

Abstract
Pulsed electron–electron double resonance (PELDOR) is an electron paramagnetic resonance (EPR) spectroscopy technique for nanometer distance measurements between paramagnetic centers such as radicals. PELDOR has been recognized as a valuable tool to approach structural questions in biological systems. In this manuscript, we demonstrate the value of distance measurements for differentiating competing structural models on the dimerization of the effector domain (ED) of the non-structural protein 1 (NS1) of the influenza A virus. Our results show NS1 to be well amenable to nanometer distance measurements by EPR, yielding high quality data. In combination with mutants perturbing protein dimerization and in silico prediction based on crystal structures, we can exclude one of two potential dimerization interfaces. Furthermore, our results lead to a viable hypothesis of a NS1 ED:ED interface which is flexible through rotation around the vector interconnecting the two native cysteines. These results prove the high value of pulse EPR as a complementary method for structural biology.
Introduction
Influenza A viruses remain a continuing threat to public health.1 In addition to seasonal epidemics, the virus has the potential to cause worldwide pandemics, such as the 2009 H1N1 “swine flu” outbreak. Recent cases of H7N9 and H5N1 avian influenza in Asia have caused widespread alarm and serve as a reminder that the next pandemic could come sooner rather than later.
The multifunctional NS1 (non-structural 1) protein of influenza A has been proposed to interact with a wide range of cellular and viral factors.2,3 Most notably, expression of NS1 suppresses stimulation of the innate immune response through interactions with cellular pathogen recognition receptors (e.g., RIG-I and TRIM25),4,5 by suppression of the host mRNA maturation via binding to the cellular and splicing processivity factor 30 (CPSF30)6 and by sequestration of double stranded RNA (dsRNA) produced during viral infection.7 Furthermore, NS1 stimulates viral growth through interactions with phosphoinositide-3-kinase (PI3K)8 and the transcription factor eIF4E.9 Added to these are a variety of other proteins to which NS1 binding has been shown but the functional relevance is unknown. NS1 is a small (26 kDa) protein comprised of two domains: an N-terminal RNA binding domain (RBD, residues 1–72), connected by a short linker to a C-terminal effector domain (ED, residues 83–203), and followed by a 27–34 residue disordered tail. Both domains form homodimeric interactions, and the full-length protein can oligomerize at higher concentrations.10−15 While the dimer interface of the RBD is unambiguous, several dimer interfaces have been proposed for the ED, based upon X-ray crystallography of the ED and full-length NS1 protein (see Figure 1).10,13,16
Figure 1.
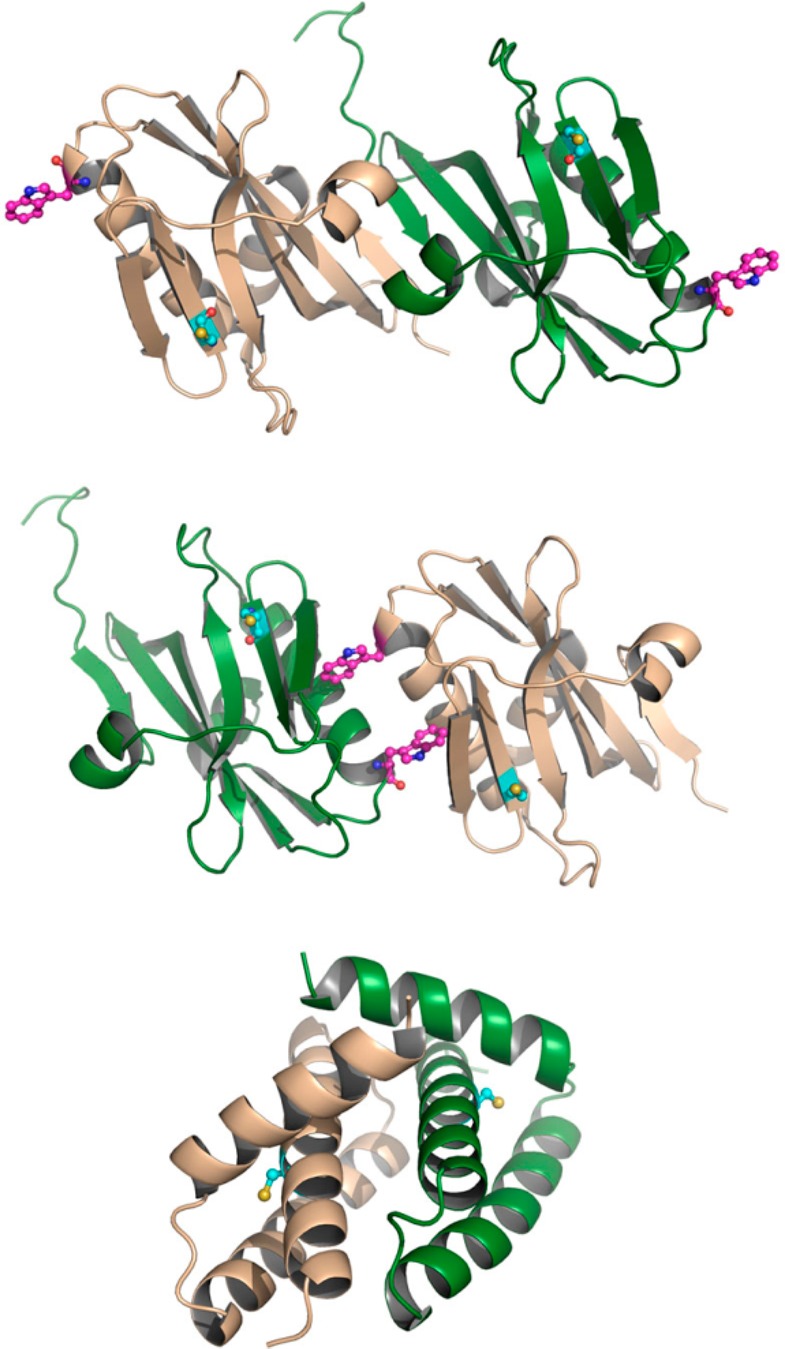
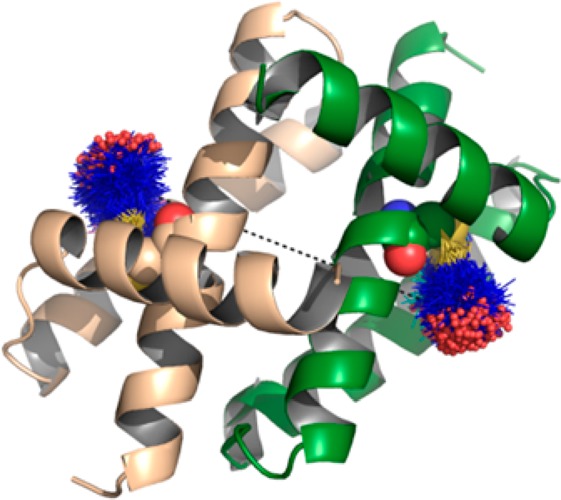
Dimerization states of the domains of NS1: (top) strand–strand ED dimer; (middle) helix–helix ED dimer; (bottom) RBD dimer. Protein shown as green and beige cartoon. In the top and middle parts, residues Cys116 and Trp187 are shown as ball-and-stick in cyan and magenta, respectively. In the bottom part, Cys13 is shown as ball-and-stick in cyan.
More specifically, from the initial crystal structure of the ED, a dimer interface mediated by β-strand interactions was proposed (termed here the strand–strand dimer).10 Subsequent crystal structures of the ED lacked this interface but contained a separate contact, also present within the crystal matrix of the first structure but initially discarded due to a slightly smaller interface surface area. In this case, the interface was mediated by contact between α-helices (termed the helix–helix dimer).13,17−19 Proponents of the stand–strand interface have suggested these differences may be due to strain-specific effects;20 however, a survey of all wild-type NS1 ED crystal structures demonstrated the helix–helix interface to be the only universally present (for details, see the Supporting Information).18 Notably, this helix–helix dimer has been described as the dominant ED:ED interaction under more physiologically relevant conditions in solution by recent NMR studies using the NS1 Udorn strain.21,22 Furthermore, incorporation of mutations (Trp187Ala or Trp187Arg) at this dimer interface was sufficient to prevent interaction as observed by both NMR and crystallography.13,18,22−24 Significantly, these mutations have been associated with decreased pathogenesis in vivo and a loss of RNA binding in vitro, further strengthening the physiological importance of ED dimerization at the helix–helix interface.18,22,25 More recently, several further interfaces have been proposed from contacts within the crystal lattice of full-length structures of NS1, although these models remain at a very early stage.16
While NMR analysis of the RBD dimer has been achieved,12 due to the size of the ED dimer, the only full NMR solution-state structure of the ED has required disrupting dimerization by incorporation of the Trp187Arg mutation.22 However, additional data from chemical shift perturbations and 15N T1/T2 relaxation times from wild-type and monomeric (Trp187Arg) EDs allowed mapping dimerization to the helix–helix interface.22 More recently, elegant studies using 19F NMR of 5-F-Trp labeled NS1 demonstrated that Trp187 is buried in the context of the isolated ED but exposed in the full-length constructs.21 Furthermore, this study supported the notion of plasticity at the helix–helix interface, as T2 relaxation times indicated a conformational change within the ED dimer 3 orders of magnitude faster than the exchange between monomer and dimer states, which could be monitored using the 19F resonance of Trp187. These studies support the hypothesis of a flexible ED dimer interface, previously postulated from variations observed within crystal structures,18 with exchange between two or more conformations on the microsecond-to-millisecond time scale.21 However, while the presence of multiple conformations is evident from 19F line broadening of the Trp187 signal, further information is needed for complete characterization. In particular, whether this heterogeneity consists of a number of discrete conformations or a spectrum of positions awaits clarification. Additionally, more information on the populations of any such conformations and the amplitudes of conformational changes in solution is needed.
Pulse EPR distance measurements26 in combination with site-directed spin labeling27 have yielded precise distance and geometry information on proteins in frozen aqueous buffer solutions. In particular, the pulsed-electron double resonance (PELDOR) method also known as double electron–electron resonance (DEER)28 has been widely employed to gather geometry constraints on soluble and membrane proteins,29 nucleic acids,30 and synthetic test samples.28 While this approach will commonly generate distance constraints too sparse for de novo structure determination, it can be extremely valuable in confirming or rejecting structures obtained by other methods or structural models.31
The aim of the present study is to investigate the molecular structures formed by domains of NS1 in solution by measurement of distances between spin labels incorporated into the domains of NS1 using PELDOR. NS1 provides an easily tractable system for PELDOR analysis, as in most strains one solvent-accessible cysteine residue is present within each domain, making them directly accessible to site-directed spin-labeling EPR. In contrast to previous studies in which conclusions have been based on measurements from a single strain, analysis was performed on constructs from three different influenza isolates, implying the results are general and not strain-specific. Importantly, these included the strain where the strand–strand dimer was initially described.10 In this study, structural interpretation was based on comprehensive in silico predictions using two different modeling approaches in combination with PELDOR analysis.
Experimental Methods
Purification and Preparation of Spin-Labeled NS1 Domains
All influenza A NS1 domain constructs were expressed in E. coli and purified as described previously.8 Briefly, His-tagged NS1 RNA binding domain (RBD) (A/Udorn/72 residues 1–72) or wild-type or mutant His-tagged NS1 effector domain (ED) [residues 72–230 (A/Puerto Rico/8/34), 72–237 (A/Udorn/72), and 83–230 (A/Brevig Mission/1/1918)] were expressed in E. coli and purified using a Ni-NTA column. After cleavage of the His-tag with TEV protease, the protein was reapplied to the column and 1 mM DTT added to the flow-through fraction. Reduced proteins were desalted using a HiPrep 26/10 desalting column (GE) prior to incorporation of the MTSSL spin label by incubation with 1 mM MTSSL for 2 h at 22 °C. Spin-labeled proteins were further purified using an S100 Sephacyl 16/60 gel filtration column (GE) equilibrated with 50 mM Tris pH 7.8, 200 mM NaCl. Sample solvent was exchanged for deuterium oxide by buffer exchange (RBD) or via lyophilization and 25% deuterated ethylene glycol added prior to freezing in liquid nitrogen for PELDOR analysis.
Theoretical Measurement of Spin–Spin Distances
In silico spin labeling, rotamer conformation searching, and distance measurements were all carried out within the software package PyMOL (www.pymol.org) using the MTSSLWizard plugin.32 Distance distributions were obtained by binning the data into 1 Å bins. The following atomic coordinates of NS1 domains were used for this modeling procedure: For the NS1 RBD, the atomic coordinates from the crystal structure of 1AIL were used; for the NS1 ED helix–helix dimer, the PDB structure 3O9S was used; and for the strand–strand dimer, the PDB structure 2GX9 was used. Similar results were obtained using NS1 RBD structures 2Z0A, 2ZKO, and 3M8A and NS1 ED structures 3O9T, 3O9U, 3M5R, 3EE8, and 3EE9. All searches were carried out with the thoroughness set to “painstaking” and the vdW restraints to “loose”. “Tight” searches did not yield any labeling for 1AIL and largely differing numbers of conformers for the individual chains in 2GX9 and 3OS9. To obtain results comparable to those from MMM after repacking side chains (see below), resulting MMM PDBs “repacked” for 1AIL, 3O9S, and 2GX9 were reanalyzed.
As a complementary approach, the modeling procedure was repeated with the software package Matlab (www.mathworks.com) using the MMM plugin.33 For the NS1 RBD, the atomic coordinates from the crystal structure of 1AIL were used; for the NS1 ED helix–helix dimer, the PDB structure 3O9S was used; and for the strand–strand dimer, the PDB structure 2GX9 was used. In addition to the predicted distance distributions, the best fit of the experimental data to any rotamer conformations, independent of their respective energies, was obtained using the “any rotamers” function. This function uses all possible rotamer pairs obtained during the site scan, independent of the populations predicted. As a further test, the “grow/repack side-chains” function of MMM, which uses the free third-party software SCWRL4,34 was used for 1AIL, 2GX9, and 3OS9, followed by the standard procedure (rotamer site scan and labeling). This function can be used to correct the conformations of side chains given by the crystal structure, which may be different in solution. All tests were performed separately, with the site scan/labeling conditions set to either cryogenic temperature (175 K) or ambient temperature (298 K).
Collection and Analysis of PELDOR Traces
All PELDOR data were recorded on ELEXSYS E580 pulsed X-band (9 GHz) or Q-band (34 GHz) EPR spectrometers including the second frequency option (E580-400U) from Bruker. Pulses were amplified by traveling wave tube (TWT) amplifiers (1 kW at X-band and 150 W at Q-band) from Applied Systems Engineering. As sample amounts were not limiting, we used an MD5 dielectric ring resonator (X-band) or TE012 cavity (Q-band) with standard flex line probe heads. The established pulse sequence π/2(νA)-τ1-π(νA)-τ1+t-π(νB)-(τ2–t)-π(νA)-τ2-echo was employed for all PELDOR experiments.28 With the following exceptions of timings and pump pulse length, the chosen settings and optimization procedures were as previously described.35 Typically, the pump pulse was set to 18–20 ns at X-band and 12 ns at Q-band, τ1 to 380 ns, τ2 to 3 μs (up to 7 μs at Q-band) and the shot repetition time to 2 ms, averaging the data for ∼14 h at X-band or <2 h at Q-band.
Raw data were fitted by a monoexponential decay in the Matlab plugin DeerAnalysis2013 to remove the background.36 All decay constants agreed with those calculated from the nominal protein concentration within 20%. Tikhonov regularization was performed in DeerAnalysis2013, and the optimum regularization parameter was chosen by the L-curve criterion.37
Results and Discussion
As spin-labeled dimers of full length NS1 can bear four spin labels for dimers and even more for oligomers leading to more challenging theoretical treatment,38 we approached the dimerization of the individual domains by performing measurements between spin labels incorporated into the individual RBD and ED dimers using PELDOR. Initially, the MTSSL spin label was incorporated into the RBD dimer (from the A/Udorn/72 strain (Udn)) at Cys13. The primary PELDOR data for this dimer reveals at least four full dipolar modulations (Figure 2, top), which is exceptional for proteins spin-labeled with MTSSL via cysteines and clearly indicates very constrained spin-label conformations giving rise to a very narrow distance distribution. The distance distribution derived by Tikhonov regularization36 is indeed in very good agreement with the uncontroversial model from crystallography (Figure 2, bottom).12,14
Figure 2.
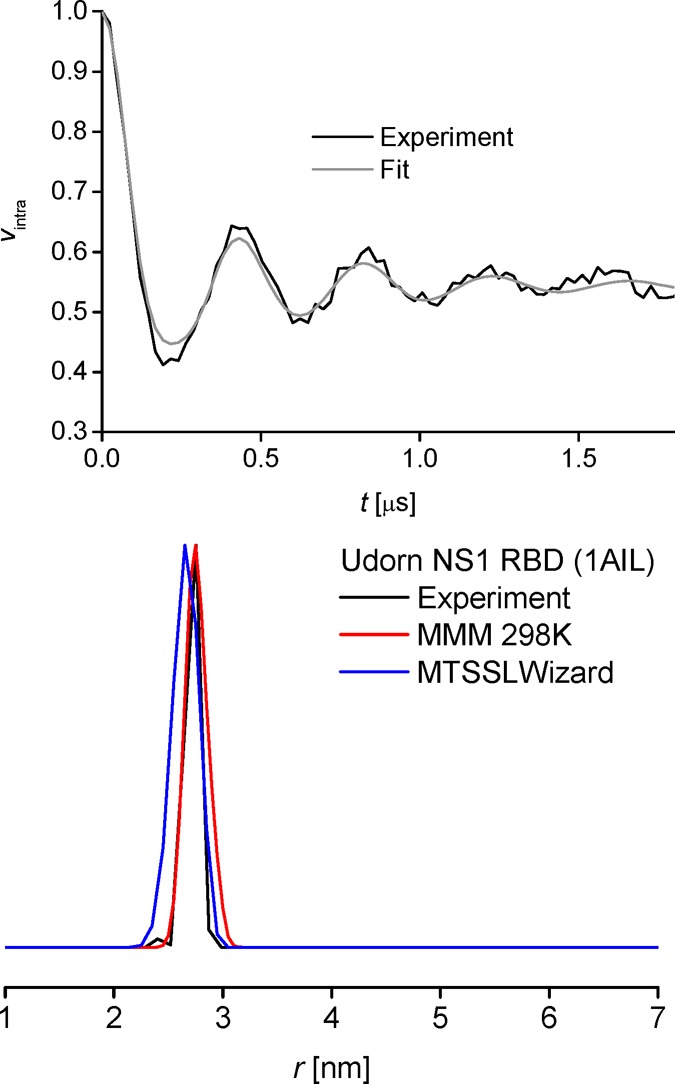
Background corrected experimental PELDOR data and fit (top) and experiment-derived distance distribution in the RBD and comparison with in silico models generated using the respective crystal structure (PDB 1AIL) (bottom).
For evaluation of structural models by PELDOR, it is essential to model the spin label, which is most often not part of the existing structure. While in a very crude approximation the spin-bearing group has to be within less than 1 nm of the respective Cα, considering the possible projections in a doubly labeled system, this would lead to an unacceptable uncertainty of several nm. More elegant approaches model the positions of spin labels explicitly by either a rotamer library approach, in which precomputed rotamers are energy-weighted for estimating their populations in a specific structure (Matlab plugin MMM),33 or a parametrized excluded volume approach stochastically generating conformations and excluding those which clash with the protein (PyMol plugin MTSSLWizard).32 We have tested both approaches for the Udn NS1 RBD and find very good agreement with the experimental data. Interestingly, MMM predicts a tight site when using ambient but not cryogenic temperature for site scan and labeling, while MTSSLWizard can only find a reasonable number of conformations at the slowest and most unconstrained search settings (see the Supporting Information), confirming our hypothesis of a very constrained label. Agreement of MMM at 175 K seems marginally worse than MTSSLWizard which becomes more evident comparing time domain data (see the Supporting Information). MMM results were slightly improved at ambient temperature, which is in line with previous studies.32,39 However, in the light of the prediction errors of both modeling programs, these rather small differences cannot be considered significant.39,40
With the results on the dimer of the wild-type RBD proving NS1 clearly amenable to our pulse EPR approach, we investigated the ED:ED dimer interface for three separate strains: A/Puerto Rico/8/34 (PR8; the strain used in the initial study proposing the strand–strand dimer),10 Udn (the strain used in recent NMR studies on ED dimerization),21,22 and A/Brevig Mission/1/1918 (1918). Evaluation of the models of NS1 ED dimerization was performed by incorporation of the spin-label MTSSL into purified NS1 EDs at the conserved Cys116. For all three EDs (PR8, Udn, 1918; Figure 3, top), the experimental data look remarkably similar. The modulation depth indicates that dimerization is not quantitative in concentrations around 100 μM, which is in good agreement with the recently reported weak binding (Kd ∼90 μM).21,22 However, the traces show clear modulation, allowing reliable distance information to be extracted.
Figure 3.
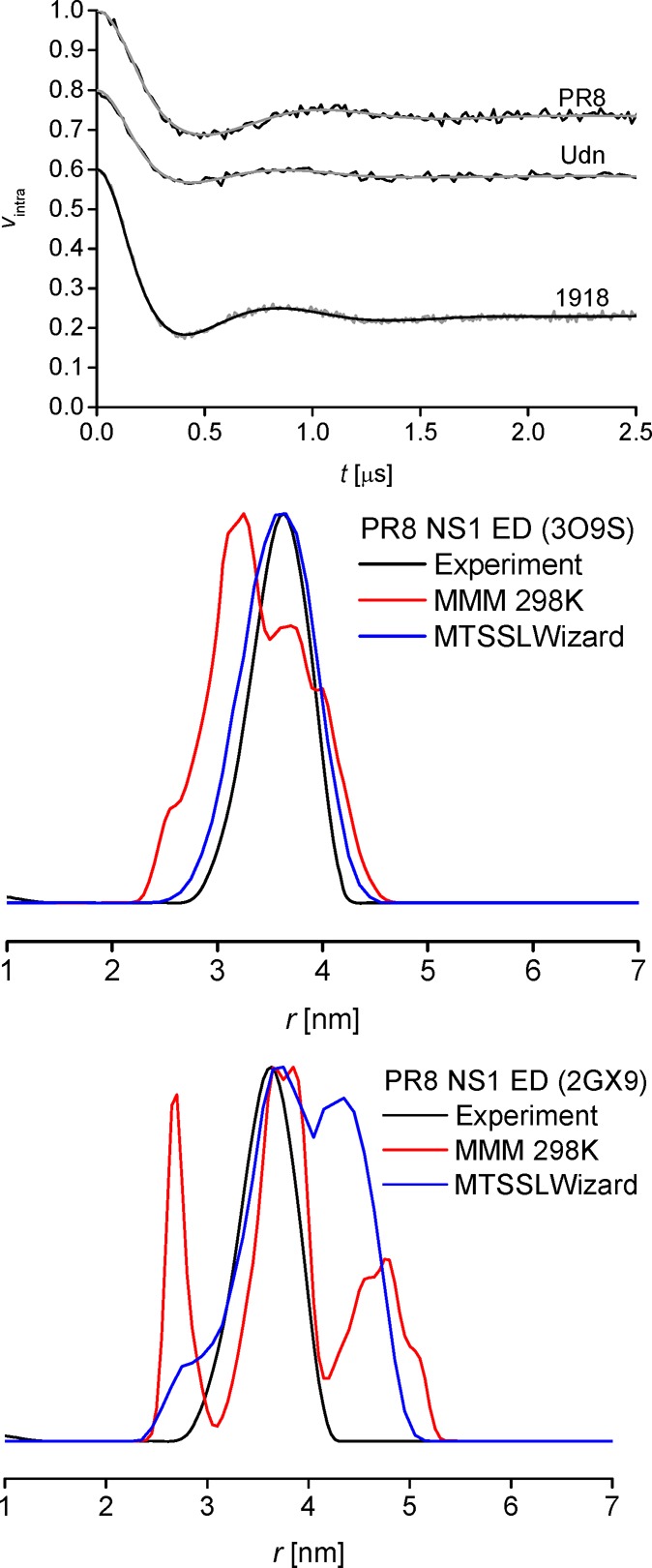
Experimental PELDOR data (black) and fits (gray) on the NS1 ED of PR8, Udn, and 1918 (top). For clarity, traces and fits of Udn and 1918 have been shifted by −0.2 and −0.4 on the y-axis, respectively. Distance distribution for PR8 in comparison with crystal helix–helix (middle) and strand–strand (bottom) dimer models.
Calculation of the expected spin–spin distances for the ED dimers was modeled using both MTSSLWizard and MMM for comparative reasons. On the basis of the strand–strand and helix–helix dimers present within the crystalline lattices of the PR8 NS1 ED structures 2GX9 and 3O9S,10,18 respectively, two distinct distance distributions emerged for both modeling approaches (Figure 3). In these calculations, the strand–strand dimer always yielded the slightly longer spin–spin distance, with a wider distribution, compared to a more compact, shorter distance within the helix–helix dimer. Interestingly, the absolute agreement between the two modeling approaches and the experiment is remarkably dependent on the settings applied, especially for MMM (see the Supporting Information). MTSSLWizard disfavors the strand–strand interface by comparison with the experimental distance distribution, and this result is mostly unaffected by further expansion of the modeling using repacked side chains34 (obtained through MMM using the free third-party software SCWRL4). Similarly, for MMM, neither using ambient instead of cryogenic temperature nor repacking the side chains nor both can improve the results obtained for the strand–strand dimer of the ED (PDB 2GX9). Significantly, the opposite is found for the helix–helix interface (PDB 3O9S), with considerable improvement of modeling results when applying both ambient temperature and side chain repacking.
Thus, MMM would also lead to favoring the helix–helix model, if the different settings were tested and directly compared with each other. This becomes evident both from the distance distributions (Figure 3) and the traces (see the Supporting Information) when comparing with the experimental data. However, even though both modeling approaches converge to favoring the helix–helix model, the observed differences between the two possible conformations may in fact be too small to be unequivocally distinguishable when taking into account the reported modeling accuracies.39,40
This reservation was further supported when we verified that the experimental data can be reproduced by an arbitrarily chosen combination of rotamers for both the helix–helix and the strand–strand PDBs using MMM. These settings explicitly use all pairs of rotamers, neglecting any clashes with the protein, and therefore merely test whether the experimental distance distribution is at all possible for the given backbone conformation. For any given setting (both ambient and cryogenic temperature; with or without repacked side chains), we found that the resulting best-fit distribution for both dimer interfaces covers the entire experimental distribution (see the Supporting Information), not rendering the strand–strand dimer model impossible. This finding from the “any rotamers” test is significant, as it deviates substantially from the MMM predictions which explicitly treat the energies of the rotamers. At least in this case, MMM predictions seem to be more affected by differences between crystal and solution structure than MTSSLWizard predictions. This is in agreement with the finding that correction of side chains significantly improves the MMM modeling results for the helix–helix dimer (see the Supporting Information). It will be very interesting to monitor the future performance of both modeling approaches for different biological systems.
In combination, comparison of the experimental data with the distance distributions obtained for PR8 with the helix–helix and strand–strand dimer models (Figure 3, middle and bottom) clearly disfavors the strand–strand interface in solution. Results for the other two strains are very similar (see the Supporting Information). Thus, our results fully agree with a solution-state study describing dimer formation of wild-type Udn ED through the helix–helix interface.22 Analyzing a set of different strains, our results confirm previous findings and provide the first direct observation of the ED helix–helix interface in NS1 strain PR8 in solution. The helix–helix conformation is also in agreement with calculations based on crystal structures from several other strains (see the Supporting Information).
Taking the reported modeling accuracies39,40 into account, our predictions obtained by MTSSLWizard and MMM disfavor, but cannot unequivocally reject, the wild-type NS1 ED strand–strand dimer. One possible approach to prove a single dimer solely by EPR distance measurements is generating a set of spin-labeled NS1 ED mutants predicted to show no overlap in distance distributions between the two potential dimer interfaces. However, an elegant disruption of the helix–helix dimer interface by incorporation of the Trp187Ala mutation has been described previously.13,21,22 We rationalized this mutant should lead to significant changes in modulation depth for the helix–helix interface only, thus generating unequivocal proof for rejecting one of the two models, by a single further EPR experiment. In contrast to data obtained from wild-type ED, PELDOR time traces from the Trp187Ala mutants contain either no evidence of dimerization (for PR8 EDs) or a significantly reduced level of dimer formation (Udn EDs), as evident from the reduction in modulation depths of the PELDOR traces in Figure 4. Thus, pulse EPR could be used to reject the strand–strand dimer interface in the NS1 PR8 strain, in which it had been initially described,10 thereby confirming and extending previous studies using the Udn strain,21,22 and others.13,18 Furthermore, we conclude that, even though the use of more than one modeling approach is strongly recommended for obtaining more reliable predictions, the unequivocal proof for one or the other conformational model may only be possible by making the effort of generating and analyzing additional informing mutants, such as the Trp187Ala mutant in this study.
Figure 4.

Experimental PELDOR data on the ED Trp187Ala mutant of PR8 and Udn. For clarity, the trace of Udn has been shifted by −0.2 on the y-axis.
NS1 is a relatively small protein, yet it is capable of interaction with a wide range of host and viral factors and how these interactions are controlled is not fully understood. However, it appears likely that the quaternary arrangements of the NS1 protein in solution play a role in the regulation of its many functions. In recent years, numerous crystal structures of truncated and full-length forms of NS1 have been published, leading to several models of NS1 structure in vivo. The evidence in this study aligns with the consensus of NMR and crystallographic studies indicating the NS1 ED forms the proposed helix–helix dimer in solution.13,21,22 However, it remains to be determined under what circumstances this interface is used and to what end. Formation of the ED dimer appears to be required for efficient interaction with dsRNA, possibly through the formation of oligomeric structures.18,22 On the other hand, it is also clear that, for some functions of NS1 (e.g., binding to CPSF30), the ED dimer must separate, as the two interactions share the same interface.41
Previous analysis of the NS1 ED dimer by crystallography18 and 19F NMR21 has indicated that the two monomers may undergo rapid conformational change between a variety of orientations at the helix–helix interface. It is perhaps surprising to note, therefore, that the spin–spin distances for the ED dimer closely align with the predicted distribution produced using a single, fixed crystal structure, implying an absence of large scale movements. This can be explained in three ways: (i) The conditions in which the EPR sample were prepared have frozen each dimer in the same conformation, by freezing out the lowest energy state of the conformational equilibrium. (ii) There are two or more conformational states with one state being dominant. The PELDOR data obtained in this study do not indicate the presence of a second distance, even when exploiting the higher sensitivity of the Q-band for the PR8 ED wild-type (see the Supporting Information). However, populations of below 20% might not be obvious from the EPR data and still by far sufficient for the observed exchange broadening in 19F NMR.21 (iii) The conformational states are aligned around an axis connecting the two spin labels, thereby only marginally affecting the actually observed spin–spin distance and thus being undetectable using PELDOR. The 19F NMR study does not inform on the interdomain orientations or their populations. On the other hand, in silico prediction of the spin–spin distances from a variety of ED crystal structures using MTSSLWizard yields similar distance distributions (see the Supporting Information). Furthermore, comparisons between the numerous ED crystal structures published to date suggest such a rotational movement to form the principal component of the differences observed (see the Supporting Information). In combination, these data make the rotation around the spin–spin vector a viable hypothesis. While we cannot fully reject the explanation of minor populations of further conformations, in our PELDOR analysis, we would anticipate detection of populations of any second conformational state over 20% of the total as long as the spin–spin distance changes significantly. Neither of these three explanations would contradict the observation of the helix–helix dimer interface by intermolecular NOEs.22
Having validated the use of PELDOR for distinguishing between different structures of NS1 domains, the next step will be the extension to the full-length protein. Analysis of intact NS1 is complicated by the propensity for oligomerization at concentrations above 40 μM,22 and thus far, most of the studies (including all crystal structures) have required the incorporation of mutations to increase solubility.11,16 Currently, the limited information we have on the relative positions of the two NS1 domains is heavily reliant on the forms observed in crystal structures, although some data from NMR have been helpful.21,22 Given the known flexibility of the interdomain linker region, such arrangements are likely to be significantly affected by the interactions formed during crystal packing. Therefore, further data from solution-state techniques such as EPR and NMR will be vital for understanding how the full-length NS1 operates in the cellular environment, with EPR techniques having the advantage of not being limited by the size or shape of the proteins under investigation. In addition, the structures formed by NS1 in complex with its many binding partners are largely unknown and it is likely that such structures and structural rearrangements upon binding could be probed by suitable in-solution methods.
Conclusions
The results of crystallographic analyses of the influenza A NS1 protein have yielded several models of NS1 function, that require further data from solution-state methods to be resolved adequately. The data presented here are of very high quality and demonstrate that measurement of distances between site-specific spin labels via PELDOR can distinguish between structural models. These differentiations become more challenging the smaller the predicted differences are. In these cases, additional information either from further mutants or from complementary models, including in silico prediction models, is invaluable. Application of this technique to the full-length NS1 protein and NS1-mediated complexes will significantly enhance our understanding of NS1 structure and function.
Acknowledgments
We gratefully acknowledge Gunnar Jeschke and Gregor Hagelueken for helpful comments on the accuracy of modeling approaches and the respective usage of MMM and MTSSLWizard. H.L.T. was supported by the University of St Andrews; S.A.J. is supported by the BBSRC, U.K.; P.S.K. is supported by the Medical Research Council, U.K.; B.E.B. is grateful for an EaStCHEM Hirst Academic Fellowship by the School of Chemistry, St Andrews, and funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme [REA 334496]. This work was supported by the Wellcome Trust [099149/Z/12/Z].
Supporting Information Available
Supplementary figures, additional modeling, additional distance data, raw PELDOR data, Q-band PELDOR data, and complete references (22) and (41). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ H.L.T.: MRC-CVR, 8 Church Street, Glasgow, G11 5JR, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Influenza Fact Sheet No. 211; World Health Organization: 2009.
- Hale B. G.; Randall R. E.; Ortin J.; Jackson D. The Multifunctional NS1 Protein of Influenza A Viruses. J. Gen. Virol. 2008, 89, 2359–2376. [DOI] [PubMed] [Google Scholar]
- Kochs G.; Garcia-Sastre A.; Martinez-Sobrido L. Multiple Anti-Interferon Actions of the Influenza A Virus NS1 Protein. J. Virol. 2007, 81, 7011–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.; Chen L. M.; Zeng H.; Gomez J. A.; Plowden J.; Fujita T.; Katz J. M.; Donis R. O.; Sambhara S. NS1 Protein of Influenza A Virus Inhibits the Function of Intracytoplasmic Pathogen Sensor, RIG-I. Am. J. Respir. Cell Mol. Biol. 2007, 36, 263–269. [DOI] [PubMed] [Google Scholar]
- Gack M. U.; Albrecht R. A.; Urano T.; Inn K. S.; Huang I. C.; Carnero E.; Farzan M.; Inoue S.; Jung J. U.; Garcia-Sastre A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff M. E.; Barabino S. M.; Li Y.; Keller W.; Krug R. M. Influenza Virus NS1 Protein Interacts with the Cellular 30 kDa Subunit of CPSF and Inhibits 3' End Formation of Cellular Pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Qian X. Y.; Chien C. Y.; Lu Y.; Montelione G. T.; Krug R. M. An Amino- Terminal Polypeptide Fragment of the Influenza Virus NS1 Protein Possesses Specific RNA Binding Activity and Largely Helical Backbone Structure. RNA 1995, 1, 948–956. [PMC free article] [PubMed] [Google Scholar]
- Hale B. G.; Jackson D.; Chen Y. H.; Lamb R. A.; Randall R. E. Influenza A Virus NS1 Protein Binds p85beta and Activates Phosphatidylinositol-3-Kinase Signaling. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14194–14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgui I.; Aragon T.; Ortin J.; Nieto A. PABP1 and eIF4GI Associate with Influenza Virus NS1 Protein in Viral mRNA Translation Initiation Complexes. J. Gen. Virol. 2003, 84, 3263–3274. [DOI] [PubMed] [Google Scholar]
- Bornholdt Z. A.; Prasad B. V. X-Ray Structure of Influenza Virus NS1 Effector Domain. Nat. Struct. Mol. Biol. 2006, 13, 559–560. [DOI] [PubMed] [Google Scholar]
- Bornholdt Z. A.; Prasad B. V. X-Ray Structure of NS1 from a Highly Pathogenic H5N1 Influenza Virus. Nature 2008, 456, 985–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C. Y.; Tejero R.; Huang Y.; Zimmerman D. E.; Rios C. B.; Krug R. M.; Montelione G. T. A Novel RNA-Binding Motif in Influenza A Virus Non-Structural Protein 1. Nat. Struct. Biol. 1997, 4, 891–895. [DOI] [PubMed] [Google Scholar]
- Hale B. G.; Barclay W. S.; Randall R. E.; Russell R. J. Structure of an Avian Influenza A Virus NS1 Protein Effector Domain. Virology 2008, 378, 1–5. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lynch P. A.; Chien C. Y.; Montelione G. T.; Krug R. M.; Berman H. M. Crystal Structure of the Unique RNA-Binding Domain of the Influenza Virus NS1 Protein. Nat. Struct. Biol. 1997, 4, 896–899. [DOI] [PubMed] [Google Scholar]
- Nemeroff M. E.; Qian X. Y.; Krug R. M. The Influenza Virus NS1 Protein Forms Multimers in Vitro and in Vivo. Virology 1995, 212, 422–428. [DOI] [PubMed] [Google Scholar]
- Carrillo B.; Choi J. M.; Bornholdt Z. A.; Sankaran B.; Rice A. P.; Prasad B. V. The Influenza A Virus Protein NS1 Displays Structural Polymorphism. J. Virol. 2014, 88, 4113–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont D. H.; Yu Y. Y. L.; Nelson C. A. Center for Structural Genomics of Infections Diseases (CSGID). Crystal Structure of Swine Flu Virus NS1 Effector Domain from H1N1 Influenza A/California/07/2009.
- Kerry P. S.; Ayllon J.; Taylor M. A.; Hass C.; Lewis A.; Garcia-Sastre A.; Randall R. E.; Hale B. G.; Russell R. J. A Transient Homotypic Interaction Model for the Influenza A Virus NS1 Protein Effector Domain. PLoS One 2011, 6, e17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Monzingo A. F.; Robertus J. D. Structure of NS1A Effector Domain from the Influenza A/Udorn/72 Virus. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt A.; Carrillo B.; Prasad B. V. V. In Influenza: Molecular Virology; Wang Q., Tao Y. J., Eds.; Caister Academic Press: Norfolk, U.K., 2010; p 15–28. [Google Scholar]
- Aramini J. M.; Hamilton K.; Ma L. C.; Swapna G. V.; Leonard P. G.; Ladbury J. E.; Krug R. M.; Montelione G. T. 19F NMR Reveals Multiple Conformations at the Dimer Interface of the Nonstructural Protein 1 Effector Domain from Influenza A Virus. Structure 2014, 22, 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramini J. M.; Ma L. C.; Zhou L.; Schauder C. M.; Hamilton K.; Amer B. R.; Mack T. R.; Lee H. W.; Ciccosanti C. T.; Zhao L.; Xiao R.; Krug R. M.; Montelione G. T. Dimer Interface of the Effector Domain of Non-structural Protein 1 from Influenza A Virus: an Interface with Multiple Functions. J. Biol. Chem. 2011, 286, 26050–26060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerry P. S.; Long E.; Taylor M. A.; Russell R. J. Conservation of a Crystallographic Interface Suggests a Role for Beta-Sheet Augmentation in Influenza Virus NS1 Multifunctionality. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 858–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia S.; Robertus J. D. X-Ray Structures of NS1 Effector Domain Mutants. Arch. Biochem. Biophys. 2010, 494, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllon J.; Russell R. J.; Garcia-Sastre A.; Hale B. G. Contribution of NS1 Effector Domain Dimerization to Influenza A Virus Replication and Virulence. J. Virol. 2012, 86, 13095–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milov A. D.; Salikov K. M.; Shirov M. D. Application of ELDOR in Electron-Spin Echo for Paramagnetic Center Space Distribution in Solids. Fiz. Tverd. Tela 1981, 23, 975. [Google Scholar]
- Altenbach C.; Flitsch S. L.; Khorana H. G.; Hubbell W. L. Structural Studies on Transmembrane Proteins. 2. Spin Labeling of Bacteriorhodopsin Mutants at Unique Cysteines. Biochemistry 1989, 28, 7806–7812. [DOI] [PubMed] [Google Scholar]
- Pannier M.; Veit S.; Godt A.; Jeschke G.; Spiess H. W. Dead-Time Free Measurement of Dipole-Dipole Interactions between Spins. J. Magn. Reson. 2000, 142, 331–340. [DOI] [PubMed] [Google Scholar]
- Jeschke G. DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 2012, 63, 419–446. [DOI] [PubMed] [Google Scholar]
- Schiemann O.; Weber A.; Edwards T. E.; Prisner T. F.; Sigurdsson S. T. Nanometer Distance Measurements on RNA Using PELDOR. J. Am. Chem. Soc. 2003, 125, 3434–3435. [DOI] [PubMed] [Google Scholar]
- Pliotas C.; Ward R.; Branigan E.; Rasmussen A.; Hagelueken G.; Huang H.; Black S. S.; Booth I. R.; Schiemann O.; Naismith J. H. Conformational State of the MscS Mechanosensitive Channel in Solution Revealed by Pulsed Electron–Electron Double Resonance (PELDOR) Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelueken G.; Ward R.; Naismith J.; Schiemann O. MtsslWizard: In Silico Spin-Labeling and Generation of Distance Distributions in PyMOL. Appl. Magn. Reson. 2012, 42, 377–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyhach Y.; Jeschke G. Prediction of Favourable Sites for Spin Labelling of Proteins. Spectroscopy 2010, 24, 651–659. [Google Scholar]
- Krivov G. G.; Shapovalov M. V.; Dunbrack R. L. Jr. Improved Prediction of Protein Side-Chain Conformations with SCWRL4. Proteins 2009, 77, 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode B. E.; Margraf D.; Plackmeyer J.; Dürner G.; Prisner T. F.; Schiemann O. Counting the Monomers in Nanometer-Sized Oligomers by Pulsed Electron–Electron Double Resonance. J. Am. Chem. Soc. 2007, 129, 6736–6745. [DOI] [PubMed] [Google Scholar]
- Jeschke G.; Chechik V.; Ionita P.; Godt A.; Zimmermann H.; Banham J.; Timmel C. R.; Hilger D.; Jung H. DeerAnalysis2006 a Comprehensive Software Package for Analyzing Pulsed ELDOR Data. Appl. Magn. Reson. 2006, 30, 473–498. [Google Scholar]
- Chiang Y.-W.; Borbat P. P.; Freed J. H. The Determination of Pair Distance Distributions by Pulsed ESR using Tikhonov Regularization. J. Magn. Reson. 2005, 172, 279–295. [DOI] [PubMed] [Google Scholar]
- Giannoulis A.; Ward R.; Branigan E.; Naismith J. H.; Bode B. E. PELDOR in Rotationally Symmetric Homo-Oligomers. Mol. Phys. 2013, 111, 2845–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G. Conformational Dynamics and Distribution of Nitroxide Spin Labels. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 72, 42–60. [DOI] [PubMed] [Google Scholar]
- Florin N.; Schiemann O.; Hagelueken G. High-Resolution Crystal Structure of Spin Labelled (T21R1) Azurin from Pseudomonas Aeruginosa: a Challenging Structural Benchmark for In Silico Spin Labelling Algorithms. BMC Struct. Biol. 2014, 14, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Ma L. C.; Xiao R.; Radvansky B.; Aramini J.; Zhao L.; Marklund J.; Kuo R. L.; Twu K. Y.; Arnold E.; Krug R. M.; Montelione G. T. Structural Basis for Suppression of a Host Antiviral Response by Influenza A Virus. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13093–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.