

Abstract
Burkholderia pseudomallei, the etiological agent for melioidosis, is an important cause of community-acquired sepsis in northern Australia and northeast Thailand. Due to the rapid dissemination of disease in acute melioidosis, we hypothesized that dendritic cells (DC) could act as a vehicle for dissemination of B. pseudomallei. Therefore, this study investigated the effect of B. pseudomallei infection on DC migration capacity and whether migration of DC enabled transportation of B. pseudomallei from the site of infection. B. pseudomallei stimulated significantly increased migration of bone marrow-derived DC (BMDC), both in vitro and in vivo, compared to uninfected BMDC. Furthermore, migration of BMDC enabled significantly increased in vitro trafficking of B. pseudomallei and in vivo dissemination of B. pseudomallei to secondary lymphoid organs and lungs of C57BL/6 mice. DC within the footpad infection site of C57BL/6 mice also internalized B. pseudomallei and facilitated dissemination. Although DC have previously been shown to kill intracellular B. pseudomallei in vitro, the findings of this study demonstrate that B. pseudomallei-infected DC facilitate the systemic spread of this pathogen.
INTRODUCTION
Melioidosis is a tropical infection caused by the intracellular bacterium, Burkholderia pseudomallei (1). In regions of northern Australia and northeast Thailand where the organism is endemic, B. pseudomallei infection is an important cause of community-acquired sepsis, which is associated with fatality rates of up to 77% (2–4). During the early phases of infection, macrophages and neutrophils provide essential innate defense against B. pseudomallei. However, macrophages also provide a potential intracellular niche for B. pseudomallei persistence and replication (5–11).
Dendritic cells (DC) are professional antigen-presenting cells that are responsible for linking the innate and adaptive immune responses (12, 13). Highly phagocytic, immature DC contribute to innate immune responses by internalizing and killing pathogens at the site of infection. DC-pathogen interaction triggers DC maturation, a process whereby DC upregulate expression of migration receptors (CC-chemokine receptor 7 [CCR7]), antigen-presenting molecules (major histocompatibility complex class I [MHC-I] and MHC-II), and T cell costimulatory molecules (CD80 and CD86) (12, 13). Mature DC also demonstrate increased production of cytokines: interleukin-6 (IL-6), IL-12, and tumor necrosis factor alpha (TNF-α) (12). Migration of mature, antigen-loaded DC from the site of infection to secondary lymphoid organs is coordinated by the CCR7 ligands, CCL19 and CCL21 (14). Within secondary lymphoid organs, elevated expression of antigen-presenting and costimulatory molecules on mature DC promotes efficient activation of naive T cells and development of pathogen-specific effector T cells (12).
Investigations of the functional responses of DC toward B. pseudomallei have shown internalization and killing of intracellular B. pseudomallei by DC occurs in vitro (15–19). DC maturation was triggered by B. pseudomallei, demonstrated by increased expression of molecules for antigen presentation and costimulation, along with increased cytokine production (15, 17). Furthermore, host susceptibility to B. pseudomallei infection was associated with altered DC functional responses (15). Despite significant maturation and IL-12 production in bone marrow-derived DC (BMDC) from B. pseudomallei-susceptible BALB/c mice, bactericidal activity against intracellular B. pseudomallei was lower than observed for BMDC from B. pseudomallei-resistant C57BL/6 mice (15). Both human and murine DC stimulated with heat-killed B. pseudomallei are capable of activating naive T cells to initiate B. pseudomallei-specific T cell responses and reactivating memory B. pseudomallei-specific T cells to produce IFN-γ in vitro (16, 19–22).
The migration of DC from the site of B. pseudomallei infection to secondary lymphoid organs is an important factor for the development of B. pseudomallei-specific adaptive immune responses. Similar to other intracellular bacteria, B. pseudomallei could potentially interfere with mechanisms driving DC migration, thereby subsequently impairing activation of B. pseudomallei-specific adaptive immune responses. The virulence of Yersinia pestis, the causative agent of the human bubonic or pneumonic plague, is in part, attributed to the bacterium's ability to impair DC migration. Cytoskeleton rearrangement is impaired in DC infected with Y. pestis, rendering them unable to adhere to surfaces and migrate toward CCL19 (23). Alternatively, other intracellular pathogens, such as Listeria monocytogenes, Francisella tularensis, and Streptococcus pneumoniae, use DC migration as a “Trojan horse” to facilitate dissemination (24–26). Blocking of DC migration or depletion of DC correlated with enhanced capacity to restrict systemic dissemination, significantly reduced bacterial loads, and improved resistance to F. tularensis and S. pneumoniae infection (25, 26). DC migration has also been shown to facilitate the rapid dissemination of Bacillus anthracis spores from the lungs to the thoracic lymph nodes (using transgenic mice developed to specifically express green fluorescent protein [GFP] in DC only [CX3CR1+/GFP]) (27, 28). Similarly, B. pseudomallei is an intracellular pathogen capable of persistence within different host cells and rapid systemic spread to multiple organs within hours of exposure (6, 29, 30). Recently, colonization of the brain in a murine model of neurological melioidosis was found to occur via the transmigration of B. pseudomallei-infected CD11b+ phagocytes across endothelial cells (31). Therefore, a mechanism whereby B. pseudomallei uses DC as a vehicle for dissemination is plausible due to the rapid progression of melioidosis from a localized infection to septicemia and involvement of multiple organs. Therefore, the current study investigated (i) the effect of B. pseudomallei infection on DC migration capacity in vitro and (ii) whether in vivo migration of DC enables transportation of B. pseudomallei from the site of infection.
MATERIALS AND METHODS
BMDC culture.
BMDC were cultured according to published methods (15, 32). Bone marrow (BM) was isolated from the femurs and tibias of C57BL/6 mice (8 to 12 weeks old; James Cook University Small Animal Breeding Facility). All animal studies were conducted according to the guidelines and institutional ethics approval provided by James Cook University Animal Ethics Committee (A1601). Isolated BM cells (2 × 105 cells/ml) were cultured in DC media (RPMI 1640 with 10% heat-inactivated fetal bovine serum [HI-FBS], 1.5 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-mercaptoethanol; Invitrogen) supplemented with 10% supernatant from Ag8653 myeloma cells transfected with the gene encoding murine GM-CSF (GM-CSF supernatant; cell line kindly provided by B. Stockinger, NMRI, Mill Hill, London, United Kingdom). On day 3, cultures were fed 10 ml of DC medium supplemented with 10% GM-CSF supernatant and, on day 6, 50% of the medium was replaced. On day 10 of culture, BMDC were harvested by gentle pipetting to remove nonadherent BMDC and then seeded into six-well plates (Nunc) at 106 cells/ml in DC medium.
Infection of BMDC.
A previously characterized clinical B. pseudomallei isolate of low virulence (NCTC 13179) was used throughout the present study for investigating the effect of B. pseudomallei infection on DC migration (30). Cultured BMDC (106 cells/ml in DC media) were infected with log-phase B. pseudomallei (cultured in tryptic soy broth (Acumedia) then washed with phosphate-buffered saline [PBS]) at a multiplicity of infection (MOI) 1:1 and incubated in 5% CO2 at 37°C as previously described (15, 33). E. coli lipopolysaccharide (LPS; 50 ng/ml; Sigma-Aldrich) was used as a positive stimulant to enable comparison and show positive DC migration (34). To determine whether B. pseudomallei isolates of high and low virulence differentially influenced the migration of BMDC in vitro, where specified, BMDC were also infected with NCTC 13178, a highly virulent clinical B. pseudomallei isolate at an MOI of 1:1 (30). Antibiotic protection of BMDC was required to enable enumeration of intracellular bacteria only and prevent uncontrolled extracellular bacterial replication from affecting BMDC viability. B. pseudomallei is resistant to a large range of antibiotics but isolates used in the present study are known to be sensitive to kanamycin (15, 35). Therefore, kanamycin (250 μg/ml; Sigma-Aldrich) was added after 4 h of infection, to eliminate extracellular bacteria.
Flow cytometric analysis of BMDC phenotype.
Expression of CD11c, CD86, MHC class II, and CCR7 on BMDC was analyzed by flow cytometry. Uninfected and B. pseudomallei-infected BMDC were fluorescently stained with a combination of anti-mouse CD11c fluorescein isothiocyanate (FITC; BD Biosciences), anti-mouse CD86 PE (BD Biosciences), anti-mouse CCR7 PerCP-Cy5.5 (eBioscience), and anti-mouse I-A/I-E PE (MHC class II; BD Biosciences). All flow cytometry was performed using fluorescence-activated cell sorting (FACS) with a FACSCalibur apparatus and CellQuest software (BD Biosciences), and postacquisition analysis was performed using FlowJo software (Tree Star, Inc.).
Assessment of intracellular B. pseudomallei within BMDC.
Intracellular B. pseudomallei were released from BMDC by lysing cells with 0.1% Triton-X in sterile deionized water. The lysates were serially diluted (10-fold) in PBS (pH 7.2), and 10-μl aliquots of each dilution were plated in triplicate on Ashdown agar. Agar plates were incubated for 48 h at 37°C prior to enumerating bacteria.
In vitro BMDC migration assay.
Mature DC expressing CCR7 migrate toward CCL19 and/or CCL21 (14, 36). To determine whether B. pseudomallei stimulated in vitro migration of BMDC, we assessed the movement of uninfected and B. pseudomallei-infected BMDC from the upper chamber of a Transwell plate (Corning; Sigma-Aldrich), through a 5-μm-pore polycarbonate membrane toward a chemokine in the lower chamber. Prior to conducting the assay, the Transwell plates were equilibrated by filling the upper and lower chambers with RPMI 1640 containing 1% HI-FBS and then incubated for 1 h at 37°C in 5% CO2. Uninfected, B. pseudomallei-infected and LPS-stimulated BMDC were seeded into the upper chamber (106 cells/well). Chemokines, CCL19 (100 ng/ml; Peprotech [Abacus ALS]) and/or CCL21 (100 ng/ml; Peprotech), were added to the lower chambers where appropriate, and cultures incubated at 37°C in 5% CO2 for 2 h (25, 36). The migrated BMDC were collected from the lower chambers, fixed with 4% paraformaldehyde (ProSciTech), washed, and resuspended in 300 μl of PBS (pH 7.2). Countbright absolute counting beads (Invitrogen) were added immediately prior to performing flow cytometric acquisition. The number of Countbright beads and cell events counted was then used to determine the cell concentration and the total number of migrated cells.
In vivo BMDC migration assay.
The in vivo migration of mature B. pseudomallei-infected BMDC from the footpad injection site to the draining popliteal lymph node (pLN) was assessed. Uninfected, B. pseudomallei-infected and LPS-stimulated BMDC were fluorescently stained with carboxyfluorescein diacetate succinimidyl ester (CFSE; 8 μM [Molecular Probes]), incubated at 37°C for 10 min with gentle inverting. The reaction was stopped by adding RPMI 1640 containing 10% HI-FBS. Washed CFSE-labeled BMDC were rested for 1 h at 37°C, and then 106 BMDC (40 μl) were injected into the left hind footpads of C57BL/6 mice. At designated time points (18, 24, or 36 h) postinjection (p.i.) of BMDC, the draining pLN was harvested, macerated, and digested with collagenase (type IV, 400 U/ml; Gibco/Life Technologies Australia Pty, Ltd.) for 30 min at 37°C to release the DC. Washed pLN cells were stained with anti-mouse CD11c biotin (BD Biosciences) and streptavidin-PE (BD Biosciences). Countbright beads were added to samples prior to analysis. BMDC that had migrated from the footpad to the pLN were identified as CFSE+/CD11cPE+ cells by flow cytometry. The percentage of CFSE+/CD11c+ BMDC that migrated to the pLN was calculated.
Assessment of B. pseudomallei dissemination by BMDC.
The dissemination of B. pseudomallei by BMDC was assessed following footpad injection of B. pseudomallei-infected BMDC or B. pseudomallei alone. Previous data indicate that BMDC internalize between 1 and 10% B. pseudomallei when infected at an MOI of 1:1. Over 24 h, BMDC exhibit killing of intracellular B. pseudomallei. Consequently, the number of intracellular bacteria within BMDC infected with B. pseudomallei at an MOI of 1:1 can range from 102 to 104 CFU (15). Therefore, to compare cell-dependent versus independent dissemination of B. pseudomallei, the left hind footpad of C57BL/6 mice was injected with 106 B. pseudomallei-infected BMDC (104 CFU of intracellular bacteria) or 104 CFU B. pseudomallei (NCTC 13179). The actual number of intracellular B. pseudomallei within 106 BMDC injected into mice was subsequently determined to be 2 × 102 CFU/106 BMDC. On day 1 p.i., mice were euthanized to harvest the draining pLN, inguinal LN (iLN), spleen, lung, and blood. Organs were macerated in PBS (pH 7.2); serial 10-fold dilutions of tissue homogenates were then performed, and aliquots were plated in triplicate on Ashdown agar. Agar plates were incubated for 48 h at 37°C prior to enumerating the CFU.
Internalization by infection site DC and dissemination of B. pseudomallei.
Internalization of B. pseudomallei by skin DC at the site of infection (footpad) and dissemination of B. pseudomallei to the draining pLN and spleen was investigated by determining the bacterial burden of DC (CD11c+) and non-DC (CD11c−) cell fractions within each organ. B. pseudomallei (NCTC 13179; 106 CFU) was injected into the left hind footpad of C57BL/6 mice. Footpads were excised at 2 and 4 h p.i. (n = 3 per time point). The draining pLN and spleen were harvested 24 and 48 h p.i. (n = 3 per time point). Each organ was macerated and digested with collagenase as described above. DC (CD11c+) and non-DC (CD11c−) fractions were prepared using anti-mouse CD11c biotin (BD Biosciences) and streptavidin-magnetic nanoparticles (BD Biosciences). Intracellular B. pseudomallei were released by lysing DC and non-DC cell fractions with 0.1% Triton-X. Lysates were serially diluted (10-fold), and aliquots were plated in triplicate on Ashdown agar. After 48 h of culture, intracellular bacteria within DC (CD11c+) and non-DC (CD11c−) cell fractions were enumerated. An aliquot of each cell fraction was reserved to assess the purity of DC (CD11c+) fractions. DC and non-DC fractions were fluorescently stained with anti-mouse CD11c FITC (BD Biosciences) and anti-mouse CD45 PerCP (BD Biosciences), and the percentage of CD45+/CD11c+ cells were determined by flow cytometry. The average purity (± the standard errors of the mean [SEM]) of DC fractions isolated from the footpad, pLN, spleen, and lung was 93.4% ± 1.9%, 85.3% ± 3.2%, 91.3% ± 1.1%, and 87.9% ± 5.3%, respectively.
Statistical analysis.
Statistical analysis of data was performed using GraphPad Prism 6 Software. Comparisons between time points and stimulation groups were tested for significance using a two-way analysis of variance (ANOVA) with Tukey's post hoc multiple-comparison test. The intracellular survival of B. pseudomallei within BMDC over time (6, 12, 18, and 24 h) was tested using a one-way ANOVA with Turkey's post hoc multiple-comparison test. Differences between tested groups were considered significant if the P value was ≤0.05, and this is indicated in the figures by asterisks (*, P ≤ 0.05; **, P ≤ 0.01).
RESULTS
Maturation and bactericidal activity of B. pseudomallei-infected BMDC.
Increased expression of DC maturation markers (MHC-II, CD86, and CCR7) on BMDC occurred following in vitro infection with B. pseudomallei (NCTC13179; Fig. 1A). CD11c was consistently expressed on uninfected and infected BMDC throughout the 24-h culture period (data not shown). After 24 h, B. pseudomallei-infected and LPS-stimulated (positive control) BMDC demonstrated significantly increased MHC-II and CD86 expression compared to uninfected BMDC (Fig. 1A). In response to B. pseudomallei infection and LPS stimulation, the level of CCR7 significantly increased on BMDC. DC maturation coincided with bactericidal activity against intracellular B. pseudomallei (NCTC 13179), with significantly decreased intracellular survival of B. pseudomallei between 6 and 24 h (Fig. 1B). After 24 h of coculture, the number of surviving bacteria was 2.18 ± 0.23 log10 CFU (2 × 102 CFU)/106 BMDC.
FIG 1.
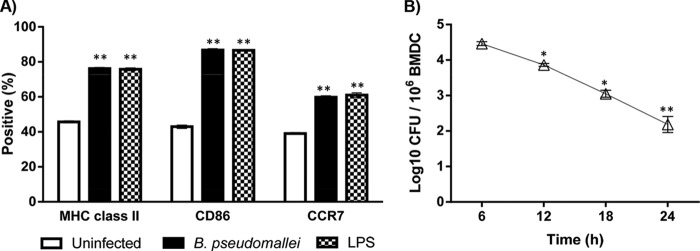
Phenotype and bacterial load of BMDC infected with B. pseudomallei. (A) The expression of MHC-II, CD86, and CCR7 on uninfected, B. pseudomallei-infected, and LPS-stimulated BMDC was determined by flow cytometry. The expression of DC maturation markers, MHC-II, CD86, and CCR7 on BMDC was significantly increased following 24 h of infection with B. pseudomallei (NCTC13179) or LPS. (B) The intracellular survival of B. pseudomallei within BMDC significantly decreased between 6 and 24 h. The data depict the mean ± SEM of two experiments. *, P ≤ 0.05; **, P ≤ 0.01.
Migrated BMDC traffic B. pseudomallei in vitro.
BMDC infected with B. pseudomallei (NCTC 13179) for 18 or 24 h demonstrated significantly increased migration compared to uninfected BMDC (Fig. 2A). B. pseudomallei-infection or LPS stimulation of BMDC elicited similar migration responses in vitro (Fig. 2B). BMDC migration in the presence of the chemokines CCL19 and CCL21 alone (Fig. 2B) or in combination (data not shown) was significantly increased compared to chemokine-free media. However, B. pseudomallei-infected BMDC migration toward CCL19 was significantly higher than CCL21. Comparison of BMDC migration in response to B. pseudomallei isolates of high (NCTC 13178) and low (NCTC 13179) virulence indicated that B. pseudomallei virulence did not affect BMDC migration (Fig. 2C). Therefore, the low-virulence B. pseudomallei isolate, NCTC 13179, was used for the in vivo migration studies. The movement of B. pseudomallei from the upper chamber to the lower chamber of an in vitro migration assay in a cell-dependent (B. pseudomallei-infected BMDC) compared to an independent (B. pseudomallei alone) manner was subsequently determined. Persistence of intracellular B. pseudomallei within migrated, mature BMDC was demonstrated in vitro, which suggests that B. pseudomallei could potentially use DC migration to facilitate dissemination in a cell-dependent manner in vivo (Fig. 2D). Overall, B. pseudomallei did not impair the in vitro migratory capacity of BMDC. Furthermore, migration of BMDC facilitated in vitro trafficking of B. pseudomallei.
FIG 2.

Migration of BMDC infected with B. pseudomallei. The in vitro migration of uninfected, B. pseudomallei-infected, and LPS-stimulated BMDC toward CCR7 ligands CCL19 and CCL21 was assessed. (A) B. pseudomallei-infected (NCTC 13179) BMDC cultured for 18 to 24 h in vitro demonstrated significantly increased migration in the presence of CCL19 compared to uninfected BMDC. (B) Significant numbers of B. pseudomallei-infected (NCTC 13179) and LPS-stimulated BMDC migrated toward the CCR7 ligands, CCL19 (100 ng/ml), and CCL21 (100 ng/ml). The migration of B. pseudomallei-infected BMDC toward CCL19 was significantly greater compared to CCL21. (C) The migration of BMDC infected with isolates of high (NCTC 13178) and low (NCTC 13179) virulence for 24 h was comparable. (D) Subsequently, the movement of B. pseudomallei from the upper chamber to the lower chamber of an in vitro migration assay in a cell-dependent (B. pseudomallei-infected BMDC) compared to an independent (B. pseudomallei alone) manner was determined. The presence of viable intracellular B. pseudomallei within BMDC that migrate in vitro demonstrates the propensity of DC to traffic B. pseudomallei. Bars in panels A to C depict mean ± SEM of three separate experiments. Bars in panel D depict the mean ± standard deviation of one experiment with six replicate wells. *, P ≤ 0.05; **, P ≤ 0.01; NS, not significant.
Mature B. pseudomallei-infected BMDC migrate from the skin to the pLN.
In vivo migration of B. pseudomallei-infected BMDC (CFSE+/CD11c+) from the skin to the draining pLN was assessed following footpad injection of mice (Fig. 3A, B, and C). BMDC infected in vitro with B. pseudomallei for 18 and 24 h prior to footpad injection, demonstrated significant in vivo migration compared to uninfected BMDC (migrated CFSE+/CD11c+ BMDC [%] at 18 h: uninfected [0.15 ± 0.06] versus B. pseudomallei-infected [0.56 ± 0.18], P ≤ 0.05; migrated CFSE+/CD11c+ BMDC [%] at 24 h: uninfected [0.08 ± 0.04] versus B. pseudomallei-infected [0.71 ± 0.02], P ≤ 0.01). Peak in vivo migration of B. pseudomallei-infected BMDC, from the injection site to the pLN, was observed at 24 h p.i. (migrated CFSE+/CD11c+ BMDC [%] at 18 h p.i. = 0.02 ± 0.01, 24 h p.i. = 0.71 ± 0.02, and 36 h p.i. = 0.18 ± 0.02). Using the optimal in vitro infection time (24 h of culture) and in vivo assay time (24 h p.i.), it was observed that B. pseudomallei-infected (NCTC 13179) and LPS-stimulated BMDC elicited similar migration responses in vivo, which were significantly higher than that observed for uninfected BMDC (Fig. 3D). Although B. pseudomallei infection caused significantly increased migration of BMDC to the pLN, <1% of the BMDC injected into the footpad were detected in the draining pLN 24 h p.i.
FIG 3.

In vivo migration of B. pseudomallei-infected BMDC and dissemination of B. pseudomallei. The in vivo migration of B. pseudomallei-infected BMDC from the site of injection to the draining pLN was assessed using CFSE labeling and flow cytometry. Representative FACS plots demonstrate increased numbers of migrated BMDC (CFSE+/CD11c+) in the draining pLN 24 h after mice were injected with uninfected (A), B. pseudomallei-infected (NCTC 13179) (B), and LPS-stimulated (C) BMDC (stimulated for 24 h in vitro). (D) The migration of B. pseudomallei-infected BMDC into pLN was significantly greater compared to uninfected BMDC at 24 h p.i. No significant difference was observed between migration of B. pseudomallei-infected and LPS-stimulated BMDC. (E) Bacterial burden of the draining pLN, iLN, spleen, lung, and blood was assessed at 24 h after footpad injection of mice with B. pseudomallei-infected BMDC (n = 5) or B. pseudomallei alone (n = 5). The bacterial burdens of the spleens and lungs were significantly higher in C57BL/6 mice that received B. pseudomallei-infected BMDC compared to B. pseudomallei alone. Bacteria were not detected in the blood of mice infected with B. pseudomallei alone. Bars depict the mean ± SEM of three experiments. *, P ≤ 0.05; **, P ≤ 0.01.
BMDC migration facilitates B. pseudomallei dissemination.
To compare cell-dependent versus independent dissemination of B. pseudomallei, the organ bacterial loads were enumerated 24 h after footpad of injection of B. pseudomallei-infected BMDC or B. pseudomallei alone (NCTC 13179). Dissemination of B. pseudomallei to the draining pLN, iLN, spleen, lung, and blood occurred after injection of B. pseudomallei-infected BMDC or B. pseudomallei alone (Fig. 3E). As shown previously, the number of intracellular B. pseudomallei within 106 BMDC injected into mice was 2 × 102 CFU/106 BMDC. Although mice injected with B. pseudomallei alone (104 CFU) actually received 50 times the number of CFU compared to B. pseudomallei-infected BMDC, the bacterial burden of the spleen and lung was significantly higher in C57BL/6 mice that received B. pseudomallei-infected BMDC compared to B. pseudomallei alone.
Infection site DC internalize B. pseudomallei and facilitate dissemination.
Internalization of B. pseudomallei by skin DC at the infection site and dissemination of B. pseudomallei to secondary lymphoid tissues was subsequently investigated. C57BL/6 mice were infected with B. pseudomallei (NCTC 13179) via the footpad. The number of intracellular B. pseudomallei within skin DC (CD11c+) and non-DC (CD11c−) fractions isolated from the footpad at 2 and 4 h postinfection were enumerated (Fig. 4A). The ratio of intracellular B. pseudomallei within infection site skin DC was significantly higher than non-DC at 2 h postinfection. Dissemination of B. pseudomallei within DC to the draining pLN (6.26 ± 4.25 B. pseudomallei/103 DC) and spleen (Fig. 4B) was observed at 24 h postinfection. The ratio of B. pseudomallei to spleen DC increased between 24 and 48 h postinfection (Fig. 4B). However, by 48 h postinfection, the bacteria were cleared from the pLN (data not shown), and no intracellular B. pseudomallei was detected within DC and non-DC fractions in the lungs (data not shown). Overall, in support of our hypothesis, skin DC at the site of infection internalized B. pseudomallei and disseminated B. pseudomallei to secondary lymphoid tissue.
FIG 4.
Internalization and dissemination of B. pseudomallei by DC. The internalization of B. pseudomallei (Bps) by skin DC at the footpad infection site and the dissemination of B. pseudomallei to the spleen was determined by quantifying the bacterial burden of DC (CD11c+) and non-DC (CD11c−) fractions for each organ. (A) Skin DC at the site of infection internalized B. pseudomallei. The ratio of intracellular B. pseudomallei within skin DC (Bps:103 cells) at the site of infection was significantly higher than non-DC at 2 h postinfection. (B) Dissemination of B. pseudomallei within DC to the spleen was observed at 24 and 48 h postinfection. At 48 h postinfection, the ratio of intracellular B. pseudomallei within DC (Bps:103 cells) was significantly higher than non-DC in the spleen. The increased ratio of B. pseudomallei to spleen DC (Bps:103 cells), at 48 h postinfection, indicates intracellular replication of B. pseudomallei within spleen DC. Bars depict mean ± SEM of three experiments. *, P ≤ 0.05.
DISCUSSION
This is the first study to investigate in vitro and in vivo migration of DC following infection with B. pseudomallei. BMDC infected with B. pseudomallei in vitro developed a mature phenotype, indicated by increased expression of MHC-II and CD86, and elicited bactericidal activity against intracellular B. pseudomallei, as shown in previous studies (15, 17). Migration of DC from the site of infection to secondary lymphoid organs requires upregulation of CCR7, enabling recognition of migratory cues by CC-chemokines, CCL19 and CCL21 (14, 36). Intracellular bacteria such as F. tularensis stimulate DC migration by triggering DC maturation and the upregulation of CCR7 expression (25). In contrast, Brucella suis infection does not activate DC maturation and increased CCR7 expression (37). In the present study, CCR7 expression was significantly increased on BMDC infected with B. pseudomallei compared to uninfected BMDC, suggesting that they have the capacity for migration. Although CCR7 expression is essential for DC migration, intracellular bacteria such as Y. pestis are known to impair the migration of CCR7+ DC via mechanisms independent from the CCR7/CCL19/CCL21 axis (23).
In vitro migration of mature B. pseudomallei-infected BMDC toward the CCR7 ligands, CCL19 and CCL21, was subsequently demonstrated. Migration of BMDC infected with B. pseudomallei isolates of high (NCTC 13178) and low (NCTC 13179) virulence was comparable in vitro. Both CCL19 and CCL21 are expressed within lymphoid organs and bind to CCR7 with similar affinities. Studies have demonstrated that CCL19 is more potent at attracting DC migration and provides directional signaling for homing DC migration to T-cell-rich areas within secondary lymphoid organs (14, 36). In contrast, initiation of DC migration is via CCL21, the only CCR7 ligand expressed in peripheral tissues on high endothelial venules and lymphatic endothelial cells (14, 38). Therefore, the migration of B. pseudomallei-infected BMDC toward both CCL19 and CCL21 was investigated. In the present study, B. pseudomallei stimulated migration of DC toward both CCR7 ligands, although the chemokine CCL19 attracted significantly higher migration of B. pseudomallei-infected BMDC compared to CCL21. Importantly, the presence of viable intracellular B. pseudomallei within in vitro-migrated BMDC demonstrates the propensity of DC to traffic B. pseudomallei.
The findings of the in vitro assays were confirmed in subsequent in vivo migration studies by injecting mice with BMDC infected in vitro with B. pseudomallei. In the present study, B. pseudomallei-infected BMDC demonstrated significant migration from the site of injection to the draining pLN in comparison to uninfected BMDC. However, <1% of B. pseudomallei-infected BMDC migrated to the draining LN, a relatively low number compared to published studies on DC migration (34). Martìn-Fontecha et al. (34) reported 3% migration for LPS-stimulated BMDC in BALB/c mice. However, differences in the in vivo migration observed between the present study and that by Martìn-Fontecha et al. may reflect variation in the migratory capacity of DC isolated from different mouse strains. In vivo migration of DC within BALB/c infected with Leishmania major was found to be 8 times higher than that observed in C57BL/6 mice (39). Comparison of the in vivo migratory response of B. pseudomallei-infected BMDC from C57BL/6 and BALB/c mice was not feasible due to the high susceptibility of BALB/c mice. The subcutaneous 50% lethal dose for BALB/c mice infected with the low-virulence strain of B. pseudomallei (NCTC 13179) used in the present study was 9 × 102 CFU compared to >108 CFU for B. pseudomallei-resistant C57BL/6 mice (30). This is further complicated by the impaired B. pseudomallei killing efficiency of BMDC derived from BALB/c mice (15). Therefore, BALB/c mice received a lethal higher dose of bacteria when injected with 106 BMDC compared to C57BL/6 mice (data not shown). Consequently, BALB/c mice may not be a suitable model for investigating the in vivo migration of BMDC infected with B. pseudomallei. There is scope for future studies to investigate the dissemination of B. pseudomallei by tissue resident DC in BALB/c mice infected with an appropriate low dose of bacteria. Overall, using the C57BL/6 model, migration of B. pseudomallei-infected BMDC exhibit in vivo migration, similar to LPS-stimulated BMDC, which was significantly increased compared to uninfected immature BMDC.
The dissemination of B. pseudomallei was facilitated by the migration of mature B. pseudomallei-infected BMDC from the site of injection. Despite relatively low numbers of migratory B. pseudomallei-infected BMDC, significant numbers of B. pseudomallei were isolated from the spleens and lungs of mice by 24 h p.i. Furthermore, the combined bacterial burden within pLNs, iLNs, spleens, and lungs of mice was higher than the original number of intracellular B. pseudomallei surviving within the injected BMDC. In the present study, total bacterial loads were determined in tissue homogenates of organs rather than within DC extracted from the organ. Therefore, it was not possible to distinguish whether B. pseudomallei replication has occurred within BMDC or after escape from DC. Regardless, these findings demonstrate that DC migration facilitated the dissemination of B. pseudomallei.
Prior to the present study, the role of skin DC at the site of B. pseudomallei infection had not been investigated. We found here that skin DC internalized B. pseudomallei within the footpad infection site and facilitated the dissemination of B. pseudomallei to the pLN and spleen. Since the percentage of DC within the footpad, pLN, and spleen ranges between 0.5 and 4% of the nucleated cell population, the ratio of B. pseudomallei per 103 cells was determined for DC and non-DC fractions from each organ (40). At the site of infection, the ratio of B. pseudomallei per 103 skin DC was significantly higher compared to non-DC at 2 h postinfection, although these numbers decreased by 4 h postinfection. Unlike the draining pLN, the ratio of B. pseudomallei per 103 spleen DC increased between 24 and 48 h. Collectively, our data demonstrate internalization of B. pseudomallei by skin DC at the site of infection and significant association of disseminated B. pseudomallei within DC in secondary lymphoid organs. Furthermore, DC within the spleen, but not the site of infection or draining LN, appears to provide an intracellular niche for B. pseudomallei replication.
A limitation of the present study is that extensive phenotyping of BMDC and tissue resident DC was not performed. Although the BMDC culture method used in the current study is well established for generating a nonadherent high CD11c+ cell with DC morphology, it does not achieve 100% purity. Furthermore, whereas high CD11c expression is distinctive for DC, CD11c is not exclusively expressed on DC (32, 41). Therefore, additional low frequencies of CD11c-expressing cell types, such as inflammatory macrophages, could potentially have been included within the tissue resident DC fractions. In addition, without extensive DC phenotyping it is not possible to distinguish migratory DC from tissue resident DC in the current study. Since it is plausible that B. pseudomallei spreads from migratory skin DC to resident spleen DC, future studies employing techniques for more extensive phenotyping will enable determination of which DC subset/s (migratory skin DC, resident spleen DC, or both) facilitate B. pseudomallei persistence in the spleen. Importantly, our findings identify CD11c+ DC migration as a mechanism for B. pseudomallei cell-dependent dissemination. These findings warrant further characterization studies, possibly using transgenic mice with specific GFP expression in DC or extensive characterization of the DC phenotype, to confirm the beneficial as opposed to the detrimental role of DC subsets and indeed other phagocytic cells involved in the in vivo trafficking of B. pseudomallei.
In summary, we investigated the effect of B. pseudomallei on the migration of DC and their potential to inadvertently facilitate systemic dissemination of the bacteria. After B. pseudomallei infection, BMDC demonstrated significant in vitro and in vivo migration compared to uninfected BMDC. Although DC are capable of killing internalized B. pseudomallei, the findings presented here indicate that the persistence of small numbers of B. pseudomallei within DC enabled the dissemination of bacteria to other organs via DC migration. Furthermore, B. pseudomallei replicated inside spleen DC in mice contradicting our in vitro studies whereby murine BMDC were capable of killing intracellular B. pseudomallei. Previous in vitro studies using human and murine DC have consistently shown that DC grown under in vitro culture conditions and protected by antibiotics are capable of killing intracellular B. pseudomallei (15, 17). However, organ-specific differences in the ability of DC to kill intracellular B. pseudomallei were observed in vivo, in particular within the spleen, where DC were a site for B. pseudomallei persistence. This highlights the need for caution when interpreting in vitro studies and the requirement for subsequent studies using animal models to determine the true in vivo relevance of in vitro observations. Further studies are warranted to determine which DC subset(s) (migrating skin DC, resident spleen DC, or both) facilitates B. pseudomallei persistence in the spleen. Notably, this is the first evidence that DC migration facilitates dissemination of B. pseudomallei to secondary lymphoid organs. The migration of mature B. pseudomallei-infected DC to secondary lymphoid organs suggests that these cells have the potential to present antigen to naive T cells. Given that DC migration is central to the development of protective adaptive immune responses to infection, clarification of the contribution of DC migration to disease progression, as opposed to the development of protection, in B. pseudomallei infection is of paramount importance and may facilitate the identification of potential novel immunomodulatory therapies and preventative strategies.
ACKNOWLEDGMENT
This study was supported by the James Cook University Graduate Research Scheme.
Footnotes
Published ahead of print 28 July 2014
REFERENCES
- 1. Cheng AC, Currie BJ. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383–416. 10.1128/CMR.18.2.383-416.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Currie BJ, Ward L, Cheng AC. 2010. The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl. Trop. Dis. 4:e900. 10.1371/journal.pntd.0000900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wiersinga WJ, Currie BJ, Peacock SJ. 2012. Melioidosis. N. Engl. J. Med. 367:1035–1044. 10.1056/NEJMra1204699 [DOI] [PubMed] [Google Scholar]
- 4. Limmathurotsakul D, Wongratanacheewin S, Teerawattanasook N, Wongsuvan G, Chaisuksant S, Chetchotisakd P, Chaowagul W, Day NP, Peacock SJ. 2010. Increasing incidence of human melioidosis in Northeast Thailand. Am. J. Trop. Med. Hyg. 82:1113–1117. 10.4269/ajtmh.2010.10-0038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Woodman ME, Worth RG, Wooten RM. 2012. Capsule influences the deposition of critical complement C3 levels required for the killing of Burkholderia pseudomallei via NADPH-oxidase induction by human neutrophils. PLoS One 7:e52276. 10.1371/journal.pone.0052276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leakey AK, Ulett GC, Hirst RG. 1998. BALB/c and C57BL/6 mice infected with virulent Burkholderia pseudomallei provide contrasting animal models for the acute and chronic forms of human melioidosis. Microb. Pathog. 24:269–275. 10.1006/mpat.1997.0179 [DOI] [PubMed] [Google Scholar]
- 7. Barnes JL, Williams NL, Ketheesan N. 2008. Susceptibility to Burkholderia pseudomallei is associated with host immune responses involving tumor necrosis factor receptor-1 (TNFR1) and TNF receptor-2 (TNFR2). FEMS Immunol. Med. Microbiol. 52:379–388. 10.1111/j.1574-695X.2008.00389.x [DOI] [PubMed] [Google Scholar]
- 8. Riyapa D, Buddhisa S, Korbsrisate S, Cuccui J, Wren BW, Stevens MP, Ato M, Lertmemongkolchai G. 2012. Neutrophil extracellular traps exhibit antibacterial activity against Burkholderia pseudomallei and are influenced by bacterial and host factors. Infect. Immun. 80:3921–3929. 10.1128/IAI.00806-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morris J, Williams N, Rush C, Govan B, Sangla K, Norton R, Ketheesan N. 2012. Burkholderia pseudomallei triggers altered inflammatory profiles in a whole-blood model of type 2 diabetes-melioidosis comorbidity. Infect. Immun. 80:2089–2099. 10.1128/IAI.00212-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rinchai D, Khaenam P, Kewcharoenwong C, Buddhisa S, Pankla R, Chaussabel D, Bancroft GJ, Lertmemongkolchai G. 2012. Production of interleukin-27 by human neutrophils regulates their function during bacterial infection. Eur. J. Immunol. 42:3280–3290. 10.1002/eji.201242526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barnes JL, Ketheesan N. 2007. Development of protective immunity in a murine model of melioidosis is influenced by the source of Burkholderia pseudomallei antigens. Immunol. Cell Biol. 85:551–557. 10.1038/sj.icb.7100084 [DOI] [PubMed] [Google Scholar]
- 12. Banchereau J, Briere F, Cauz C, Davoust J, Lebecque S, Liu Y-J, Pulendran B, Palucka K. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. 10.1146/annurev.immunol.18.1.767 [DOI] [PubMed] [Google Scholar]
- 13. Shortman K, Naik SH. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7:19–30. 10.1038/nri1996 [DOI] [PubMed] [Google Scholar]
- 14. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. 2013. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 24:269–283. 10.1016/j.cytogfr.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 15. Williams NL, Kloeze E, Govan BL, Korner H, Ketheesan N. 2008. Burkholderia pseudomallei enhances maturation of bone marrow-derived dendritic cells. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S71–S75. 10.1016/S0035-9203(08)70019-1 [DOI] [PubMed] [Google Scholar]
- 16. Charoensap J, Engering A, Utaisincharoen P, van Kooyk Y, Sirisinha S. 2008. Activation of human monocyte-derived dendritic cells by Burkholderia pseudomallei does not require binding to the C-type lectin DC-SIGN. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S76–S81. 10.1016/S0035-9203(08)70020-8 [DOI] [PubMed] [Google Scholar]
- 17. Horton RE, Morrison NA, Beacham IR, Peak IR. 2012. Interaction of Burkholderia pseudomallei and Burkholderia thailandensis with human monocyte-derived dendritic cells. J. Med. Microbiol. 61:607–614. 10.1099/jmm.0.038588-0 [DOI] [PubMed] [Google Scholar]
- 18. Hodgson KA, Morris JL, Feterl ML, Govan BL, Ketheesan N. 2011. Altered macrophage function is associated with severe Burkholderia pseudomallei infection in a murine model of type 2 diabetes. Microbes Infect. 13:1177–1184. 10.1016/j.micinf.2011.07.008 [DOI] [PubMed] [Google Scholar]
- 19. Elvin SJ, Healey GD, Westwood A, Knight SC, Eyles JE, Williamson ED. 2006. Protection against heterologous Burkholderia pseudomallei strains by dendritic cell immunization. Infect. Immun. 74:1706–1711. 10.1128/IAI.74.3.1706-1711.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Charoensap J, Utaisincharoen P, Engering A, Sirisinha S. 2009. Differential intracellular fate of Burkholderia pseudomallei 844 and Burkholderia thailandensis UE5 in human monocyte-derived dendritic cells and macrophages. BMC Immunol. 10:20–27. 10.1186/1471-2172-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Healey GD, Elvin SJ, Morton M, Williamson ED. 2005. Humoral and cell-mediated adaptive immune responses are required for protection against Burkholderia pseudomallei challenge and bacterial clearance postinfection. Infect. Immun. 73:5945–5951. 10.1128/IAI.73.9.5945-5951.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tippayawat P, Pinsiri M, Rinchai D, Riyapa D, Romphruk A, Gan YH, Houghton RL, Felgner PL, Titball RW, Stevens MP, Galyov EE, Bancroft GJ, Lertmemongkolchai G. 2011. Burkholderia pseudomallei proteins presented by monocyte-derived dendritic cells stimulate human memory T cells in vitro. Infect. Immun. 79:305–313. 10.1128/IAI.00803-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Velan B, Bar-Haim E, Zauberman A, Mamroud E, Shafferman A, Cohen S. 2006. Discordance in the effects of Yersinia pestis on the dendritic cell functions manifested by induction of maturation and paralysis of migration. Infect. Immun. 74:6365–6376. 10.1128/IAI.00974-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pron B, Boumaila C, Jaubert F, Berche P, Milon G, Geissmann F, Gaillard JL. 2001. Dendritic cells are early cellular targets of Listeria monocytogenes after intestinal delivery and are involved in bacterial spread in the host. Cell Microbiol. 3:331–340. 10.1046/j.1462-5822.2001.00120.x [DOI] [PubMed] [Google Scholar]
- 25. Bar-Haim E, Gat O, Markel G, Cohen H, Shafferman A, Velan B. 2008. Interrelationship between dendritic cell trafficking and Francisella tularensis dissemination following airway infection. PLoS Pathog. 4:e1000211. 10.1371/journal.ppat.1000211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosendahl A, Bergmann S, Hammerschmidt S, Goldmann O, Medina E. 2013. Lung dendritic cells facilitate extrapulmonary bacterial dissemination during pneumococcal pneumonia. Front. Cell Infect. Microbiol. 3:21. 10.3389/fcimb.2013.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cleret A, Quesnel-Hellmann A, Vallon-Eberhard A, Verrier B, Jung S, Vidal D, Mathieu J, Tournier JN. 2007. Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J. Immunol. 178:7994–8001. 10.4049/jimmunol.178.12.7994 [DOI] [PubMed] [Google Scholar]
- 28. Brittingham KC, Ruthel G, Panchal RG, Fuller CL, Ribot WJ, Hoover TA, Young HA, Anderson AO, Bavari S. 2005. Dendritic cells endocytose Bacillus anthracis spores: implications for anthrax pathogenesis. J. Immunol. 174:5545–5552. 10.4049/jimmunol.174.9.5545 [DOI] [PubMed] [Google Scholar]
- 29. Hoppe I, Brenneke B, Rohde M, Kreft A, Haussler S, Reganzerowski A, Steinmetz I. 1999. Characterization of a murine model of melioidosis: comparison of different strains of mice. Infect. Immun. 67:2891–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barnes JL, Ketheesan N. 2005. Route of infection in melioidosis. Emerg. Infect. Dis. 11:638–639. 10.3201/eid1104.041051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu PJ, Chen YS, Lin HH, Ni WF, Hsieh TH, Chen HT, Chen YL. 2013. Induction of mouse melioidosis with meningitis by CD11b+ phagocytic cells harboring intracellular B. pseudomallei as a Trojan horse. PLoS Negl. Trop. Dis. 7:e2363. 10.1371/journal.pntd.0002363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lutz MB, Kukutsch N, Ogilvie ALJ, Robner S, Koch F, Romani N, Schuler G. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223:77–92. 10.1016/S0022-1759(98)00204-X [DOI] [PubMed] [Google Scholar]
- 33. Williams NL, Morris JL, Rush C, Govan BL, Ketheesan N. 2011. Impact of streptozotocin-induced diabetes on functional responses of dendritic cells and macrophages toward Burkholderia pseudomallei. FEMS Immunol. Med. Microbiol. 61:218–227. 10.1111/j.1574-695X.2010.00767.x [DOI] [PubMed] [Google Scholar]
- 34. Martìn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198:615–621. 10.1084/jem.20030448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feterl M, Govan BL, Ketheesan N. 2008. The effect of different Burkholderia pseudomallei isolates of varying levels of virulence on Toll-like-receptor expression. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S82–S88. 10.1016/S0035-9203(08)70021-X [DOI] [PubMed] [Google Scholar]
- 36. Ricart BG, John B, Lee D, Hunter CA, Hammer DA. 2011. Dendritic cells distinguish individual chemokine signals through CCR7 and CXCR4. J. Immunol. 186:53–61. 10.4049/jimmunol.1002358 [DOI] [PubMed] [Google Scholar]
- 37. Billard E, Dornand J, Gross A. 2007. Brucella suis prevents human dendritic cell maturation and antigen presentation through regulation of tumor necrosis factor alpha secretion. Infect. Immun. 75:4980–4989. 10.1128/IAI.00637-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Toebak MJ, Gibbs S, Bruynzeel DP, Scheper RJ, Rustemeyer T. 2009. Dendritic cells: biology of the skin. Contact Dermatitis 60:2–20. 10.1111/j.1600-0536.2008.01443.x [DOI] [PubMed] [Google Scholar]
- 39. Misslitz AC, Bonhagen K, Harbecke D, Lippuner C, Kamradt T, Aebischer T. 2004. Two waves of antigen-containing dendritic cells in vivo in experimental Leishmania major infection. Eur. J. Immunol. 34:715–725. 10.1002/eji.200324391 [DOI] [PubMed] [Google Scholar]
- 40. Duriancik DM, Hoag KA. 2009. The identification and enumeration of dendritic cell populations from individual mouse spleen and Peyer's patches using flow cytometric analysis. Cytometry A 75:951–959. 10.1002/cyto.a.20794 [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto D, Miller J, Merad M. 2011. Dendritic cell and macrophage heterogeneity in vivo. Immunity 35:323–335. 10.1016/j.immuni.2011.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]