
Figure 1.
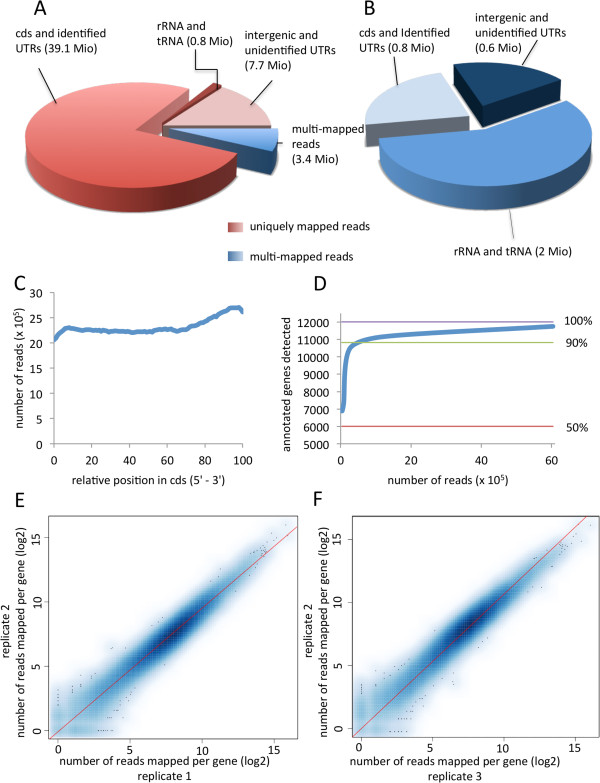
Statistics and analysis of the quality of RNA-Seq data. A. Distribution of mapped reads over exons, introns, intergenic and untranslated regions (UTRs). About 93% of all reads mapped to unique locations in the genome of C. graminicola, of which 82.5% matched coding sequences (cds) and identified UTRs, 15.9% to intergenic sequences and unidentified UTRs, and 1.6% to sequences encoding rRNA/tRNA. B. The remaining 7% of reads mapped to multiple locations, of which 56.8% matched sequences encoding rRNA/tRNA, 23.3% to cds and identified UTRs, and 19.9% to intergenic sequences and unidentified UTRs. The read numbers of each category are given in brackets. C. Total coverage of cds by RNA-Seq reads. The cds were divided into 100 equal windows. D. Gene coverage vs. sequencing depth. Approx. 87% of C. graminicola genes were detected at about 2.5 million uniquely mapped RNA-Seq reads, and coverage reaches a plateau afterwards despite the increasing sequencing depth. E-F. Scatter plot analysis of RNA-Seq data of biological repeats 1 vs. 2 (E) and 2 vs. 3 (F). Each repeat consists of a combined data set of six individual RNA samples taken at 0, 12, 24, 48, 72, 120 hours postinoculation. Each blue point represents a particular gene. The color shading correlates with the density of these points.