

Abstract
We report a case of type-A Coffin-Siris syndrome (CSS) with a unique constellation of congenital heart defects. A 17-year-old Indian boy was referred to our hospital for central cyanosis with features of right heart failure. The cardiac abnormalities included biventricular outflow tract obstruction, small atrial septal defect (ASD), subaortic ventricular septal defect, drainage of left superior venacava to left atrial appendage, and aortic arch anomaly. Patient underwent successful right ventricular infundibular resection, subaortic membrane resection, closure of atrial and ventricular septal defect, rerouting left superior vena cava to left pulmonary artery and aortic valve replacement.
Keywords: Atrial septal defect (ASD), arteria lusoria, Adden Brooks Cognitive Examination (ACE), Coffin-Siris syndrome (CSS), Infundibular stenosis, Persistent left superior venacava (LSVC), subaortic membrane (subAS), ventricular septal defect (VSD)
INTRODUCTION
Congenital heart diseases associated with Coffin-Siris syndrome (CCS) are seen in an average of 30% of reported patients.[1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22] Cardiac defects are extremely heterogeneous in this syndrome. None of the defect is sine-qua-non for the syndrome. The common defects include atrial or ventricular level of septal defects, patent ductus arteriosus and Tetralogy of Fallot. This case report for the first time in literature would describe a rare case of CSS with multiple unusual cardiac (atrial septal defect, ventricular septal defect, subaortic membrane, right ventricular infundibular stenosis) and vascular malformation (persistent left superior vena cava and aortic arch anomaly) successfully managed by surgery.
CASE REPORT
A 17-year-old boy presented with an episode of exertional syncope and gradually worsening exertional breathlessness for last 6 months. The proband was not of consanguineous parents and he was the youngest amongst his four siblings. Mother had not used alcohol or hydantoin in antenatal period. Neonatal history was suggestive of small for gestational age (SGA) with birth weight of 2.25 kg, Apgar score of 7/10 and respiratory support was needed for first 7 days of life. The failure to thrive (FTT) was apparent.
The developmental milestones record showed delayed onset of walking. His school performance was fair and had completed tenth class. For the first time, he had one episode of giddiness and syncope at the age of 13 years while playing at school but went on without evaluation. Morphometric analysis showed normal adrenarche, height of 156 cm (≤2SD), weight of 37 Kg, lower BMI of 16 Kg/m2 , head circumference of 51 cm (-3 to -4 SD) and upper segment/lower segment was 0.9. The coarse facial feature includes thin and long face, the right eye ptosis, up slanting palpebral fissure, prominent arched long eyebrows, synophres, thick eyelashes, hypoplastic nasal bridge, bulbous nose, wide philtrum, thick and prominent lips, maloccluded teeth, and scalp hair was sparse [Figure 1]. Pectus excavatum and scoliosis were both apparent [Figure 2]. The left little finger was hypoplastic, devoid of nail and terminal phalanx [Figure 3]; and absent nail of little and fourth toe of both feet [Figure 4]. Adden Brooks Cognitive Examination (ACE) showed IQ of ACE III-86/100, memory of 21/26, fluency 10/14, language 25/26, visiospastic of 13/16 and affection of 17/18. Ophthalmology consultation revealed ptosis in the right eye with normal vision. Hearing was normal. Central cyanosis was consistent with room air SpO2 of 63% and pan digital clubbing. Jugular venous pulse was elevated with prominent “a” wave and there were bilateral outflow tract murmur of grade IV/VI.
Figure 1.
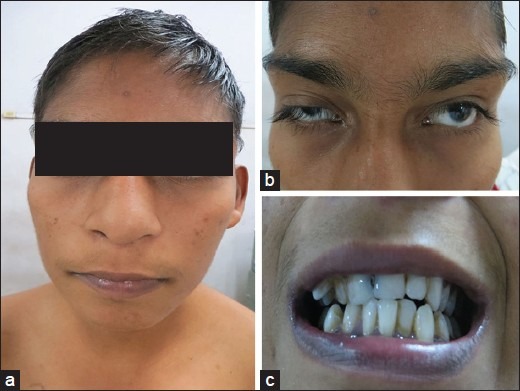
Frontal view photograph showed (a) typical coarse faces (microcephaly, thick and long eye brows, wide mouth, and bulbous nose); (b) synophres; (c) malocclusion
Figure 2.
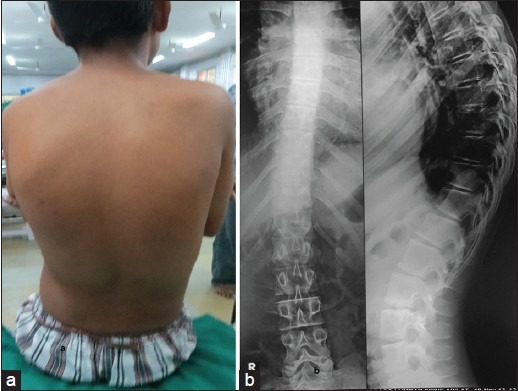
(a) Photograph of back showed scoliosis causing a sideways curve of thoracic spine in form of S-shaped curve and (b) The radiograph of spine in anterior posterior view confirms scoliosis
Figure 3.
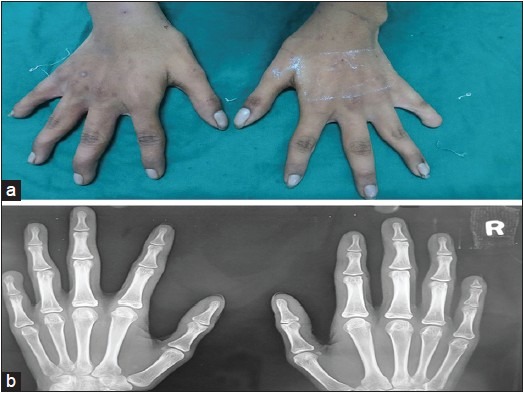
(a) The photograph of hands showed the left hand had shorter little finger with absence nail and (b) Radiographs of left hand showed absence of distal phalange of left little finger
Figure 4.
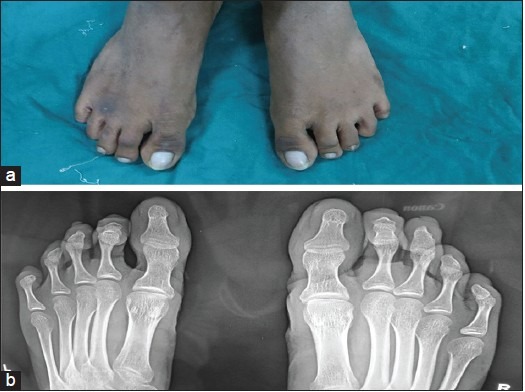
(a) The photograph of both right and left feet showed hypoplastic nails in both the fourth toes and (b) Radiographs of the feet normal
Twelve lead electrocardiogram (ECG) showed sinus rhythm, biventricular hypertrophy, right atrial hypertrophy, and right axis deviation. Chest radiograph showed cardiothoracic ratio of 70% with oligemic lung fields, right ventricular apex, right atrial enlargement, concave pulmonary bay, left aortic arch, normal rib cage, and scoliosis [Figure 2]. Transthoracic echocardiography (TTE) showed significant right ventricular infundibular stenosis of gradient of 160 mm Hg, discrete subAS with significant obstruction of gradient of 70 mm Hg, mild aortic regurgitation, good left ventricular function, and mildly decreased right ventricular function with significant biventricular hypertrophy. Right atrium was dilated with mild tricuspid regurgitation. Situs was normal with no other apparent cardiac defects. However the TTE could not yield any anatomical reason except suspicion of right to left shunt through ASD because of high right atrial pressure. Transesophagial echocardiography (TEE) confirmed discrete subAS and right ventricular infundibular obstruction [Video 1]; agitated saline contrast follow up from left antecubetal vein showed early filling of left atrium and left ventricle and delayed filling of right atrium and right ventricle, conforming the persistence of left superior vena cava (LSVC) draining in to left atrium [Video 2]. 64 Slice computerized tomography (CT) of chest with pulmonary angiogram showed normal lung parenchyma and pulmonary arteries. Routine evaluation showed raised hemoglobin of 19.5 gm/dl and packed cell volume (PCV) of 61.5% and non-elevated level of thyroid stimulating hormone (TSH) and alkaline phosphatases (AP). Abdominal ultrasonography was normal. Plain CT scan of brain was normal.64 slice CT with contrast enhancement confirmed right superior venacava without innominate connection, persistent left superior vena cava connection to left atrium, left common carotid from innominate artery, and arteria lusoria [Figure 5].
Figure 5.
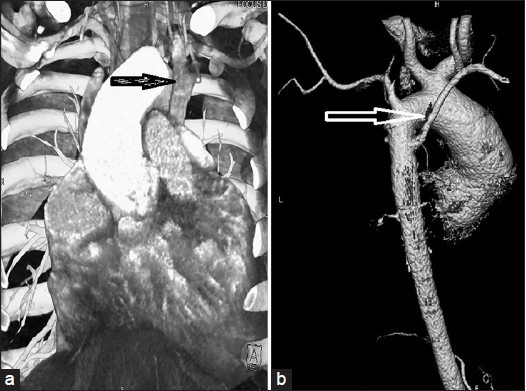
64 Slice Computerized Tomography (CT): The left-sided superior vena cava (a); Variant of aortic arch branching, the left common carotid artery originates separately from the innominate artery and the aberrant right subclavian artery arises from the aorta distal to the origin of the left subclavian artery (arteria lusoria) shown by black arrow (b)
Cardiac catherization showed saturation data of superior vena cava (SVC) to be 47.3%, inferior vena cava (IVC) 48.8%, right atrium (RA) 50.3%, right ventricle (RV) 48%, left ventricle (LV) 60%, femoral artery (FA) 65%, MVO2 47.7%, recorded pressure in various cardiac chamber includes right atrial pressure (RAP) of 3 mmHg, RV of170/0-10 mmHg, FA of 120/80 mmHg, LV pressure of 190/0-6 mmHg, pull back left ventricular outflow tract (LVOT) gradient of 70 mmHg. RV angiogram in shallow left anterior oblique (LAO) cranial and right lateral showed significant infundibular obstruction, mild tricuspid regurgitation and hypertrophied hypo contractile RV [Figure 6 and Videos 3 and 4]. LV angiogram in LAO view (LAXO) view confirmed discrete sub aortic membrane [Figure 7 and Video 5]. Aortic root angiogram confirmed Sellers’ grade II aortic regurgitation (AR), dilated aortic root and subaortic ventricular septal defect. Selective coronary angiogram showed normal origin and course of coronaries with dominant left system. Left antecubetal vein contrast follow through confirmed the left superior vena cava draining into left atrium without innominate continuation [Video 6].
Figure 6.
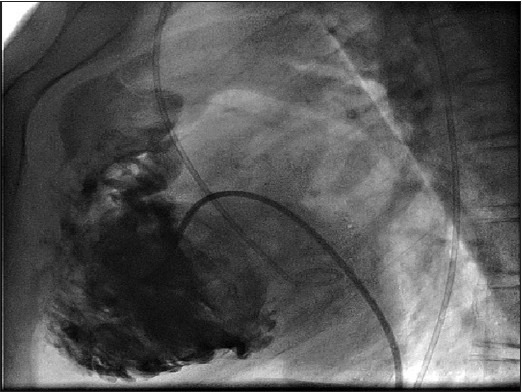
Right ventricular angiogram shows significant hypertrophied trabeculated right ventricle, infundibular stenosis,and smooth inflow and mild tricuspid valve regurgitation
Figure 7.
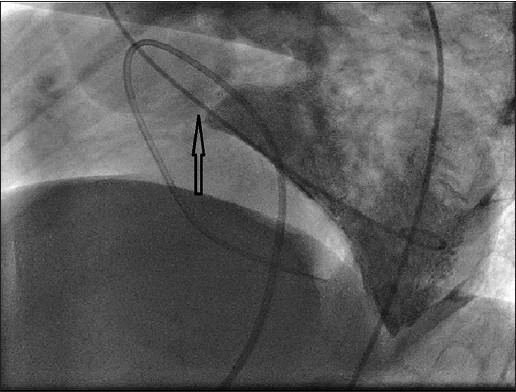
Left ventricular angiogram in left anterior oblique-Cranial (60-30 degree) shows discrete sub aortic membrane
Skeletal radiography revealed scoliosis [Figure 2]. Bone age was normal. Karyotype study was normal.
Intra cardiac repair of this was quite challenging. The surgical pathology altogether included seven cardiac defects (ASD, VSD, direct opening coronary vein into right atrium, infundibular stenosis; subaortic membrane), and extra cardiac defects (left superior venacava connected to left atrial appendage and arteria lusoria). The operation stretched over 6 hours [Figure 8]. This successful surgical note incudes small ASD closure, small subaortic VSD closure [Figure 9], massive infundibular resection, subAS resection with aortic valve replacement (St. Jude Medical, size of 17 mm) [Figure 10], rerouting of left superior venacava to left lower pulmonary artery. Final sternal closure was done after 8 hours for hemostatic security purpose. One month follow up is uneventful. He did not need any physical or mental rehabilitation.
Figure 8.
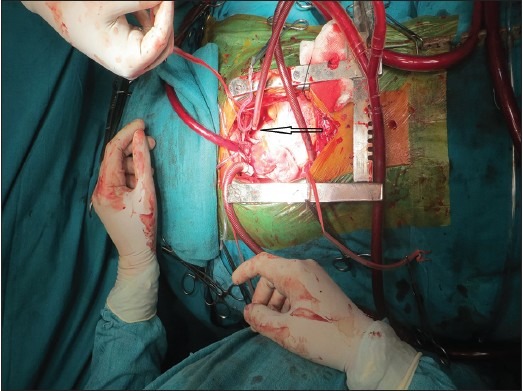
Left superior vena cava connected to left atrial appendage marked by black arrow
Figure 9.
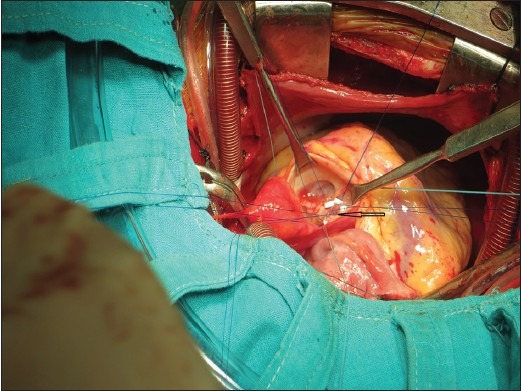
Subaortic ventricular septal defect (5 mm) was closed using Dacron patch indicated black arrow
Figure 10.
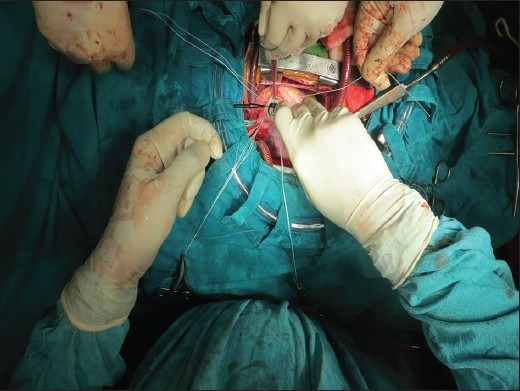
Aortic valve replacement was done using 17 mm St. Jude aortic valve prosthesis after subaortic membrane resection, is marked by black arrow
DISCUSSION
The diagnosis of this condition is purely based on the most frequent clinical features. The role of genetic evaluation is complementary. We used the clinical criteria most recently framed by Schrier SA et al., 2012[19] to conclude that ours is a case of CSS type A [Table 1]. This criteria requires the presence of all the three of the major findings and one of each of the three categories of minor findings. Major findings are fifth digit nail and distal phalanx hypoplasia/aplasia, developmental or cognitive delay and facial features. The three categories of minor findings are: ectodermal (hirsutism/hypertrichosis, sparse scalp hair, dental anomalies); constitutional (microcephaly, intrauterine growth retardation, short stature, failure to thrive, frequent infections), and organ related (feeding difficulties, cardiac, neurological, gastrointestinal, genitourinary, hearing and vision related). The clinical profile of our case includes all the three major criteria in form of 1.absence of nail and distal phalanx of left little finger and hypoplastic nails of both fourth toes, 2.borderline developmental or cognitive delay and 3.coarse facial features. The three categories of minor findings are: ectodermal (sparse scalp hair, malocclusion of right lower canine and premolar); constitutional (microcephaly, failure to thrive), organ related (cardiac defects). The close differential diagnosis of this case has been discussed [Table 2]. In near future, to confirm the diagnosis of CSS, it would be necessary to perform molecular genetic testing.
Table 1.
Confirming the diagnosis of Coffin-Siris syndrome in our case using Schrier's criteria

Table 2.
The seven close differential diagnoses of Coffin-Siris Syndrome
PubMed, Medline, and MeSH data search provided us nearly 100 cases of this rare disorder by December 2013 as described below. Congenital cardiovascular anomalies associated with this syndrome are quite heterogeneous and incidence is in an average up to 30%. The acyanotic cardiac defects like atrial or ventricular level septal defects, patent ductus arteriosus, patent foramen ovale are more frequent than cyanotic conditions like Tetralogy of Fallot, left superior vena cava and other unspecified defects. Among cyanotic congenital cardiac defects, Tetralogy of Fallot is most frequently reported. Peripheral vascular malformation includes hemangioma and arch anomalies. Left superior vena with unroofed coronary sinus with positional worsening of cyanosis successfully managed by percutaneous closure has been reported before.
Literature review shows the following congenital cardiac and extra cardiac defects associated with Coffin-Siris syndrome from 1966 to 2013 as given below
1970: Coffin GS [1] and Bartsocas CS [2]-First time reported Coffin-Siris syndrome in association with cardiovascular malformation in the form of cutaneous hemangioma.
1978: Carey JC [3] - Two of their six cases had Tetralogy of Fallot (TOF), proved by cardiac catheterization.
1979: Schinzel A [4]-One case of Tetralogy of Fallot (TOF) proved by cardiac catheterization
1980: Ueda K [5]-One case of ventricular septal defect
1988: Alembic Y [6]-One case of hypertrophic cardiomyopathy
1991: Pillar Levy [7] -This review involves a total 20 cases, out of which 10 cases had cardiac defects. Author had two of his own in this review without cardiac defect. Photographs were available in nine cases. The lesions were: A patent foramen ovale (1), tetralogy of Fallot (1), atrial septal defect (4), patent ductus arteriosus (3), ventricular septal defect (2), pulmonary stenosis (1), and persistent left superior vena cava.
1991: Rabe Petra [8] - One of two sisters had ventricular septal defect.
1992: Kirel B [9] -One case operated for Patent ductus arteriosus.
1995: Imaizumi, Kiyoshi [10]-one Small atrial septal defect and one small ventricular septal defect.
1996: Coulibaly, Bema B [11]-One Male still born with cardiac defect (not specified).
1998: Ounap K [12]-One of two sisters had ventricular septal defect (VSD), atrial septal defect (ASD) and patent ductus arteriosus (PDA).
1999: Geggel RL [13]-Left superior vena cava (LSVC) connection to unroofed coronary sinus closed by device.
2000: D H Karunathilka [14]-Perimembranous ventricular septal defect, mitral valve prolapse with mitral regurgitation, dilated coronary sinus and a partial anomalous pulmonary venous drainage were diagnosed by Echocardiography.
2001: Fleck BJ [15]-This original observation includes a total of 18 cases. Nine had congenital heart diseases in form of VSD/ASD/TOF/PDA. Six underwent surgical repair.
2006: Flynn, Maureen A [16]-11 months baby girl had surgical closure of VSD
2007: Keller Mayer R [17]-One case with PDA and ligated at 1-month of age
2009: Brautbar, Ariel [18]-Large ostium primum/ cleft mitral valve with mitral regurgitation .This case had undergone surgery.
2012: Schrier SA [19]-Original study describes 15 cases: Patent foramen ovale (PFO)-2, ASD-3, VSD-3, PDA-1, pericarditis-1 and eight cases had no congenital cardiac defects.
2013: Ng D [20]-first time described associated Non compaction of left ventricle; Wieczorek D [21]-one case with Patent ductus arteriosus; Santen, Gijs W.E [22]-Dextrocardia in two patients with SMARCB1 mutation; Kosho, tomoki {23}-One case had VSD with SMARCB1 mutation, One case had VSD/PDA with SMARCA4 and 1 had mitral atresia/PFO/Single ventricle of right ventricular morphology with ASD/PDA.
Our case is the first reported case of CSS that is associated with a rare constellation of many a congenital cardiac defects.
The management of this case is a big challenge as there was severe systemic desaturation because of persistent left superior vena cava connecting left atrium, biventricular outflow tract obstruction, significant right ventricular dysfunction and delayed presentation.
CONCLUSION
This is the first time in medical literature we are reporting a case of type A Coffin-Siris syndrome associated with constellation of extremely rare multiple cardiovascular defects and successful surgical management.
Limitation
Our study does not have genetic mutation study support.
Video available on www.annalspc.com
ACKNOWLEDGMENT
I am extremely thankful to Dr. Prajnya Ranganth, MD (Medical Genetics) affiliated to Nizam's Institute of Medical Sciences, Hyderabad-500082, India for her kind help for genetic counselling of this patient.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Coffin GS, Siris E. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970;119:433–9. doi: 10.1001/archpedi.1970.02100050435009. [DOI] [PubMed] [Google Scholar]
- 2.Bartsocas CS, Tsiantos AK. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970;120:493–4. doi: 10.1001/archpedi.1970.02100100153030. [DOI] [PubMed] [Google Scholar]
- 3.Carey JC, Hall BD. The Coffin-Siris syndrome: Five new cases including two siblings. Am J Dis Child. 1978;132:667–71. doi: 10.1001/archpedi.1978.02120320027005. [DOI] [PubMed] [Google Scholar]
- 4.Schinzel A. The Coffin-Siris syndrome. Acta Paediatr Scand. 1979;68:449–52. doi: 10.1111/j.1651-2227.1979.tb05037.x. [DOI] [PubMed] [Google Scholar]
- 5.Ueda K, Saito A, Nakano H, Iinuma K. The Coffin-Siris syndrome: A case report. Helv Paediatr Acta. 1980;35:385–90. [PubMed] [Google Scholar]
- 6.Alembic Y, Ray E, Hirsh E. Coffin siris syndrome with lenoux fgerhout syndrome and hypertrophic cardiomyopathy. Annals of Paediatrics. 1988 [Google Scholar]
- 7.Pilar L, Baraitser M. Coffin-Siris syndrome. J Med Genet. 1991;28:338–41. doi: 10.1136/jmg.28.5.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petra R, Haverkamp F, Emons D, Rosskamp R, Zerres K. Sisters with possible coffin siris syndrome. Am J Med Genet. 1991;41 doi: 10.1002/ajmg.1320410317. [DOI] [PubMed] [Google Scholar]
- 9.Kirel B, Kural N, Yakut A, Adapinar B. Triplet with growth failure, microcephaly, mental retardation, nail hypoplasia and corpus callosum agenesis; Is it a variant of CSS or a new syndrome. Turk J Pediatr. 2000;42:171–6. [PubMed] [Google Scholar]
- 10.Imaizumi K, Nakamura M, Masuno M, Makita Y, Kuroki Y. Hypoglycemia in Coffin-Siris syndrome. Am J Med Genet. 1995;59 doi: 10.1002/ajmg.1320590111. [DOI] [PubMed] [Google Scholar]
- 11.Coulibaly, Bema B, Sigaudy, Sabine S, Girard, Nadine N. Coffin siris syndrome with multiple congenital malformations and intrauterine death. Eur J Med Genet. 2010;53:318–21. doi: 10.1016/j.ejmg.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Ounap K, Justus I, Lipping-Sitska M. Two sisters with growth failure, microcephaly, peculiar facies and apical dystrophy: The presentation of brachymorphism-onychodysplasia-dysphalangism syndrome? Clin Dysmorphol. 1998;7:45–50. [PubMed] [Google Scholar]
- 13.Geggel RL, Perry SB, Blume ED, Baker CM. Left superior vena cava connection to unroofed coronary sinus associated with positional cyanosis: Successful transcatheter treatment using gianturco-grifka vascular occlusion device. Cathet Cardiovasc Intervent. 1999;48:369–73. doi: 10.1002/(sici)1522-726x(199912)48:4<369::aid-ccd9>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Karunathilka DH, Amaratunga GW, Perera KD. Coffin-Siris syndrome in a twelve year old girl. Sri Lanka J Child Health. 2000;29:57. [Google Scholar]
- 15.Fleck BJ, Pandya A, Vanner L, Kerkering K, Bodurtha J. Coffin-Siris Syndrome: Review and presentation of new cases from a questionnaire study. Am J Med Genet. 2001;99:1–7. doi: 10.1002/1096-8628(20010215)99:1<1::aid-ajmg1127>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Maureen AF, Milunsky, Jeff M. Autosomal dominant syndrome resembling coffin siris syndrome. Am J Med Genet. 2006;140A doi: 10.1002/ajmg.a.31287. [DOI] [PubMed] [Google Scholar]
- 17.Kellermayer R, Kitagawa S, Redel CA, Cass DL, Belmont JW, Klish W. Upper gastrointestinal malformations in Coffin-Siris syndrome. Am J Med Genet A. 2007;143A:1519–21. doi: 10.1002/ajmg.a.31865. [DOI] [PubMed] [Google Scholar]
- 18.Brautbar A, Ragsdale J, Shinawi M. Is this the Coffin-Siris syndrome or the BOD syndrome? Am J Med Genet. 2009;149A doi: 10.1002/ajmg.a.32671. [DOI] [PubMed] [Google Scholar]
- 19.Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MD, D’avanzo MG, et al. The Coffin-Siris Syndrome: A proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet. 2012;Part A 158A:1865–76. doi: 10.1002/ajmg.a.35415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng D, Bouhlal Y, Ursell PC, Shieh JT. Monoamniotic monochorionic twins discordant for noncompaction cardiomyopathy. Am J Med Genet A. 2013;161A:1339–44. doi: 10.1002/ajmg.a.35925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wieczorek D, Bögershausen N, Beleggia F, Steiner-Haldenstätt S, Pohl E, Li Y, et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22:5121–35. doi: 10.1093/hmg/ddt366. [DOI] [PubMed] [Google Scholar]
- 22.Kosho T, Okamoto N, Ohashi H. Clinical correlations of mutations affecting six components of SWI/SNF complex. Detailed description of 21 patients and review of literature. Am J Med Genet. 2013;161 doi: 10.1002/ajmg.a.35933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.