
Fig. 4.
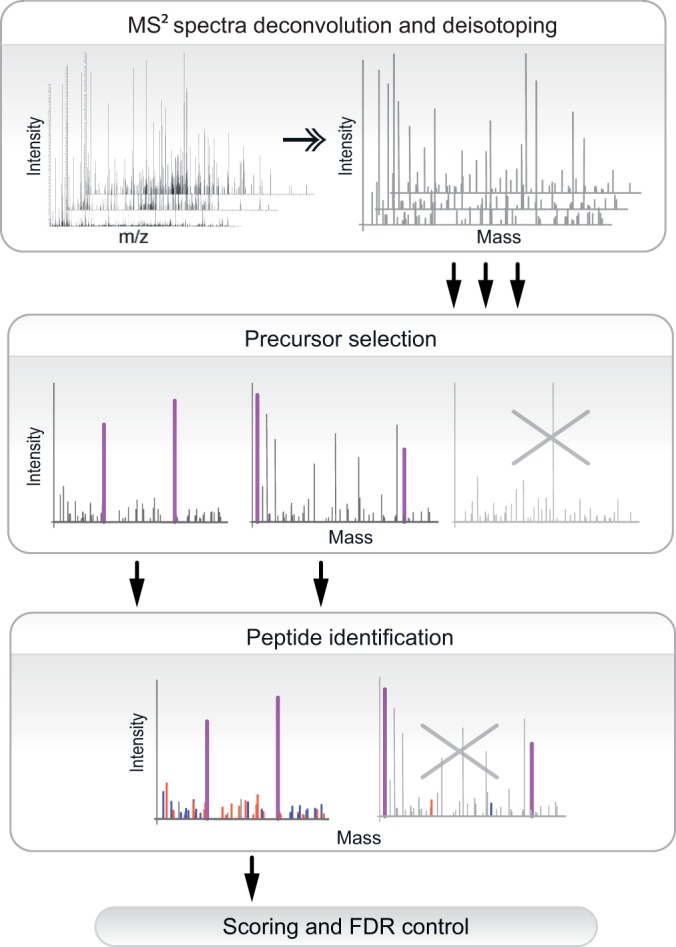
Schematic outline of SlinkS workflow for the identification of interpeptide disulfide bridges. Raw files are deconvoluted, deisotoped, converted to peak list files, and subjected to SlinkS analysis. SlinkS first performs precursor mass selection to obtain the monoisotopic mass of each disulfide-cleaved peptide, and spectra that do not contain any candidate peptide pairs are discarded. Next, all MS2 ions are matched against in silico fragments of each candidate peptide, and spectra that do not have sufficient matched fragment ions are omitted. Finally, the n-score and FDR are calculated, and identification results are filtered based on the user-defined n-score and FDR cutoff value.