish in differentiation medium. Dead cells were excluded using 7‐aminoactinomycin D (Invitrogen) and OP9 cells were excluded from Flk1+ MPCs by gating in flow cytometry. Analyses and sorting were performed by FACS Aria II (Beckton Dickinson).
Immunofluorescence Staining and Visualization of Cardiomyocytes
We performed Immunofluorescence staining at day 10.5 and at day 12.5 after αMHC‐GFP+ cardiomyocyte FACS sorting. The cells were fixed with 2% paraformaldehyde and blocked with 5% goat (or donkey) serum in PBST (0.1% Tween 20 in PBS) for 1 hour at room temperature. The cells were stored overnight at 4°C with the following primary antibodies: anti‐mouse cTnT (Clone 13‐11, Thermo Scientific) monoclonal antibody, anti‐mouse CD144 (clone 11D4.1, BD Pharmingen), and anti‐mouse CD45 (clone 30‐F11, BD Pharmingen). After being washed with PBST 3 times, the cells were incubated for 2 hours at room temperature with the following secondary antibodies: Cy3 or FITC conjugated anti‐mouse IgG antibody (Invitrogen). After being stained with the antibodies, the cells were mounted in fluorescent mounting medium (DAKO). Nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI, Invitrogen). Immunocytochemistry‐stained images were obtained using an LSM510 confocal fluorescence microscope (Carl Zeiss). Live images of αMHC‐GFP+ cardiomyocytes were obtained using Axiovert 200M microscope (Carl Zeiss) equipped with AxioCam MRm (Carl Zeiss). Images were analyzed using Image J software (http://imagej.nih.gov/ij/, 1.47V, National Institutes of Health [NIH]). Phase‐contrast images including beating foci of cardiomyocytes and sorted single cardiomyocyte were obtained using an Infinity X digital camera and DpxView LE software (DeltaPix). Co‐localization between Mitotracker and cTnT+ signal was evaluated and illustrated using Matlab R2011b (7.13; MathWorks, Inc.). Immunofluorescence signal intensity was normalized to a value between 0 and 1 according to the minimum and maximum level of each channel, respectively. When both the normalized intensity values of Mitotracker and cTnT+ were above 0.5 at the same pixel, co‐localized index was obtained by multiplying these 2 normalized intensity values. Otherwise, co‐localized index was set as 0. Co‐localized index is illustrated by a pseudocolor spectrum and by the height in color‐coded 3‐dimensional mesh.
Mitochondrial Function Assays
Mitochondrial assays were performed at day 10.5. The mPTP opening was measured by monitoring Calcein AM fluorescent dye (Invitrogen) in the presence of CoCl2 (Invitrogen).38 Calcein AM fluorescent dye can move freely between cytosol and mitochondria via the opening of mPTP. CoCl2 acts as a quencher which eliminates Calcein AM fluorescence. If the mPTP is closed, the CoCl2 cannot quench the Calcein AM trapped inside of mitochondria. Because of these unique properties, Calcein AM fluorescence after CoCl2 treatment reflects the degree of mPTP closure. In brief, cells were washed twice with PBS and resuspended at 1×106 cells/mL in pre‐warmed Hanks' balanced salt solution (HBSS) containing 2 mmol/L Ca2+. The cells were then loaded with 2 μmol/L Calcein AM for 15 minutes at 37°C in the presence of 2 mmol/L CoCl2. After washing away excess stain and quenching reagent, the cell pellets were resuspended in 400 μL HBSS containing 2 mmol/L Ca2+ and analyzed for Calcein AM fluorescence by flow cytometry. Mitochondrial membrane potential (Δψm) was measured by tetramethylrhodamine methyl esters (TMRM) fluorescent dye (Invitrogen) and mitochondrial Ca2+ was measured by Rhod‐2 AM fluorescent dye (Invitrogen). Cells were loaded with fluorescent dye for 30≈60 minutes at 37°C in serum free medium. Then, the cells were harvested with 0.25% trypsin‐EDTA and washed twice using PBS/2% FBS. Then the cell pellets were resuspended in 400 μL PBS/2% FBS and analyzed by flow cytometry. Dead cells were excluded using 7‐aminoactinomycin D (Invitrogen) and OP9 cells were excluded from Flk1+ MPCs by gating in flow cytometry. Analysis was performed by FACS Aria II (Beckton Dickinson). Data were analyzed using FlowJo Version 7.5.4 software (TreeStar). Immunofluorescence staining of mitochondria was performed using MitoTracker Orange CMTMRos probe (Invitrogen) and cells were incubated with probe for 30≈60 minutes at 37°C in serum free medium before fixation.
Electron Microscopic Analysis of H9C2 Cells
The cells were fixed in 2.5% glutaraldehyde in PBS solution at 4°C overnight, and then with 1% osmium tetroxide in PBS for 2 hour. The tissues were washed, dehydrated, and embedded, and then semi‐thin sections were cut (0.5 to 1 μm). Further ultra‐sectioning (60 to 90 nm) was performed and then the slices were double stained with uranyl acetate and lead citrate and imaged using a JEM 1200 EX2 electron microscope (Jeol). Developed images were scanned on a flatbed scanner (Umax PowerLook 1100) and analyzed using Image J software (http://imagej.nih.gov/ij/, 1.47V, NIH).
Quantitative Real Time PCR
Total RNA was extracted using Trizol RNA extraction kit (Invitrogen) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using GoScript™ cDNA synthesis system (Promega). cDNA was applied for quantitative real‐time PCR using FastStart SYBR Green Master mix (Roche) and Bio‐rad S1000 Thermocycler with the indicated primers (Table 1). Beta‐actin was used as a reference gene and the results were presented as relative expression to control using the ΔΔCt method.
Table 1.
Primers for Real Time PCR
| Mouse tbx5 | Forward | 5′‐CGCCTCTGGAGCCTGATTCCAAAG‐3′ |
| Reverse | 5′‐GTGCCCACTTCGTGGAACTTCAGC‐3′ | |
| Mouse nkx2.5 | Forward | 5′‐CACGCCTTTCTCAGTCAAAGACATCC‐3′ |
| Reverse | 5′‐CTGGGAAAGCAGGAGAGCACTTGG‐3′ | |
| Mouse tnnt2 | Forward | 5′‐GACCTGTGTGCAGTCCCTGTTCAG‐3′ |
| Reverse | 5′‐CTTGCTCGTCCTCCTCTTCTTCAC‐3′ | |
| Mouse pgc1α | Forward | 5′‐GCGCCGTGTGATTTACGTT‐3′ |
| Reverse | 5′‐AAAACTTCAAAGCGGTCTCTCAA‐3′ | |
| Mouse pparα | Forward | 5′‐GGCATGGCAGCAATATCAGAGGTAG‐3′ |
| Reverse | 5′‐CTGCCTGGATAGCGAAGTCACTTTTG‐3′ | |
| Mouse nrf1 | Forward | 5′‐GGAAAGAAAGCTGCAAGCCTATCTGG‐3′ |
| Reverse | 5′‐CTGAAGGTCTGTACTACGGTCTGTG‐3′ | |
| Mouse beta actin | Forward | 5′‐GCTCTTTTCCAGCCTTCCTT‐3′ |
| Reverse | 5′‐CTTCTGCATCCTGTCAGCAA‐3′ |
PCR indicates polymerase chain reaction.
Measurement of Oxygen Consumption
After the cells were harvested, oxygen consumption rates (OCR) were measured using NeoFox Phase Measurement system (Ocean Optics) with a 400 μL chamber at room temperature, in an air‐saturated (220 nmol/O2 per milliliter) culture medium. OCR is expressed as nmol O2/min per milligram total protein.
ATP Assay
ATP concentration was analyzed by ATP bioluminescent somatic cell assay kit (Sigma). In brief, 100 μL of ATP assay mix working solution, 100 μL of somatic cell ATP releasing reagent, and 50 μL of ultrapure water were added and mixed in each of the assay vials. Cells were harvested and then 1×105 cells were added to each assay vials. Luminescence was detected by SpectraMax M2 (Molecular device). ATP concentration was calculated by using standard curve with known concentration (0, 10, 100, and 200 μmol/L) of ATP standard.
Measurement of ROS Levels
ROS levels were measured by 2′,7′‐dichlorodihydrofluorescein diacetate (H2DCFDA) fluorescent dye (Invitrogen). In brief, cells were loaded with 1.5 μmol/L of fluorescent dye for 30 minutes at 37°C in PBS. Then, the cells were harvested with 0.25% trypsin‐EDTA and washed using PBS/2% FBS twice. The cell pellets were re‐suspended in 400 μL PBS/2% FBS and analyzed by flow cytometry. Data were analyzed using FlowJo Version 7.5.4 software (TreeStar).
Electrophysiology
We performed the patch clamp study at day 14.5 after αMHC‐GFP+ cardiomyocyte FACS sorting. Action potentials (APs) and ion currents were recorded from beating cardiomyocytes placed onto the recording chamber of microscope by using Axopatch 200B amplifier (Axon Instrument) at room temperature (23±1°C). Patch clamp recordings were carried out in whole‐cell configuration where normal Tyrode (NT) solution was used for perfusion of cardiomyocytes and a K+‐rich solution was used for pipette filling. The NT solution contained (in mmol/L): 143 NaCl, 5.4 KCl, 0.5 MgCl2, 1.8 CaCl2, 5.5 glucose, and 5 N‐[2‐hydroxyethyl]piperazine‐N‐[2‐ethanesulfonic acid] (HEPES). pH was adjusted to 7.4 with 1 mol/L NaOH. The K+‐rich pipette filling solution contained (in mmol/L): 140 KCl, 1 MgCl2, 5 MgATP, 5 EGTA, 5 glucose, and 5 HEPES, titrated to pH 7.2 with 1 mol/L KOH. Patch pipettes were pulled from thin‐walled borosilicate capillaries (Clark Electromedical Instruments) using a PP‐83 vertical puller (Narishige). GΩ seal formation and membrane rupture were achieved by applying negative pressure onto the membrane patch and only whole‐cell patches with series resistance <5 MΩ were selected for recording. All the recordings were carried out at least 5 minutes after achieving whole‐cell configuration to allow cells being completely dialyzed with pipette‐filling solution. Spontaneous APs were recorded in current‐clamp mode while ion currents were recorded in voltage‐clamp mode. The voltage and current signals were filtered at 10 kHz, 4‐pole Bessel type low‐pass filter and sampled at a rate of 4 kHz for voltage and 27 kHz for ion current. All experimental parameters, such as pulse generation and data acquisition, were controlled using our own software (PatchPro). The liquid‐junction potentials between bathing and pipette filling solution, which were calculated based on ionic mobility, were <5 mV. Kharche's model39 was employed and modified to simulate 3 different types of AP in ESC‐derived αMHC‐GFP+ cardiomyocytes.
Statistical Analysis
Values presented are means±standard deviation (SD). The assumption of normality was evaluated using Shaprio‐Wilk test. Significant differences between means were determined by unpaired Student t test or analysis of variance with 1‐way ANOVA followed by the Student‐Newman‐Keuls test. The Mann‐Whitney test and Kruskal‐ Wallis ANOVA were performed when data were not normally distributed. Statistical significance was set at P<0.05 or 0.01.
Results
CsA Promotes Differentiation and Expansion of Cardiomyocytes From Flk1+ MPCs Derived From Mouse PSCs in OP9 Co‐Culture System
To monitor cardiomyocyte differentiation in a live condition, we used mouse EMG7 ESC line,11 which has a transgene of cardiac specific αMHC promoter‐driven enhanced GFP. At day 4.5 after the start of mesodermal induction without LIF in the ESCs, Flk1+ MPCs were sorted and were co‐cultured with MMC‐treated OP9 feeder cell layer in differentiation medium,11 and the cardiomyocyte differentiation was analyzed at day 10.5 unless otherwise indicated (Figure 1A). Self‐beating cardiomyocytes, αMHC‐GFP+ and cTnT+ appeared at day 8.5 (Figure 1B and 1C), and almost maximally expanded until day 10.5. In range of 0.5 to 4 μg/mL of CsA, 2 μg/mL of CsA showed the maximal effects on differentiation and expansion of cardiomyocytes (Figure 1D and 1E). In fact, 2 μg/mL of CsA (hereafter named as CsA) increased number of self‐beating foci (8.6‐fold), αMHC‐GFP+ area (12.8‐fold), and the population of cTnT+ cardiomyocytes (≈7‐ to 9‐fold) compared with control vehicle (Figures 1F through 1M, Videos S1 and S2). CsA also increased the population of cTnT+ cardiomyocytes (2.9‐fold) from the mouse iPSCs (Figures 1N and 1O). In contrast, CsA reduced differentiation of ECs (26.0%) and HCs (14.4%) (Figures 2A through 2F). Interestingly, CsA was superior to previously known cardiomyocyte differentiation agents including Wnt signaling inhibitors IWP‐2 and IWP‐4, and transforming growth factor beta (TGFβ) signaling inhibitor SB431542 in the cardiomyocyte differentiation and expansion (Figures 2G and 2H). These findings led us to investigate how CsA promotes differentiation and expansion of cardiomyocytes (hereafter called “cardiomyogenic effect”) from the PSC‐derived Flk1+ MPCs.
Figure 1.

CsA promotes differentiation and expansion of cardiomyocytes from the mouse PSC‐derived Flk1+ MPCs in OP9 co‐culture system. A, Protocol for differentiation of cardiomyocytes in OP9 co‐culture system. B, Live cell image showing αMHC‐GFP+ cardiomyocytes in differentiating Flk1+ MPCs. C, Image showing cTnT+ sarcomeres and DAPI+ nuclei of differentiating Flk1+ MPCs. D, Relative total cell number incubated with indicated concentrations of CsA. Population of total cells without CsA was regarded as 100%. E, Percentage of cTnT+ cardiomyocyte population incubated with indicated concentrations of CsA. F, Phase‐contrast image showing beating cardiomyocyte foci (black arrow head) incubated with control vehicle (Con) and CsA (2 μg/mL). G, Comparison of the number of beating foci generated by control vehicle and CsA. H, Live cell images showing αMHC‐GFP+ cardiomyocytes. I, Percentage of αMHC‐GFP+ area in a live cell image. J, Images showing cTnT+ cardiomyocytes and DAPI+ nuclei. K, Comparison of cTnT+ area (%). L and M, Representative FACS analysis and the percentage of ESC‐derived cTnT+ cardiomyocytes. N and O, Representative FACS analysis and the percentage of iPSC‐derived cTnT+ cardiomyocytes. All scale bars, 100 μm. *P<0.01 vs 0 or Con. CsA indicates cyclosporin A; cTnT, cardiac Troponin T; DAPI, 4,6‐diamidino‐2‐phenylindole; ESC, embryonic stem cell; FACS, fluorescence‐activated cell sorting; GFP, green fluorescent protein; iPSC, induced pluripotent stem cell; LIF, leukemia inhibitory factor; MHC, myosin heavy chain; MPCs, mesodermal precursor cells; SSC, side scatter.
Figure 2.

CsA reduces the differentiation of ECs and HCs from Flk1+ MPCs and has more cardiomyogenic effect compared to other cardiomyogenic compounds. A and B, Representative FACS analysis and the percentage of CD31+ ECs and cTnT+ cardiomyocytes incubated with control vehicle (Con) and CsA (2 μg/mL). C, Immunofluorescence images showing VE‐cadherin+ ECs in differentiating Flk1+ MPCs. D and E, Representative FACS analysis and the percentage of CD45+ HCs and cTnT+ cardiomyocytes at day 10.5. F, Immunofluorescence images showing CD45+ HCs in differentiating Flk1+ MPCs. G and H, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes incubated with control vehicle (Con), CsA (2 μg/mL), IWP‐2 (5 μmol/L), IWP‐4 (5 μmol/L) and SB431542 (20 μmol/L). All scale bars, 100 μm. *P<0.01 and #P<0.05 vs Con. CMs indicates cardiomyocytes; CsA, cyclosporin A; cTnT, cardiac Troponin T; ECs, endothelial cells; HCs, hematopoietic cells; IWP, inhibitors of Wnt production; MPCs, mesodermal precursor cells.
The Cardiomyogenic Effect of CsA Mainly Resulted From mPTP Inhibition Rather Than From Calcineurin Inhibition
CsA binding to calcineurin results in its well‐known immunosuppressive effect through the nuclear factor of activated T‐cells (NFAT).18 Another target protein for CsA is cyclophilin D, which is the matrix protein component of mPTP.18 To delineate how CsA induces the cardiomyogenic effect, we compared the effects of CsA with FK506, which blocks calcineurin activity via its receptor FKBP12 but has no effect on mPTP and NIM811, which blocks mPTP but has no effect on calcineurin.18 Compared with control vehicle, NIM811 (3.6 μg/mL) increased the cTnT+ cardiomyocyte population (6.8‐fold), whereas FK506 (100 ng/mL) only slightly increased the cTnT+ cardiomyocyte population (2.0‐fold). Treatment of both NIM811 and FK506 showed an additive effect in increasing the cTnT+ cardiomyocyte population (9.0‐fold) (Figures 3A and 3B). Accordingly, NIM811 markedly increased the number of self‐beating foci (10.5‐fold) and αMHC‐GFP+ area (12.4‐fold), whereas FK506 had only minor effects (3.3‐fold and 2.7‐fold) (Figures 3C through 3F and Videos S3 and S4). Thus, the cardiomyogenic effect of CsA was mainly due to mPTP inhibition rather than calcineurin inhibition.
Figure 3.

The cardiomyogenic effect of CsA mainly resulted from mPTP inhibition rather than from calcineurin inhibition. A and B, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes incubated with control vehicle (Con), CsA (2 μg/mL), NIM811 (3.6 μg/mL), FK506 (100 ng/mL) and a combination of NIM811 and FK506. C, Phase‐contrast image showing beating cardiomyocyte foci (black arrow head) incubated with Con, NIM811 and FK506. D, Comparison of the number of beating foci. E, Live cell images showing αMHC‐GFP+ cardiomyocytes. F, Percentage of αMHC‐GFP+ area in live cell image. All scale bars, 100 μm. *P<0.01, #P<0.05 and NS (not significant) vs Con or each group. CsA indicates cyclosporin A; cTnT, cardiac Troponin T; GFP, green fluorescent protein; MHC, myosin heavy chain; mPTP, mitochondrial permeability transition pore.
Inhibition of mPTP by CsA Directly Increases Mitochondrial Function, Which is Closely Related to Cardiomyogenesis
To alternatively confirm the inhibition of mPTP by CsA, a CoCl2/Calcein AM quenching method38 was used. Indeed, CsA increased the mean fluorescence intensity of Calcein AM (1.9‐fold) in the Flk1+ MPCs compared with control vehicle (Figure 4A), indicating that CsA inhibits mPTP leading to the prevention of CoCl2 entry into the mitochondria. To clarify whether CsA directly inhibited the mPTP of cardiomyocytes, TMRM fluorescent dye that shows high signal in beating cardiomyocytes40 was employed. Through this we observed that CsA induced a co‐localization of TMRM with Calcein AM (Figure 4B), indicating that CsA directly inhibits mPTP of cardiomyocytes.
Figure 4.

Inhibition of mPTP by CsA directly increases mitochondrial function, which is closely related to cardiomyogenesis. A, Representative FACS analysis of Calcein AM fluorescence intensity and the percentage of relative mean fluorescence intensity of Calcein AM incubated with control vehicle (Con) and CsA (2 μg/mL). B, Live cell images showing TMRM+ and Calcein AM+ cells. C, Representative FACS analysis of Rhod‐2 AM fluorescence intensity and the percentage of relative mean fluorescence intensity of Rhod‐2 AM incubated with Con, CsA (2 μg/mL), NIM811 (3.6 μg/mL) and FK506 (100 ng/mL). D, Representative FACS analysis of TMRM fluorescence intensity and the percentage of relative mean fluorescence intensity of TMRM incubated with Con, CsA, NIM811 and FK506. E, Live cell images showing TMRM+ and αMHC‐GFP+ cells. F and G, Representative FACS analysis and the percentage of TMRM+/αMHC‐GFP+ co‐positive cells. H, Representative FACS analysis of TMRM fluorescence intensity and the percentage of relative mean fluorescence intensity of TMRM in differentiating ESCs at day 4.5 and Flk1+ MPCs incubated with control vehicle and CsA (2 μg/mL) at day 10.5. I, Relative mRNA expression level of cardiomyocyte specific and mitochondrial function related genes in differentiating Flk1+ MPCs. All scale bars, 100 μm. *P<0.01, #P<0.05 and NS (not significant) vs Con or each group. AM indicates acetoxymethyl ester; CsA, cyclosporin A; ESCs, embryonic stem cells; GFP, green fluorescent protein; MHC, myosin heavy chain; MPCs, mesodermal precursor cells; mPTP, mitochondrial permeability transition pore; TMRM, tetramethylrhodamine methyl esters.
Increase in mitochondrial Ca2+ and Δψm are also reliable markers of mitochondrial function and mPTP inhibition.38 In differentiating Flk1+ MPCs at day 10.5, Rhod‐2 AM fluorescent dye assay revealed that CsA (1.4‐fold) and NIM811 (1.4‐fold) increased the mitochondrial Ca2+ level compared with control vehicle, while FK506 did not (Figure 4C). Moreover, the TMRM staining revealed that CsA (2.2‐fold) and NIM811 (1.7‐fold) increased the Δψm level compared to control vehicle, whereas FK506 showed no difference (Figure 4D). Live cell imaging and FACS analysis showed that CsA increased TMRM fluorescence in the αMHC‐GFP+ cardiomyocytes and the percentages of TMRM+/αMHC‐GFP+ co‐positive cells (2.1‐fold) compared to control vehicle (Figures 4E through 4G). These data suggest that CsA directly increases mitochondrial function in PSC‐derived cardiomyocytes. Additionally, Δψm of differentiating ESCs in day 4.5 is lower than in day 10.5 (Figure 4H). These data indicate that mitochondrial function is increased during cardiomyocyte differentiation from ESCs.
Next, we examined the mRNA expression levels of the genes related to mitochondrial function (pgc1α, pparα and nrf1) and cardiomyocyte differentiation (tbx5, nkx2.5 and tnnt2) in the differentiating Flk1+ MPCs. The expression levels of all these genes were variably up‐regulated by CsA compared with control vehicle (Figure 4I), suggesting that CsA regulates the cardiomyogenic effect by alterations of the gene transcriptions related to mitochondrial function.
Observation of individual cardiomyocytes after FACS sorting using αMHC‐GFP revealed that the shape, beating rate, mitochondria content, and cTnT+ sarcomere structure of each cardiomyocyte was quite varied (Video S5, Figure 5A). Interestingly, cardiomyocytes with loosely organized sarcomere structure also showed less developed, fragmented mitochondria that are located in the perinuclear region (Figure 5A‐1 and 5B‐1), while cardiomyocytes with densely organized sarcomere structure contained well‐developed, elongated mitochondria which are distributed along the sarcomere (Figures 5A‐2 and 5B‐2). These data indicate that mitochondrial development and function are closely related to cardiomyogenesis.
Figure 5.

Mitochondrial development and function are closely related to cardiomyogensis. A, Immunofluorescence images showing Mitotracker+ and cTnT+ cardiomyocytes and DAPI+ nuclei in sorted αMHC‐GFP+ cardiomyocytes at day 11.5. 1 and 2 are magnified views of square‐dotted area in the left side. Scale bar represent 100 μm in the left side and 50 μm in the right side. B, Co‐localized signals of Mitotracker+ and cTnT+ in reconstructed image of (A). cTnT indicates cardiac Troponin T; DAPI, 4,6‐diamidino‐2‐phenylindole; GFP, green fluorescent protein; MHC, myosin heavy chain.
To confirm whether the effect of CsA to mitochondria in differentiating Flk1+ MPCs is a direct effect rather than a secondary effect due to cardiomyogenesis, we treated CsA to H9C2 cardiac cell line (Figure 6A). Consistent with the data from differentiating Flk1+ MPCs, CsA increased the mean fluorescence intensity of Calcein AM (4.0‐fold), mitochondrial Ca2+ (1.5‐fold) and Δψm (1.8‐fold) compared with control vehicle in FACS analysis (Figures 6B through 6D). CsA also increased the fluorescence of Calcein AM, TMRM and Mitotracker in live cell and immunofluorescence images (Figures 6E and 6F). Additionally, electron microscope images revealed that CsA increased the mitochondrial size (1.7‐fold) and yielded more matured cristae structure (Figures 7A and 7B). These data suggest that an increase of mitochondrial function is a direct effect of CsA rather than a secondary effect due to cardiomyogenesis in differentiating Flk1+ MPCs.
Figure 6.

Inhibition of mPTP by CsA directly increases mitochondrial function in H9C2 cardiac cell line. A, Protocol for proliferation and differentiation of H9C2 cells. H9C2 cells were incubated for 2 days in growth medium and then incubated with control vehicle (Con) and CsA (1 μg/mL) in differentiation medium for 4 days. B, Representative FACS analysis of Calcein AM fluorescence intensity and the percentage of relative mean fluorescence intensity of Calcein AM. C, Representative FACS analysis of Rhod‐2 AM fluorescence intensity and the percentage of relative mean fluorescence intensity of Rhod‐2 AM. D, Representative FACS analysis of TMRM fluorescence intensity and the percentage of relative mean fluorescence intensity of TMRM. E, Live cell images showing Calcein AM+ and TMRM+ H9C2 cells. Scale bars represent 100 μm. F, Immunofluorescence images showing Mitotracker+ H9C2 cells and DAPI+ nuclei. The bottom panel is a magnified view of upper panel. Scale bar represent 100 μm in the upper panel and 50 μm in the bottom panel. *P<0.01 and #P<0.05 vs Con. CsA indicates cyclosporin A; DAPI, 4,6‐diamidino‐2‐phenylindole; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; mPTP, mitochondrial permeability transition pore; TMRM, tetramethylrhodamine methyl esters.
Figure 7.
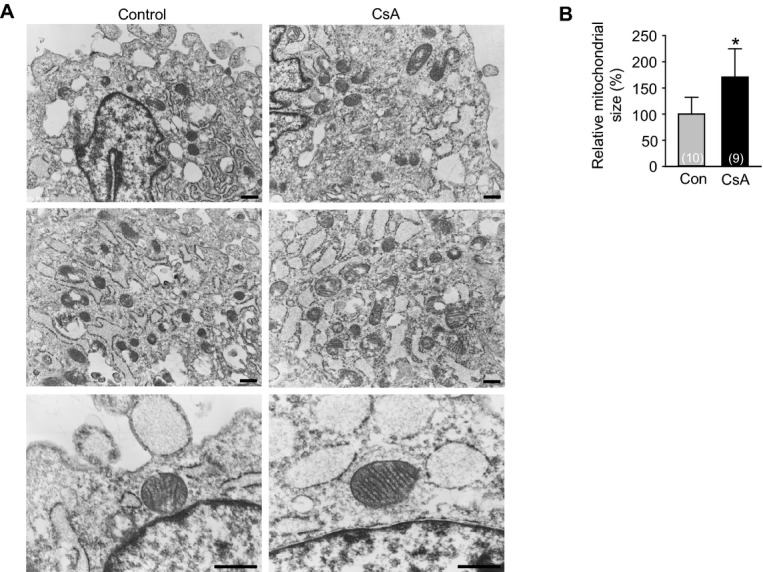
Inhibition of mPTP directly increases mitochondrial maturation in H9C2 cardiac cell line. A, Electron microscope images showing the mitochondrial morphology and cristae incubated with control vehicle (Con) and CsA (1 μg/mL). Scale bars represent 500 nm. B, Quantification of mitochondrial size in electron microscope images. *P<0.01 vs Con. CsA indicates cyclosporin A; mPTP, mitochondrial permeability transition pore.
Activation of Mitochondrial Oxidative Metabolism via mPTP Inhibition Promotes Cardiomyogenesis
The aforementioned findings led us to investigate whether CsA has an effect on activation of mitochondrial oxidative metabolism by measurements of OCR and ATP levels.41 Compared with control vehicle, CsA increased the OCR and ATP levels in the differentiating Flk1+ MPCs (1.7‐fold and 6.6‐fold respectively) and H9C2 cardiac cell line (2.0‐fold and 4.9‐fold respectively) (Figures 8A through 8C and 8F through 8H), suggesting that CsA directly activates mitochondrial oxidative metabolism through mPTP inhibition. Addition of FCCP (3 μmol/L), an uncoupler of mitochondrial oxidative phosphorylation, reduced the CsA‐induced cardiomyogenic effect by 54.5%, while FCCP alone did not significantly change the cardiomyocyte differentiation (Figures 8D and 8E). This result clearly indicates that the CsA‐induced cardiomyogenic effect is partially caused by activation of mitochondrial oxidative metabolism.
Figure 8.

Activation of mitochondrial oxidative metabolism via mPTP inhibition promotes cardiomyogenesis. A, Representative graph of relative oxygen consumption rate from differentiating Flk1+ MPCs incubated with control vehicle (Con) and CsA (2 μg/mL). B, Quantification of oxygen consumption rate. C, Relative ATP level from differentiating Flk1+ MPCs. D and E, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes incubated with or without CsA and FCCP (3 μmol/L). F, Representative graph of relative oxygen consumption rate in H9C2 cells with control vehicle (Con) and CsA (1 μg/mL). G, Quantification of oxygen consumption rate in H9C2 cells. H, Relative ATP level in H9C2 cells. *P<0.01, #P<0.05 and NS (not significant) vs Con or each group. CsA indicates cyclosporin A; cTnT, cardiac Troponin T; FCCP, carbonyl cyanide‐p‐trifluoromethoxy phenylhydrazone; MPCs, mesodermal precursor cells; mPTP, mitochondrial permeability transition pore.
Dual Modulation of mPTP and Redox Signaling Synergistically Promotes Cardiomyogenesis
mPTP inhibition by CsA or NIM811 increased ROS generation during cardiomyocyte differentiation (Figure 9A). Therefore, to confirm the role of redox signaling for cardiomyocyte differentiation under mPTP inhibition, we added either the antioxidants Trolox (100 to 1600 μmol/L, water‐soluble analog of vitamin E) or NAC (100 to 1600 μg/mL), or the pro‐oxidant tBHP (2 to 32 μmol/L) with CsA. Surprisingly, the addition of either Trolox or NAC synergistically enhanced the CsA‐induced cardiomyogenesis (160.6% to 330.5% or 144.9% to 239.7%) in a dose‐dependent manner, while Trolox or NAC alone did not significantly enhance the cardiomyocyte differentiation (Figures 9B and 9C) even though the antioxidants indeed blunted the ROS levels (Figure 9E). However, the addition of pro‐oxidant tBHP did not enhance the cardiomyocyte differentiation, regardless of CsA treatment (Figures 9C and 9D) even though it exhibited the pro‐oxidant effect (Figure 9F). These results indicate that the cardiomyogenic effect of antioxidants is dependent on CsA, and that the redox signaling is a cardiomyogenesis regulatory factor lying downstream of mPTP inhibition. In addition, these data suggest that the reduction of ROS and oxidative stress is critical for efficient cardiomyogenesis under mPTP inhibition.
Figure 9.

Dual modulation of mPTP and redox signaling synergistically promotes cardiomyogenesis. A, Representative FACS analysis of H2DCFDA fluorescence intensity and the percentage of relative mean fluorescence intensity of H2DCFDA incubated with control vehicle (Con), CsA (2 μg/mL), NIM811 (3.6 μg/mL), and FK506 (100 ng/mL). B, Dose‐response curves of Trolox (100 to 1600 μmol/L) and NAC (100 to 1600 μg/mL) with or without CsA (2 μg/mL) for the percentage of cTnT+ cardiomyocytes. C, Representative FACS analysis of cTnT+ cardiomyocytes incubated with control vehicle, CsA (2 μg/mL), Trolox (1600 μmol/L), NAC (1600 μg/mL), tBHP (32 μmol/L), CsA+Trolox (1600 μmol/L), CsA+NAC (1600 μg/mL) and CsA+tBHP (32 μmol/L). D, Dose‐response curves of tBHP (2 to 32 μmol/L) with or without CsA (2 μg/mL) for the percentage of cTnT+ cardiomyocytes. E, Representative FACS analysis of H2DCFDA, 2′,7′‐dichlorodihydrofluorescein diacetate fluorescence intensity and the percentage of relative mean fluorescence intensity of H2DCFDA incubated with control vehicle (Con), CsA (2 μg/mL), Trolox (1600 μmol/L), NAC (1600 μg/mL), CsA+Trolox (1600 μmol/L) and CsA+NAC (1600 μg/mL). F, Representative FACS analysis of H2DCFDA fluorescence intensity and the percentage of relative mean fluorescence intensity of H2DCFDA incubated with control vehicle (Con), CsA (2 μg/mL), tBHP (32 μmol/L) and CsA+tBHP (32 μmol/L). *P<0.01, #P<0.05 and NS (not significant) vs Con or each group. CsA indicates cyclosporin A; cTnT, cardiac Troponin T; H2DCFDA, 2′,7′‐dichlorodihydrofluorescein diacetate; mPTP, mitochondrial permeability transition pore; NAC, N‐acetyl‐L‐cysteine; tBHP, tert‐butyl hydroperoxide.
Cardiomyocyte Induced by CsA has Electrophysiologically Functional Properties
To examine whether the single beating αMHC‐GFP+ cardiomyocytes, induced by CsA (Video S5) indeed retain APs and ion currents characteristic of a cardiomyocyte, whole‐cell patch clamp recordings were carried out in both current‐clamp and voltage‐clamp modes. Constant and robust firings of spontaneous APs were identified from 16 different cardiomyocytes (Figures 10A through 10C). All the results are summarized in Table 2. Among the types of AP, the nodal type was most frequently recorded according to our criteria (Figure 10D). To identify whether those beating cardiomyocytes have functional ion currents, voltage‐clamp recordings were carried out in various pulse protocols. Depolarizing test pulses between −30 and +50 mV from a holding potential of ‐40 mV induced large inward currents (Figure 10E). The current‐voltage (IV) relation curve was bell shaped with its peak at 0 mV, which is typical of L‐type Ca2+ currents. Hyperpolarizing test pulses between −130 and −60 mV induced slowly developing inward currents (Figure 10F). The extrapolation of IV relation indicates the reversal potential is similar to that of hyperpolarization activated current (IHCN). Depolarizing test pulses between −70 mV and +50 mV from a holding potential of −80 mV induced either rapidly activating (Figure 10G) or slowly developing (Figure 10H) outward currents, which are considered to be transient outward K+ current (Ito) or slowly activating delayed rectifier K+ current (IKs). These results indicate that ESC‐derived cardiomyocytes induced by CsA are electrophysiologically functional cardiomyocytes. Simulation was also carried out as shown in right panel of each Figure 10 (indicated as blue) to gain insight into which ion channels are responsible for differentiation of APs among cardiomyocytes. Our simulation study suggests that the difference in inactivation kinetics of L‐type Ca2+‐channel among cardiomyocytes might be a key determinant for their different shapes of APs.
Figure 10.

Cardiomyocyte induced by CsA has electrophysiologically functional properties. A through C, Nodal type (A), atrial type (B), and ventricular type (C) APs recorded from different cardiomyocytes in current‐clamp mode. Each AP trace in the middle panel corresponds to a single AP denoted with an asterisk in the left panel. Each AP trace in the right panel corresponds to a simulated AP. Dotted lines indicate zero voltage level. D, Percentile distribution of 3 different types of APs in cardiomyocytes. E through H, Membrane currents recorded from cardiomyocytes (left panel) and simulation (middle panel) in voltage‐clamp mode. Dotted lines indicate zero current level. Current‐voltage relationship is compared between experimental recording and simulation (right panel) (E) Time‐dependent inward currents activated by depolarizing test pulses between −30 and +50 mV from a holding potential of −40 mV at 10 second interval. F, Time‐dependent inward current activated by hyperpolarizing test pulses between −130 and −60 mV from a holding potential of −50 mV. G, Rapidly activating outward currents by depolarizing test pulses. H, Slowly activating outward currents by depolarizing test pulses. APs indicates action potentials; CsA, cyclosporin A.
Table 2.
Action Potential Profile of 3‐Different Types of Cardiomyocytes
| APs (n=16) | Peak (mV) | MDP (mV) | Amp (mV) | Interval (second) | dV/dtmax (V/second) | APD10 (ms) | APD30 (ms) | APD90 (ms) | APD10‐30 /APD90 |
|---|---|---|---|---|---|---|---|---|---|
| Nodal type (n=8) | 18.2±1.5 | −46.5±1.2 | 64.7±2.2 | 2.5±0.5 | 2.6±0.4 | 25.6±5.5 | 57.8±9.6 | 135.9±15.3 | 0.23±0.01 |
| Atrial type (n=3) | 29.6±6.0 | −59.3±1.9 | 89.0±4.1 | 2.1±1.1 | 4.3±0.9 | 38.0±3.3 | 90.2±16.4 | 155.3±28.3 | 0.33±0.03 |
| Ventricular type (n=5) | 27.4±3.2 | −60.7±2.6 | 88.1±2.9 | 2.1±0.5 | 4.9±0.6 | 50.2±10.4 | 133.2±36.8 | 212.4±0.4 | 0.37±0.02 |
Amp indicates amplitude; APD10‐30/APD90, ratio of APD10‐30 to APD90; APDX, APD at X% repolarization; APs, action potentials; dV/dtmax, maximal rate of depolarization; MDP, maximum diastolic potential.
CsA Promotes Cardiomyogenesis From Mouse and Human ESCs in Feeder‐Free Condition
To examine whether CsA can exert its cardiomyogenic effect independently of OP9 feeder cells, we treated CsA to both feeder‐free ESC monolayer and EBs during mouse ESC differentiation. Only few (2.4% in the monolayer and 1.7% in the EBs) cTnT+ cardiomyocytes were detected in control vehicle group. On the other hand, CsA increased the area of αMHC‐GFP+ (2.2‐fold) and the percentages of cTnT+ cardiomyocytes (2.0‐fold) in the monolayer, while it increased the area of αMHC‐GFP+ (3.7‐fold) and the percentages of cTnT+ cardiomyocytes (2.0‐fold) in the EBs (Figure 11A through 11J). CsA also increased the area of self‐beating cardiomyocytes in the EBs compared to control vehicle (Videos S6 and S7). Moreover, CsA expanded the area (3.5‐fold) and percentages (2.2‐fold) of cTnT+ cardiomyocytes in the differentiating human ESCs (Figures 11K through 11O). These data indicate that CsA is directly capable of promoting cardiomyogenesis in both mouse and human PSCs.
Figure 11.

CsA promotes cardiomyogenesis from mouse and human ESCs in feeder‐free condition. A, Protocol for differentiation of cardiomyocytes in the monolayer without feeder cells. B, Live cell images showing αMHC‐GFP+ cardiomyocytes in the monolayer incubated with control vehicle (Con) and CsA (2 μg/mL). Scale bar, 100 μm. C, Percentage of αMHC‐GFP+ area in live cell image. D and E, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes in the monolayer. F, Protocol for differentiation of cardiomyocytes in the EBs. G, Live cell images showing αMHC‐GFP+ cardiomyocytes in the EBs incubated with Con and CsA (1 μg/mL). Scale bar, 100 μm. H, Percentage of αMHC‐GFP+ area in live cell image. I and J, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes in the EBs. K, Protocol for human ESC‐derived cardiomyocyte differentiation in feeder‐free condition. L, Immunofluorescence images showing cTnT+ cardiomyocytes incubated with Con and CsA (2 μg/mL). Scale bar, 200 μm. M, Comparison of cTnT+ area (%). N and O, Representative FACS analysis and the percentage of cTnT+ cardiomyocytes. *P<0.01 and #P<0.05 vs Con. bFGF indicates basic fibroblast growth factor; BMP4, bone morphogenetic protein 4; CsA, cyclosporin A; cTnT, cardiac Troponin T; DAPI, 4,6‐diamidino‐2‐phenylindole; Dkk1, dickkopf‐related protein 1; EBs, embryoid bodies; FSC, forward scatter; GFP, green fluorescent protein; LIF, leukemia inhibitory factor; mESCs, mouse embryonic stem cells; MHC, myosin heavy chain; VEGF, vascular endothelial growth factor.
Discussion
Obtaining ample differentiated cardiomyocytes from PSCs with a simple and efficient method is a critical component in achieving stem cell therapy for cardiac regeneration. To date, various small molecules for cardiomyogenesis have been tested, most of which are related to signaling pathways such as BMP, TGFβ, activin, nodal, Wnt, and FGF.7,42 Commonly used as an immunosuppressant for the prevention of organ transplantation rejection, CsA by comparison intriguingly promotes the differentiation of cardiomyocytes from PSCs.15–16 Here, we demonstrate that the effect of CsA in cardiomyogenesis from PSCs mainly results from mPTP inhibition rather than from calcineurin inhibition.
mPTP is an emerging therapeutic target for treating pathologic conditions including cardiac and neurodegenerative diseases.18 Pathologic conditions such as oxidative stress and calcium overload can trigger the opening of the mPTP, which leads to the collapse of Δψm followed by ATP depletion, mitochondrial swelling, and ultimately cell death.18 In fact, mPTP inhibition by CsA has a cardioprotective effect in ischemia‐reperfusion injury in both animal studies18,43 and clinical trial.44 However, only a few publications address mPTP inhibition during the development and differentiation of stem cells. Hom et al recently reported that pharmacological and genetic inhibition of mPTP promotes structural and functional maturation of mitochondria, which aids in the differentiation of cardiomyocytes; these results are similar to our findings, but are based on work with primary cultured cardiomyocytes derived from embryonic heart.45 Thus, our findings from PSC‐derived cardiomyocytes present a practical possibility in the field of regenerative medicine.
This study also demonstrates that mPTP inhibition by CsA directly increases mitochondrial function in differentiating Flk1+ MPCs and H9C2 cardiac cells, and that the mitochondrial function determines the fate of Flk1+ MPCs toward the cardiac lineage. In a mature heart, mitochondrial oxidative metabolism is a major source of energy. In contrast to mature cardiomyocytes, energy metabolism during cardiomyocyte development is altered along with the progression of differentiation.46 During differentiation from undifferentiated ESCs to differentiated cardiomyocytes, mitochondrial function and morphology undergo dramatic changes; in particular, cellular energy metabolism switches from glycolysis to mitochondrial oxidative metabolism.20,47 Morphologically, previous studies and our data show that mitochondria in ESCs or immature cardiomyocytes are spherical and located in the perinuclear area with poorly developed cristae, but become elongated and relocate to the cytoplasm with abundant and organized cristae in mature cardiomyocytes.20,45 In addition, mitochondrial biogenesis and oxidative metabolism‐related genes are increased during cardiomyocyte development.48 These findings might be considered as a consequence, rather than a cause of cardiomyocyte differentiation, merely reflecting increasing contraction and thus the metabolic demands of cardiomyogenesis. However, some evidence suggests that mitochondrial development precedes cardiomyogenesis, and that mitochondrial function and maturation control cardiomyogenesis. A previous study showed that the induction of mitochondrial biogenesis begins shortly before cardiomyocytes beat during EB differentiation.40 Additionally, mitochondrial electron transport chain (ETC) inhibitors block cardiomyocyte differentiation from ESCs,20,48 and complex III of the mitochondrial ETC activity is necessary for cardiomyocyte differentiation via intracellular Ca2+ oscillations.48 Similarly, our results show that CsA directly increases the mitochondrial Ca2+ level during cardiomyocyte differentiation. Furthermore, the differentiation potential of cardiac progenitors into cardiomyocytes relies on mitochondrial content and the capacity for oxidative metabolism.24 Hom et al reported that inhibition of mPTP promotes mitochondrial maturation, which supports cardiomyocyte differentiation in primary cultured cardiomyocytes derived from embryonic heart.45 Our data show that mPTP inhibition by CsA also increases mitochondrial oxidative metabolism and thus cardiomyocyte differentiation; on the other hand, the addition of FCCP with CsA significantly reduces cardiomyocyte differentiation from PSCs. Recently, it was shown that mitochondrial fusion directs cardiomyocyte differentiation via Ca2+, calcineurin, and Notch signaling, independently of mitochondrial function.49 Therefore, the minimal cardiomyogenic effect of calcineurin inhibition in our data might be related to mitochondrial dynamics and Notch signaling. Taken together, the results of previous reports and our data indicate that mitochondria‐related signaling has direct implications for cardiomyogenesis and stem cell fate determination.
Our data collectively showed that antioxidants enhance the cardiomyogenic effect of CsA. ROS are important regulators of intracellular signaling50 and regulate the differentiation of ESCs into cardiomyocytes. Many previous groups have reported that ROS generated from NADPH (nicotinamide adenine dinucleotide phosphate) oxidases mediate cardiomyocyte differentiation26–28,30 while antioxidant treatment inhibits cardiomyocyte differentiation from ESCs,26,28,51 in contrast with our findings. However, one study suggested that valproic acid, which significantly induces ROS generation, delays cardiomyocyte differentiation while further treatment with antioxidant treatment restores it.52 Recent work indicates that antioxidant treatment promotes myocyte differentiation from primary cultured cardiomyocytes derived from embryonic heart while pro‐oxidant treatment inhibits it.45 The most recent report showed that lowering ROS levels increases the Ca2+ transient amplitude, like mature adult cardiomyocyte in human ESC‐derived cardiomyocyte.53 These conflicting findings might depend on cell type and ROS levels: relatively modest concentrations of ROS promote cardiomyogenesis, while high or low concentrations inhibit it.31–32,50,54 Our results showed that antioxidant with CsA treatment augmented cardiomyogenesis but that antioxidants alone or supplementing with a pro‐oxidant did not yield any significant changes. Our interpretation of these findings is that mPTP inhibition by CsA increases mitochondrial oxidative metabolism that induce cardiomyogenesis and the generation of ROS, but that a reduction of ROS and oxidative stress is also critical for efficient cardiomyogenesis under mPTP inhibition (Figure 12A).32,50,54
Figure 12.
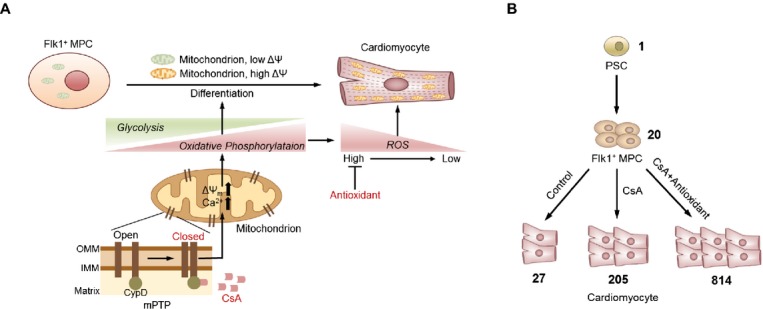
Schematic diagram of mitochondrial metabolic and redox signaling pathways during cardiomyocyte differentiation. A, During differentiation from Flk1+ MPCs to cardiomyocytes, cellular metabolism is changed from glycolysis to oxidative phosphorylation. CsA binds to cycophilin D which is a mitochondrial matrix protein that inhibits mPTP. Inhibition of mPTP activates the mitochondrial oxidative metabolism and promotes cardiomyocyte differentiation from Flk1+ MPCs. Addition of antioxidant with CsA profoundly augments cardiomyocyte differentiation. B, Estimation and comparison of the absolute number of cardiomyocytes generated from one ESC differentiated under control vehicle, CsA and CsA with antioxidants treatment. CsA indicates cyclosporin A; ESC, embryonic stem cell; IMM, inner mitochondrial membrane; MPCs, mesodermal precursor cells; mPTP, mitochondrial permeability transition pore; OMM, outer mitochondrial membrane; PSC, pluripotent stem cell; ROS, reactive oxygen species.
Taken together, our results show that the activation of mitochondrial oxidative metabolism via mPTP inhibition by CsA enhances differentiation of functional cardiomyocytes from PSCs. We also found that antioxidant treatment augments the cardiomyogenic effect of CsA. Because CsA and antioxidants are widely used in clinical practice, supplementation of CsA and antioxidants would be a simple, efficient, and safe method for obtaining an ample amount of PSC‐derived cardiomyocytes (Figure 12B). Additionally, these data provide new insights into the mitochondrial metabolic and redox signaling pathways that are crucial targets for cardiac stem cell therapy in the field of regenerative medicine.
Supplementary Material
Supplemental Video 1. Beating cardiomyocyte colonies incubated with control vehicle in OP9 co-culture system at day 10.5.
Supplemental Video 2. Beating cardiomyocyte colonies incubated with CsA (2 μg/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 3. Beating cardiomyocyte colonies incubated with NIM811 (3.6 μg/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 4. Beating cardiomyocyte colonies incubated with FK506 (100 ng/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 5. Beating cardiomyocytes after αMHC-GFP⁺ FACS sorting at day 10.5.
Supplemental Video 6. Beating cardiomyocyte colonies incubated with control vehicle in the EBs at day 10.5.
Supplemental Video 7. Beating cardiomyocyte colonies incubated with CsA (1 μg/mL) in the EBs at day 10.5.
Sources of Funding
This work was supported by the grant from Korea Healthcare Technology R&D project, Ministry of Health & Welfare of Korea (A120275, Koh) and from the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2012R1A2A1A03007595).
Disclosures
None.
Acknowledgments
We thank Su Jin Seo and Sam Mi Yoo for their technical assistance. We also thank Intae Park and Joon Seo Lim for proofreading our manuscript.
References
- 1.Hansson EM, Lindsay ME, Chien KR. Regeneration next: toward heart stem cell therapeutics. Cell Stem Cell. 2009; 5:364-377 [DOI] [PubMed] [Google Scholar]
- 2.Segers VF, Lee RT. Stem‐cell therapy for cardiac disease. Nature. 2008; 451:937-942 [DOI] [PubMed] [Google Scholar]
- 3.Passier R, van Laake LW, Mummery CL. Stem‐cell‐based therapy and lessons from the heart. Nature. 2008; 453:322-329 [DOI] [PubMed] [Google Scholar]
- 4.Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet. 2012; 379:933-942 [DOI] [PubMed] [Google Scholar]
- 5.Zaruba MM, Soonpaa M, Reuter S, Field LJ. Cardiomyogenic potential of C‐kit(+)‐expressing cells derived from neonatal and adult mouse hearts. Circulation. 2010; 121:1992-2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011; 473:326-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012; 10:16-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soonpaa MH, Rubart M, Field LJ. Challenges measuring cardiomyocyte renewal. Biochim Biophys Acta. 2013; 1833:799-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ban K, Wile B, Kim S, Park HJ, Byun J, Cho KW, Saafir T, Song MK, Yu SP, Wagner M, Bao G, Yoon YS. Purification of cardiomyocytes from differentiating pluripotent stem cells using molecular beacons that target cardiomyocyte‐specific mRNA. Circulation. 2013; 128:1897-1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K. Flk1‐positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000; 408:92-96 [DOI] [PubMed] [Google Scholar]
- 11.Yamashita JK, Takano M, Hiraoka‐Kanie M, Shimazu C, Peishi Y, Yanagi K, Nakano A, Inoue E, Kita F, Nishikawa S. Prospective identification of cardiac progenitors by a novel single cell‐based cardiomyocyte induction. FASEB J. 2005; 19:1534-1536 [DOI] [PubMed] [Google Scholar]
- 12.Joo HJ, Kim H, Park SW, Cho HJ, Kim HS, Lim DS, Chung HM, Kim I, Han YM, Koh GY. Angiopoietin‐1 promotes endothelial differentiation from embryonic stem cells and induced pluripotent stem cells. Blood. 2011; 118:2094-2104 [DOI] [PubMed] [Google Scholar]
- 13.Joo HJ, Choi DK, Lim JS, Park JS, Lee SH, Song S, Shin JH, Lim DS, Kim I, Hwang KC, Koh GY. ROCK suppression promotes differentiation and expansion of endothelial cells from embryonic stem cell‐derived Flk1(+) mesodermal precursor cells. Blood. 2012; 120:2733-2744 [DOI] [PubMed] [Google Scholar]
- 14.Narazaki G, Uosaki H, Teranishi M, Okita K, Kim B, Matsuoka S, Yamanaka S, Yamashita JK. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation. 2008; 118:498-506 [DOI] [PubMed] [Google Scholar]
- 15.Yan P, Nagasawa A, Uosaki H, Sugimoto A, Yamamizu K, Teranishi M, Matsuda H, Matsuoka S, Ikeda T, Komeda M, Sakata R, Yamashita JK. Cyclosporin‐A potently induces highly cardiogenic progenitors from embryonic stem cells. Biochem Biophys Res Commun. 2009; 379:115-120 [DOI] [PubMed] [Google Scholar]
- 16.Fujiwara M, Yan P, Otsuji TG, Narazaki G, Uosaki H, Fukushima H, Kuwahara K, Harada M, Matsuda H, Matsuoka S, Okita K, Takahashi K, Nakagawa M, Ikeda T, Sakata R, Mummery CL, Nakatsuji N, Yamanaka S, Nakao K, Yamashita JK. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin‐A. PLoS One. 2011; 6:e16734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012; 111:1237-1247 [DOI] [PubMed] [Google Scholar]
- 18.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther. 2009; 330:670-678 [DOI] [PubMed] [Google Scholar]
- 19.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005; 434:658-662 [DOI] [PubMed] [Google Scholar]
- 20.Chung S, Dzeja PP, Faustino RS, Perez‐Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007; 4suppl 1:S60-S67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012; 11:596-606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA. Metabolic regulation in pluripotent stem cells during reprogramming and self‐renewal. Cell Stem Cell. 2012; 11:589-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, Jung HJ, McCaffery JM, Kurland IJ, Reue K, Lee WN, Koehler CM, Teitell MA. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011; 30:4860-4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.San Martin N, Cervera AM, Cordova C, Covarello D, McCreath KJ, Galvez BG. Mitochondria determine the differentiation potential of cardiac mesoangioblasts. Stem Cells. 2011; 29:1064-1074 [DOI] [PubMed] [Google Scholar]
- 25.Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013; 18:325-332 [DOI] [PubMed] [Google Scholar]
- 26.Sauer H, Rahimi G, Hescheler J, Wartenberg M. Role of reactive oxygen species and phosphatidylinositol 3‐kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett. 2000; 476:218-223 [DOI] [PubMed] [Google Scholar]
- 27.Ding L, Liang XG, Hu Y, Zhu DY, Lou YJ. Involvement of p38MAPK and reactive oxygen species in icariin‐induced cardiomyocyte differentiation of murine embryonic stem cells in vitro. Stem Cells Dev. 2008; 17:751-760 [DOI] [PubMed] [Google Scholar]
- 28.Sharifpanah F, Wartenberg M, Hannig M, Piper HM, Sauer H. Peroxisome proliferator‐activated receptor alpha agonists enhance cardiomyogenesis of mouse ES cells by utilization of a reactive oxygen species‐dependent mechanism. Stem Cells. 2008; 26:64-71 [DOI] [PubMed] [Google Scholar]
- 29.Crespo FL, Sobrado VR, Gomez L, Cervera AM, McCreath KJ. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells. 2010; 28:1132-1142 [DOI] [PubMed] [Google Scholar]
- 30.Buggisch M, Ateghang B, Ruhe C, Strobel C, Lange S, Wartenberg M, Sauer H. Stimulation of ES‐cell‐derived cardiomyogenesis and neonatal cardiac cell proliferation by reactive oxygen species and NADPH oxidase. J Cell Sci. 2007; 120:885-894 [DOI] [PubMed] [Google Scholar]
- 31.Puceat M. Role of Rac‐GTPase and reactive oxygen species in cardiac differentiation of stem cells. Antioxid Redox Signal. 2005; 7:1435-1439 [DOI] [PubMed] [Google Scholar]
- 32.Drenckhahn JD. Heart development: mitochondria in command of cardiomyocyte differentiation. Dev Cell. 2011; 21:392-393 [DOI] [PubMed] [Google Scholar]
- 33.Hirai H, Ogawa M, Suzuki N, Yamamoto M, Breier G, Mazda O, Imanishi J, Nishikawa S. Hemogenic and nonhemogenic endothelium can be distinguished by the activity of fetal liver kinase (Flk)‐1 promoter/enhancer during mouse embryogenesis. Blood. 2003; 101:886-893 [DOI] [PubMed] [Google Scholar]
- 34.Kodama H, Nose M, Niida S, Nishikawa S. Involvement of the C‐kit receptor in the adhesion of hematopoietic stem cells to stromal cells. Exp Hematol. 1994; 22:979-984 [PubMed] [Google Scholar]
- 35.Cho HJ, Lee CS, Kwon YW, Paek JS, Lee SH, Hur J, Lee EJ, Roh TY, Chu IS, Leem SH, Kim Y, Kang HJ, Park YB, Kim HS. Induction of pluripotent stem cells from adult somatic cells by protein‐based reprogramming without genetic manipulation. Blood. 2010; 116:386-395 [DOI] [PubMed] [Google Scholar]
- 36.Uosaki H, Fukushima H, Takeuchi A, Matsuoka S, Nakatsuji N, Yamanaka S, Yamashita JK. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS One. 2011; 6:e23657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menard C, Pupier S, Mornet D, Kitzmann M, Nargeot J, Lory P. Modulation of L‐type calcium channel expression during retinoic acid‐induced differentiation of H9C2 cardiac cells. J Biol Chem. 1999; 274:29063-29070 [DOI] [PubMed] [Google Scholar]
- 38.Dedkova EN, Blatter LA. Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol. 2012; 52:48-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kharche S, Yu J, Lei M, Zhang H. A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am J Physiol Heart Circ Physiol. 2011; 301:H945-H963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattori F, Chen H, Yamashita H, Tohyama S, Satoh YS, Yuasa S, Li W, Yamakawa H, Tanaka T, Onitsuka T, Shimoji K, Ohno Y, Egashira T, Kaneda R, Murata M, Hidaka K, Morisaki T, Sasaki E, Suzuki T, Sano M, Makino S, Oikawa S, Fukuda K. Nongenetic method for purifying stem cell‐derived cardiomyocytes. Nat Methods. 2010; 7:61-66 [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Nuebel E, Wisidagama DR, Setoguchi K, Hong JS, Van Horn CM, Imam SS, Vergnes L, Malone CS, Koehler CM, Teitell MA. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc. 2012; 7:1068-1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verma V, Purnamawati K, Manasi, Shim W. Steering signal transduction pathway towards cardiac lineage from human pluripotent stem cells: a review. Cell Signal. 2013; 25:1096-1107 [DOI] [PubMed] [Google Scholar]
- 43.Cour M, Loufouat J, Paillard M, Augeul L, Goudable J, Ovize M, Argaud L. Inhibition of mitochondrial permeability transition to prevent the post‐cardiac arrest syndrome: a pre‐clinical study. Eur Heart J. 2011; 32:226-235 [DOI] [PubMed] [Google Scholar]
- 44.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, Andre‐Fouet X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008; 359:473-481 [DOI] [PubMed] [Google Scholar]
- 45.Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA., Jr The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell. 2011; 21:469-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopaschuk GD, Jaswal JS. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol. 2010; 56:130-140 [DOI] [PubMed] [Google Scholar]
- 47.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K. Distinct metabolic flow enables large‐scale purification of mouse and human pluripotent stem cell‐derived cardiomyocytes. Cell Stem Cell. 2013; 12:127-137 [DOI] [PubMed] [Google Scholar]
- 48.Spitkovsky D, Sasse P, Kolossov E, Bottinger C, Fleischmann BK, Hescheler J, Wiesner RJ. Activity of complex III of the mitochondrial electron transport chain is essential for early heart muscle cell differentiation. FASEB J. 2004; 18:1300-1302 [DOI] [PubMed] [Google Scholar]
- 49.Kasahara A, Cipolat S, Chen Y, Dorn GW, II, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and notch signaling. Science (New York, N Y). 2013; 342:734-737 [DOI] [PubMed] [Google Scholar]
- 50.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012; 48:158-167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi T, Lord B, Schulze PC, Fryer RM, Sarang SS, Gullans SR, Lee RT. Ascorbic acid enhances differentiation of embryonic stem cells into cardiac myocytes. Circulation. 2003; 107:1912-1916 [DOI] [PubMed] [Google Scholar]
- 52.Na L, Wartenberg M, Nau H, Hescheler J, Sauer H. Anticonvulsant valproic acid inhibits cardiomyocyte differentiation of embryonic stem cells by increasing intracellular levels of reactive oxygen species. Birth Defects Res A Clin Mol Teratol. 2003; 67:174-180 [DOI] [PubMed] [Google Scholar]
- 53.Birket MJ, Casini S, Kosmidis G, Elliott DA, Gerencser AA, Baartscheer A, Schumacher C, Mastroberardino PG, Elefanty AG, Stanley EG, Mummery CL. PGC‐1alpha and reactive oxygen species regulate human embryonic stem cell‐derived cardiomyocyte function. Stem Cell Reports. 2013; 1:560-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dennery PA. Effects of oxidative stress on embryonic development. Birth Defects Res C Embryo Today. 2007; 81:155-162 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1. Beating cardiomyocyte colonies incubated with control vehicle in OP9 co-culture system at day 10.5.
Supplemental Video 2. Beating cardiomyocyte colonies incubated with CsA (2 μg/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 3. Beating cardiomyocyte colonies incubated with NIM811 (3.6 μg/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 4. Beating cardiomyocyte colonies incubated with FK506 (100 ng/mL) in OP9 co-culture system at day 10.5.
Supplemental Video 5. Beating cardiomyocytes after αMHC-GFP⁺ FACS sorting at day 10.5.
Supplemental Video 6. Beating cardiomyocyte colonies incubated with control vehicle in the EBs at day 10.5.
Supplemental Video 7. Beating cardiomyocyte colonies incubated with CsA (1 μg/mL) in the EBs at day 10.5.