

Abstract
Fanconi’s Anemia is primarily an autosomal recessive genetic disorder characterized by congenital abnormalities, defective haematopoiesis leading to bone marrow failure and increased risk of development of Myelodysplastic syndrome, acute myeloid leukemia and solid tumours. Chromosomal instability can be demonstrated by breakage caused by alkylating agents and forms the basis of diagnosis. Our patient presented with structural deformities associated with features of bone marrow failure in form of pancytopenia. Bone marrow analysis and flow cytometry done on aspirate was suggestive of MDS. He subsequently progressed to frank acute myeloid leukemia and succumbed to the illness. The case is being reported for its rarity especially, Fanconi’s Anemia associated with monosomal karyotype (one monosomy plus one more structural abnormality).
Keywords: Fanconi’s anemia, Chromosomal breakage syndrome, AML, Chromosomal anomalies
Introduction
Fanconi’s Anemia is an inherited Bone marrow failure syndrome characterized by varying degree of bone marrow failure, birth defects and predisposition to Myelodysplastic syndrome (MDS) and acute leukemia [1]. First description of Fanconi’s anemia (FA) dates back to 1927 when Fanconi described three brothers who had pancytopenia and birth defects. Since then, more than 1,000 cases of Fanconi’s Anemia have been reported in World’s literature. Exact frequency in India is not known. The prevalence of FA is estimated to be 1–5 per million, and heterozygous carrier frequency is estimated to be 1 in 300, although the true frequency is probably higher. It is a chromosome instability syndrome with cellular hypersensitivity to inter-strand DNA crosslinking agents such as cisplatin, mitomycin C(MMC), diepoxybutane (DEB), and melphalan [2, 3]. Treatment with these agents causes increased chromosome breakage in cells derived from patients with FA [4]. Here we present a rare case of Fanconi’s anaemia who developed MDS followed by frank leukemia and was detected to have complex cytogenetic abnormalities.
Case Report
A five year old boy, product of a non-consanguineous marriage was referred to our tertiary care centre with complaints of easy fatigability, progressively increasing pallor and gum bleeding of six months duration. There was no associated history of fever, jaundice or blood transfusion. Examination revealed a conscious, cooperative child with normal vital parameters. He had severe pallor. However there was no icterus, lymphadenopathy or pedal edema. The child had a triangular face with mongoloid slant, frontal bossing, epicanthal fold in both eyes, depressed nasal bridge and partial syndactyly (both sides second and third toes fused) (Fig. 1a, b). There was hyper pigmented patch over right shoulder (Café-au-lait) (Fig. 1c). On anthropometry the child was found to be in 3rd percentile for height and head circumference and 50th percentile for weight. Systemic examination was essentially within normal limits. Investigations revealed Hb- 5.7 g/dl, MCV- 95 fl (femtoliters), RDW-14.5 %, Reticulocyte-1.5 %,TLC- 3,800/µl, DLC- N20L79M01 and Platelets- 40,000/µl. Peripheral blood smear showed macrocytes with normocytic normochromic red cells, leukopenia with lymphocytosis and thrombocytopenia. His biochemical parameters were within normal limits with serum LDH- 368 U/l (normal range 200–400 IU/l). Bone marrow aspiration yielded cellular smears with dyserythropoiesis (nuclear budding, multinuclearity and megaloblastoid nuclei in 35 % of erythroid cells) and dysmyelopoisis (hypogranularity and nuclear hyposegmentation in 35 % of myeloid cells). Megakaryocytes were reduced. Blasts constituted 13 % of all nucleated cells and on cytochemistry were myeloperoxidase positive. Final opinion on bone marrow was Myelodysplastic Syndrome—Refractory anemia with excess Blasts (MDS—RAEB- II) (High grade Myelodysplastic Syndrome) (Fig. 2a). A flow cytometric evaluation (Beckman Coulter FC 500, Miami, USA) was done which showed two population of blasts, one with monocytic differentiation and another were myeloblast. The myeloblast were positive for CD45, CD34, CD13, HLA-DR and MPO. The blasts with monocytic differentiation were positive for CD34, CD45, CD14, Cd13, CD11c and HLA-DR. Gating strategy was CD45 versus SSC.
Fig. 1.
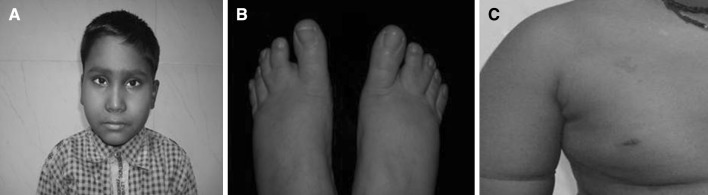
a–c Photograph of the child showing epicanthal fold, simple partial syndactyly of both feet (fusion of second and third toes) and Café au lait spot on the right shoulder
Fig. 2.

a Cellular bone marrow aspirate smears showing myeloblasts with erythroid precursors and a unilobate megakaryocyte (BMA Leishman Giemsa stain × 100×). b Hypercellular bone marrow aspirate showing moderate to large sized blasts with high N: C ratio, greyish blue cytoplasm, cleaved nucleus and prominent nucleoli (BMA Leishman Giemsa stain × 100×). (Color figure online)
The chromosomal breakage study was performed as per the International Fanconi Anemia Recommendation. The reporting was done on peripheral blood lymphocytes after exposure to Di-epoxybutane exposure (DEB). Karyotype showed increase in chromatid breaks in the form of gaps/endoreduplications/radials and the number of breaks per cell ranged from 16 to 25 %/cell (normal control range was 0–0.05 %/cell) (Fig. 3a). The conventional karyotype was performed on peripheral blood of the patient using standard protocol and Roswell Park Memorial Institute medium (RPMI media). The cultures were studied after minimum of 72 h and 20 metaphases were analysed. Aneuploidy was found in 12 out of 20 metaphases in form of monosomy 7. Trisomy 21 was seen in at least eight out of the 20 metaphases [Karyotype -47, XY, +21, −7] (Fig 3b).
Fig. 3.

a Metaphase spread from Fanconi Anemia patient exhibiting multiple chromosomal breaks and radial forms (some indicated by arrow). b G-band metaphase chromosome analysis revealed abnormalities in chromosome 7 and 21 [Karyotype -47, XY, +21, −7]
A final diagnosis of Fanconi’s anaemia with clonal chromosomal abnormalities and MDS (RAEB-II) was given. Patient was managed conservatively and was kept under close follow up, however within a short span of two months he converted to acute myeloid leukemia (AML-M4) (Fig 2b). Cytochemistry was done on the peripheral smear and >3 % blasts revealed MPO (Myeloperoxidase) positivity and at least 20 % blasts showed positivity in combined dual esterase test. The bone marrow showed more 60 % blasts of myelomonocytic lineage and flow cytometry showed strong positivity with CD33 and CD14 (a strong monocyte lineage marker).
Repeat cytogenetics could not be done.
He succumbed to his disease within a week thereafter before treatment for leukemia could commence.
Discussion
Patients with FA show extreme clinical heterogeneity. The median age at diagnosis is 6.5 years for male patients, and 8 years for female patients, although the age at diagnosis ranges from birth to 48 years. The median survival age has improved to 30 years in patients reported between 1991 and 2000. The common congenital defects seen in patient with FA includes short stature (51 %), abnormalities of the skin (55 %), of upper extremities (43 %), head (26 %), eyes (23 %), kidneys (21 %), and ears (11 %) and developmental disability (11 %) [3]. However the physical abnormalities may be absent or subtle. The most common haematological malignancies seen in patients with FA are myelodysplastic syndrome and AML. Our patient had dysmorphic facial features, syndactyly and Café-au-lait spot who developed MDS followed by acute myeloid leukemia.
Patients with FA are also susceptible to solid tumors such as head and neck squamous cell carcinoma (HNSCC), gynecologic squamous cell carcinoma (SCC), oesophageal carcinoma, liver tumors, brain tumors, skin tumors, and renal tumors [5].
Many hematologists are familiar with Fanconi anemia (FA) as a cause of childhood-onset aplastic anemia. The hematologic complications of FA have also been extensively reviewed. Patients with FA develop bone marrow failure typically during the first decade of life. The actuarial risk of developing bone marrow failure is 90 % by 40 years of age. The median age of patients who develop cancer is 14 years for acute myelogenous leukemia (AML), 13 years for liver tumors, and 26 years for all solid tumors. AML is the initial presentation in approximately one third of published cases of FA with AML [6].
Our patient presented with pancytopenia with evidence of MDS-RAEB-II who subsequently transformed into AML-M4 within a period of two months.
The most frequently reported chromosomal abnormalities in FA-associated AML are monosomy 7, gain of 1q and gain of 3q while t(8;21), t(6;9), inv(16) (p13q22) and trisomy 8 are sole findings in de novo AML [7, 8]. A study identified a high incidence of chromosome 3q abnormalities in bone marrow cells of patients with FA. Gain of 3q is strongly associated with a poor prognosis. In our patient peripheral blood analysis for chromosomal breakage exhibited tri-radial and complex figures and cytogenetic studies showed monosomy 7 and trisomy 21.
Treatment of FA cells with DNA cross-linking agents, such as DEB causes increased chromosome breakage and marked G2 accumulation. The DEB-induced chromosome breakage assay (DEB test) is widely used as a standard diagnostic test for suspected cases of FA and should be preferred over mitomycin-C.
The management of patients of FA has also been a medical challange. For BM failure, androgens and hematopoietic growth factors are effective in many cases but most patients with FA become refractory to these agents. For such patients hematopoietic stem cell transplantation (SCT) is performed, provided a donor is available. The outcome of SCT has improved over the years [9, 10]. Stem cell transplantation is often effective treatment in bone marrow failure in FA. In the patients with MDS/AML, it is best to proceed to a SCT rather than chemotherapy. As a new modality of therapy, gene therapy will be an option in the future [11].
Conclusion
This case has been reported here due to the unusual presentation of Fanconi’s anemia presenting with features of MDS and progressing to AML within a short period of two months. Morever he was found to have two congenital genetic abnormalities i.e. monosomy 7 and trisomy21. In such cases haematopoietic stem cell transplant (HSCT) is the only successful treatment option available.
Acknowledgments
Conflict of interest
Authors declare no conflict of interest.
Consent Statement
Consent of the patients parents has been taken and it has been explained to them that since no masking is being done identity of child will be revealed. All Human studies have been approved by the appropriate ethical comittee and therefore have been performed in accordance with the ethical standards laid down in 1964 Declaration of Helsinki and its later amendments.
Contributor Information
H. Rama, Phone: 08967596474, Email: rama.siddarth@gmail.com
Devika Gupta, Phone: 9158984335, Email: devikalives5h@gmail.com.
Tathagata Chatterjee, Phone: 08551910936, Email: ctathagat@hotmail.com.
Srishti Gupta, Phone: 9910760125, Email: srishtigpt10@gmail.com.
References
- 1.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auerbach AD, Buchwald M, Joenje H (2001) Fanconi anemia. In: Scriver CR, Sly WS, Childs B et al (eds) The metabolic and molecular bases of inherited disease, vol 1, 8th ed. McGraw-Hill, New York, pp. 753–768
- 3.Alter BP (2003) Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT (eds) Nathan and Oski’s hematology of infancy and childhood, vol 1, 6th ed. Saunders, Philadelphia, pp. 280–365
- 4.Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21:731–733. [PubMed] [Google Scholar]
- 5.Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 6.Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97:425–440. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 7.Rochowski A, Olson SB, Alonzo TA, et al. Patients with Fanconi anemia and AML have different cytogenetic clones than de novo cases of AML. Pediatr Blood Cancer. 2012;59(5):922–924. doi: 10.1002/pbc.24168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tonnies H, Huber S, Kuhl JS, et al. Clonal chromosome aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood. 2003;101:3872–3874. doi: 10.1182/blood-2002-10-3243. [DOI] [PubMed] [Google Scholar]
- 9.Tischkowitz M, Dokal I. Fanconi anaemia and leukaemia— clinical and molecular aspects. Br J Haematol. 2004;126:176–191. doi: 10.1111/j.1365-2141.2004.05023.x. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg PS, Alter BP, Socie G, et al. Secular trends in outcomes for Fanconi anemia patients who receive transplants: implications for future studies. Biol Blood Marrow Transplant. 2005;11:672–679. doi: 10.1016/j.bbmt.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Croop JM. Gene therapy for Fanconi anemia. Curr Hematol Rep. 2003;2:335–340. [PubMed] [Google Scholar]