

Abstract
ProBMP4 is generated as a latent precursor that is sequentially cleaved at two sites within the prodomain to generate an active ligand. An initial cleavage occurs adjacent to the ligand domain, which generates a non-covalently associated prodomain/ligand complex that is subsequently dissociated by cleavage at an upstream site. An outstanding question is whether the two sites need to be cleaved sequentially and in the correct order to achieve proper control of BMP4 signaling during development. In the current studies, we demonstrate that mice carrying a knock-in point mutation that causes simultaneous rather than sequential cleavage of both prodomain sites show loss of BMP4 function and die during mid-embryogenesis. Levels of mature BMP4 are severely reduced in mutants, although levels of precursor and cleaved prodomain are unchanged compared with wild type. Our biochemical analysis supports a model in which the transient prodomain/ligand complex that forms during sequential cleavage plays an essential role in prodomain-mediated stabilization of the mature ligand until it can acquire protection from degradation by other means. By contrast, simultaneous cleavage causes premature release of the ligand from the prodomain, leading to destabilization of the ligand and loss of signaling in vivo.
Keywords: Bone morphogenetic protein 4, Prodomain, Proprotein convertase
INTRODUCTION
Bone morphogenetic protein 4 (BMP4) is a secreted signaling molecule that plays an essential role in the development of most organs. BMP4 activates distinct cellular responses in a dose-dependent manner and proper regulation of ligand levels is crucial for normal development. For example, null mutations in Bmp4 lead to early embryonic lethality (Winnier et al., 1995), whereas hypomorphic or tissue-specific mutations cause a spectrum of ocular, skeletal, urogenital, cardiovascular and brain anomalies in both mice (Bragdon et al., 2011) and humans (Bakrania et al., 2008; Reis et al., 2011; Suzuki et al., 2009; Tabatabaeifar et al., 2009; Weber et al., 2008). Conversely, mutations that result in gain of BMP function lead to embryonic lethality in animal models and bone disorders in humans (Shore et al., 2006; Walsh et al., 2010). Thus, BMP activity must be strictly regulated to prevent congenital anomalies.
BMP4 is generated as inactive precursor protein that dimerizes in the endoplasmic reticulum (ER) and is then cleaved to generate prodomain and mature domain fragments (Bragdon et al., 2011). Both of these cleavage products are secreted but only the mature domain is able to bind to and activate BMP receptors. Although the prodomain lacks signaling activity, it is essential for the generation of an active ligand. Point mutations or deletions within the prodomain lead to loss of BMP4 function in animal models (Cui et al., 2001; Degnin et al., 2004; Goldman et al., 2006; Kunnapuu et al., 2009; Sopory et al., 2010) and in humans (Bakrania et al., 2008; Suzuki et al., 2009; Tabatabaeifar et al., 2009; Weber et al., 2008).
BMP4 is cleaved by members of the proprotein convertase (PC) family of endoproteases (Constam and Robertson, 1999; Cui et al., 1998), seven of which have been characterized in mammals (Seidah et al., 2008). Among these, furin has been identified as an endogenous BMP4 convertase (Kim et al., 2012), although PC6 and PC7 also contribute to cleavage in some tissues (Nelsen and Christian, 2009). Furin preferentially cleaves substrates containing the optimal consensus motif -R-X-R/K-R-, but can also cleave following the minimal recognition motif -R-X-X-R- (Molloy et al., 1992; Nakayama, 1997).
BMP4 is sequentially cleaved at two sites: initially at an optimal furin recognition motif (S1) adjacent to the mature domain and subsequently at an upstream minimal motif (S2) within the prodomain (illustrated in Fig. 1A). Mature BMP4 remains non-covalently attached to the prodomain following cleavage at the S1 site, whereas subsequent cleavage at the S2 site disrupts the complex, freeing the mature ligand from the prodomain (Degnin et al., 2004). Whereas the prodomain/mature BMP4 complex is preferentially targeted for lysosomal degradation, free BMP4 is more stable. Analyses in Drosophila and mice have shown that cleavage at the S2 site is essential for normal development, and that this site is selectively cleaved in a tissue-specific fashion (Goldman et al., 2006; Kunnapuu et al., 2009; Sopory et al., 2010). These studies demonstrate that cleavage of the S2 site provides a tissue-specific mechanism to regulate the stability, and thus the signaling intensity, of mature BMP4.
Fig. 1.

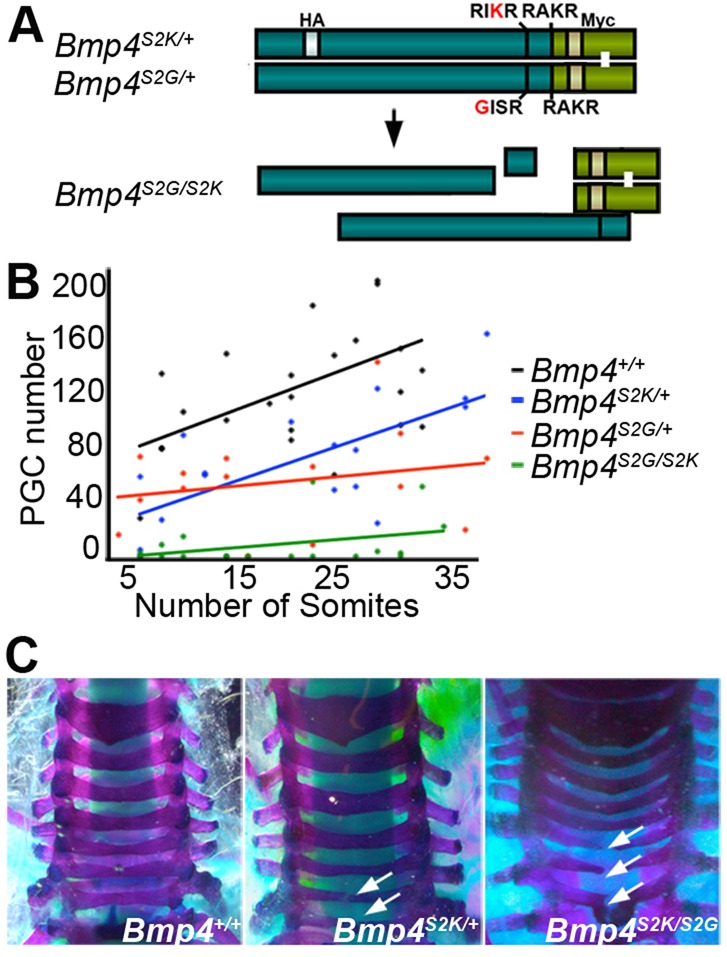
Phenotypic defects in Bmp4S2KHAmyc mutant embryos. (A) Schematic illustration and cleavage patterns of BMP4 proteins expressed by Bmp4HAmyc and Bmp4S2KHAmyc mice. Prodomain, dark green bar; mature domain, light green bar. (B) E10.5 littermates from Bmp4S2KHAMyc/+ or Bmp4HAMyc/+ intercrosses. White arrow, ballooned heart; black arrows, hemorrhage; black arrowhead, small eye. (C) Dorsal view of alkaline phosphatase-stained E9.5 embryos (top panel) and graph showing PGC number versus somite pairs (bottom panel). PGCs are indicated by white arrows.
Kinetic analysis of in vitro cleavage of recombinant BMP4 suggests that ordered cleavage is driven, at least in part, by the presence of optimal and minimal furin consensus motifs at the S1 and S2 sites, respectively. Introduction of an optimal furin consensus motif at the S2 site (BMP4S2K; illustrated in Fig. 1A) allows both sites to be cleaved simultaneously such that mature BMP4 is generated and released from the prodomain complex more rapidly (Cui et al., 1998; Degnin et al., 2004). Overexpression of BMP4S2K in Xenopus embryos generates higher ectopic BMP activity than that induced by the same level of wild-type precursor (Cui et al., 2001), suggesting that the minimal furin motif at the S2 site acts to dampen BMP4 activity in vivo, perhaps by slowing production of fully cleaved mature BMP4 that is free of its prodomain.
In order to test whether sequential cleavage of BMP4 is essential to regulate growth factor activity under physiological conditions, we generated mice carrying a knock-in point mutation (Bmp4S2KHAMyc) that converts the S2 site from a minimal to an optimal furin motif. By contrast to the gain of function phenotype that is observed when BMP4 is overexpressed (Cui et al., 2001), we found instead that Bmp4S2KHAMyc is a severe hypomorphic allele, due to selective loss of cleaved BMP4 ligand.
RESULTS
Introduction of an optimal furin motif at the S2 site of BMP4 leads to embryonic lethality
To test whether the presence of a minimal furin motif at the S2 site of BMP4 is required for proper regulation of BMP4 activity in vivo, we generated a cleavage mutant ‘knock-in’ mouse (Bmp4S2KHAMyc, the S2K allele) by conventional gene targeting (supplementary material Fig. S1A,B). These mice have sequences encoding HA and Myc epitope tags knocked in to the Bmp4 allele, as well as a single point mutation that changes the amino acid sequence of the S2 cleavage site from a minimal furin motif (-RISR-) to an optimal furin motif (-RIKR-) (Fig. 1A). Whereas wild-type BMP4 is sequentially cleaved at the S1 and then the S2 site, the S2K mutation allows for simultaneous cleavage of both sites (Cui et al., 2001; Degnin et al., 2004) (illustrated in Fig. 1A). A control mouse line that carries the same HA and Myc epitope tags at the wild-type Bmp4 locus (Bmp4HAMyc) was generated in parallel. The epitope tags do not interfere with proper folding or function of BMP4 when ectopically expressed in Xenopus embryos (supplementary material Fig. S1C).
In initial studies, Bmp4S2KHAMyc/+ and Bmp4HAMyc/+ mice were intercrossed to determine viability at weaning. Whereas Bmp4HAMyc homozygotes were viable, fertile and appeared grossly normal, virtually all Bmp4S2KHAMyc homozygotes died before weaning (Tables 1 and 2). To determine when mutants were dying, we established timed matings between Bmp4S2KHAMyc/+ mice and analyzed embryos at various developmental ages. As detailed in Table 1, Bmp4S2KHAMyc/S2KHAMyc embryos were recovered at the expected frequency between embryonic day (E) 8 and E11.5, but were recovered at less than one-third of the expected frequency by E12.5. Visual analysis of Bmp4S2KHAMyc homozygotes recovered at E10.5 or later revealed multiple gross abnormalities relative to wild-type or Bmp4S2KHAMyc/+ littermates (Fig. 1B, top four panels). Bmp4S2KHAMyc/S2KHAMyc embryos were severely runted, had abnormal hearts (Fig. 1B, white arrow), hemorrhages (black arrows) and/or small eyes (black arrowhead). By contrast, Bmp4HAmyc homozygotes could not be distinguished from heterozygotes (data not shown) or wild-type littermates (Fig. 1B, bottom two panels). Bmp4HAmyc homozygotes were adult viable, grossly normal and did not show the skeletal or eye defects that are observed in Bmp4 null heterozygotes (Dunn et al., 1997). These results demonstrate that a minimal cleavage motif at the S2 site is essential for BMP4 function in development. Notably, because both loss and gain of BMP function can lead to embryonic lethality and patterning defects, these results alone do not indicate whether the S2K mutation causes upregulation or downregulation of BMP4 activity.
Table 1.
Progeny from Bmp4S2KHAMyc/+ intercrosses
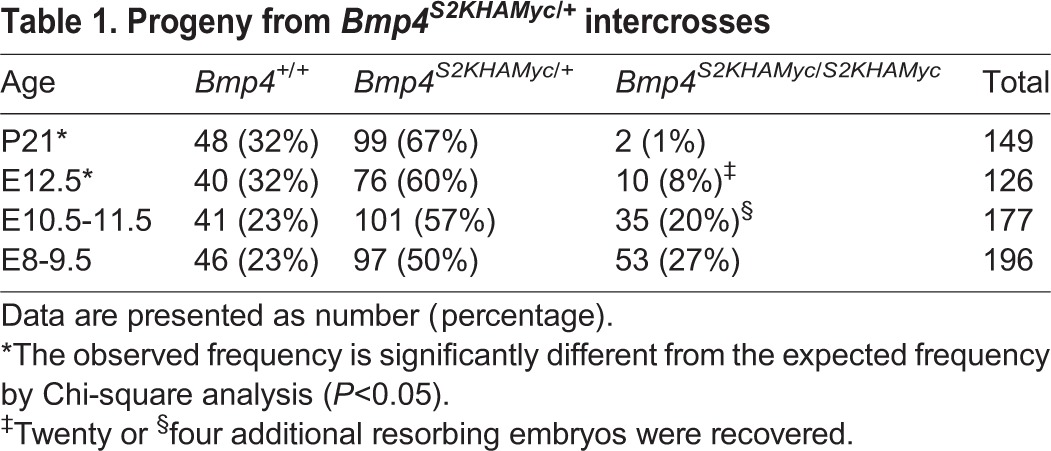
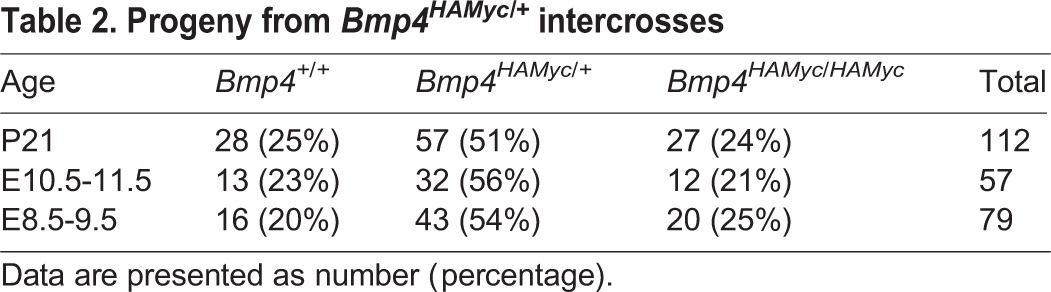
Table 2.
Progeny from Bmp4HAMyc/+ intercrosses
To test whether the S2K mutation causes a loss or gain of BMP function, we analyzed primordial germ cell (PGC) number, which is a sensitive indicator of Bmp4 dosage. Bmp4 null heterozygotes show a 50% reduction, and null homozygotes show complete loss of PGCs (Lawson et al., 1999). As shown in Fig. 1C, Bmp4S2KHAMyc heterozygotes and homozygotes showed a 50 and 100% reduction in PGC number, respectively. This reduction is comparable to that observed in Bmp4 null heterozygotes and homozygotes, respectively, consistent with this being a severe hypomorphic allele. PGC number was slightly reduced in Bmp4HAMyc heterozygotes (85-90% of control) and homozygotes (70-80% of control) (supplementary material Fig. S2A), indicating that the epitope tags cause some reduction in activity when measured in this highly sensitive assay. However, Bmp4HAMyc heterozygotes and homozygotes have substantially higher levels of PGCs than age-matched Bmp4S2KHAMyc heterozygotes (supplementary material Fig. S2B), indicating that the epitope tags do not account for the reduction in PGCs in mice carrying the S2K mutation.
Bmp4S2KHAMyc is a hypomorphic allele
If Bmp4S2KHAMyc is a hypomorphic allele, then a reduction in Bmp4 dosage should exacerbate the phenotype. By contrast, if these defects represent a gain of function, then they should be partially rescued by a reduction in gene dosage. To distinguish between these possibilities, we reduced Bmp4 gene dosage in all tissues by intercrossing Bmp4S2KHAMyc/+ and Bmp4lacZ/+ mice [a null allele in which exon 3 is replaced with lacZ (Lawson et al., 1999)] to generate compound heterozygotes. Bmp4S2KHAMyc/+ and Bmp4lacZ/+ mice were recovered and appeared grossly indistinguishable from controls at weaning, whereas no live born Bmp4S2KHAMyc/lacZ compound heterozygotes were observed (Table 3). Bmp4S2KHAMyc/lacZ embryos were recovered at the expected frequency between E10.5 and E11.5 but were severely runted, deformed and/or beginning to resorb relative to wild-type, Bmp4S2KHAMyc/+ or Bmp4+/lacZ littermates (Fig. 2). Thus, in a haploinsufficient background, the S2K allele does not support viability or normal development at the same level as the wild-type allele.
Table 3.
Progeny from Bmp4S2KHAMyc/+ and Bmp4lacZ/+ intercrosses
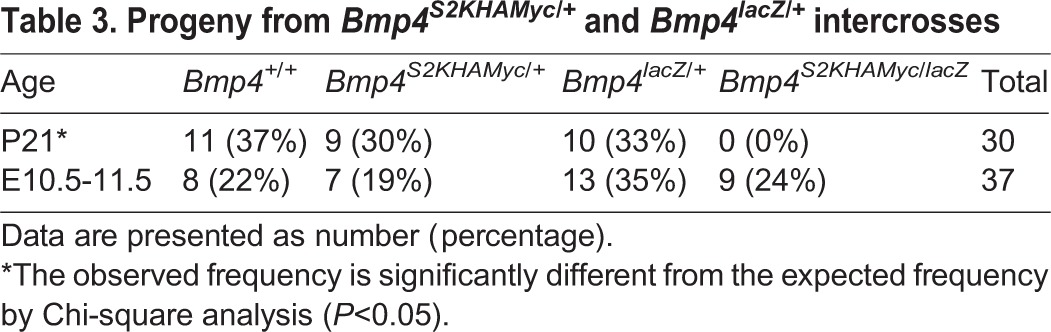
Fig. 2.
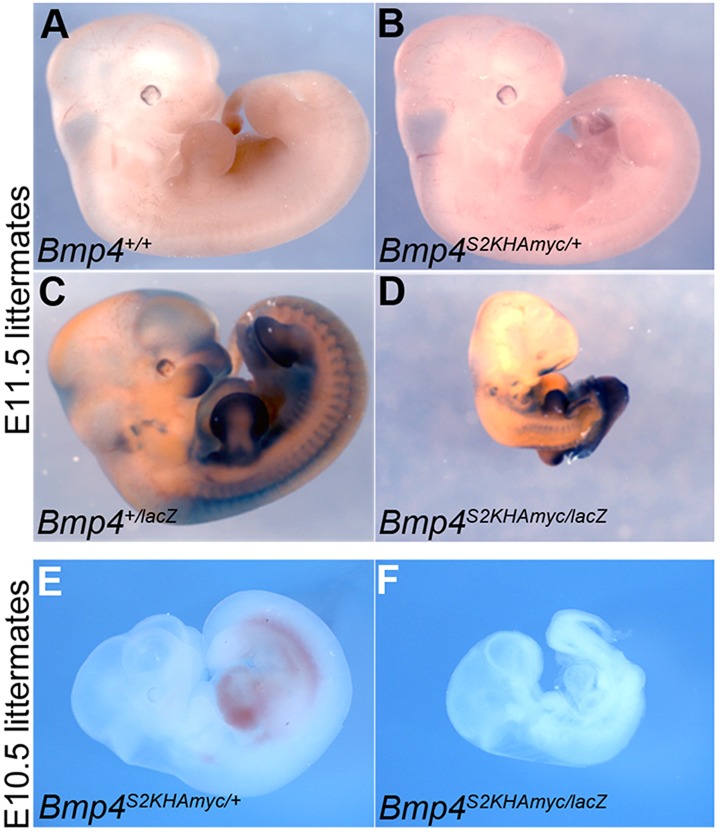
Removal of one copy of Bmp4 enhances phenotypic defects in Bmp4S2KHAmyc mutants. (A-D) Bmp4+/lacZ and Bmp4S2K/+ mice were intercrossed, and representative E11.5 embryos from a single litter were stained for β-galactosidase, which marks the Bmp4lacZ allele. (E,F) Embryos from a second litter were photographed without staining at E10.5.
To test further whether the S2K allele leads to a gain or loss of BMP4 function, we analyzed BMP activity in BRE:lacZ transgenic embryos. This transgene contains a BMP-responsive element coupled to lacZ (Monteiro et al., 2004) that serves as an in vivo reporter of total BMP signaling downstream of all BMP ligands and not just BMP4. To narrow down the sites of BMP activity that might be attributed to BMP4, we first analyzed expression of Bmp4 by β-galactosidase staining of Bmp4lacZ/+ embryos, in which lacZ marks the site of transcription of endogenous Bmp4 (Lawson et al., 1999). At E9.5, Bmp4 transcripts are highly expressed in the ventroposterior mesoderm (Fig. 3A, black arrows), branchial arches (black arrowheads), the eye (open arrowhead) and the roof of the midbrain (black asterisk). Endogenous BMP activity was detected in each of these tissues (Fig. 3B-E) and in the heart (white asterisk) as shown by β-galactosidase staining of E9.5 BRE:lacZ transgenic embryos. Bmp4S2KHAMyc/S2KHAMyc embryos exhibited a reproducible reduction in BMP activity in the ventroposterior mesoderm (Fig. 3B,C, black arrows), the eye (open arrowhead) and branchial arches (black arrowheads, boxed insets) relative to heterozygous (not shown) or wild-type siblings. No differences in activity were noted in the atria or ventricles of the heart (white asterisk), which is expected as Bmp4 is not expressed in these tissues at E 9.5 (Fig. 3A). Bmp4 is expressed in the brain (Fig. 3A), but we saw little or no decrease in activity in the brain of S2K mutants. As other BMP family members (e.g. Bmp2 and Bmp7) are co-expressed with Bmp4 in the brain (Danesh et al., 2009), it is probable that these other BMPs can function redundantly to compensate for reduced BMP4 signaling in the brain of Bmp4S2KHAMyc homozygotes. No differences in BMP activity were detected in any tissues of Bmp4HAMyc/HAMyc embryos relative to wild-type littermates (Fig. 3D,E). Collectively, these data indicate that the presence of an optimal rather than a minimal furin cleavage motif at the S2 site of BMP4 leads to a reduction in BMP4 activity in vivo.
Fig. 3.
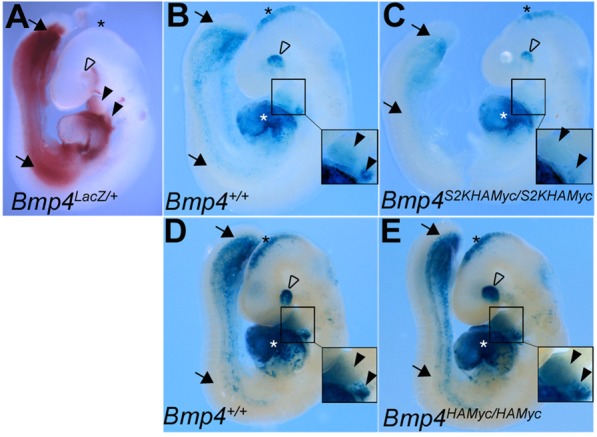
Bmp4S2KHAmyc/S2KHAmyc mutants show reduced BMP activity in a subset of tissues where endogenous Bmp4 is expressed. (A) E9.5 Bmp4lacZ/+ embryo stained with Red-Gal to detect sites of endogenous Bmp4 expression. (B-E) E9.5 Bmp4+/+ and Bmp4S2KHAmyc/S2KHAmyc (B,C) or Bmp4+/+ and Bmp4HAmyc/HAmyc littermates (D,E) carrying a BRE-lacZ transgene were stained with X-Gal to detect endogenous BMP activity. Embryos from a single litter were stained for an identical period of time under identical conditions. Closed black arrows, ventroposterior mesoderm; white asterisk, left atria and ventricle of heart; open arrowhead, eye; black asterisk, brain; closed arrowheads, branchial arches. Insets in B-E show enlarged view of branchial arches.
The S2K mutation does not disrupt precursor stability or cleavage but leads to a selective loss of cleaved ligand
Equivalent levels of proBMP4 and cleaved prodomain were detected on western blots of lung lysates from Bmp4HAMyc/+ and Bmp4S2KHAMyc/+ mice probed with anti-HA antibodies in three independent experiments (Fig. 4A). This finding shows that the loss of function in Bmp4S2KHAMyc/S2KHAMyc mice is not due to protein misfolding caused by the amino acid substitution, as misfolding would cause the precursor to be retained in the ER, where it could not be cleaved and/or would be degraded. Thus, the S2K point mutation does not disrupt folding or trafficking of proBMP4 out of the ER.
Fig. 4.
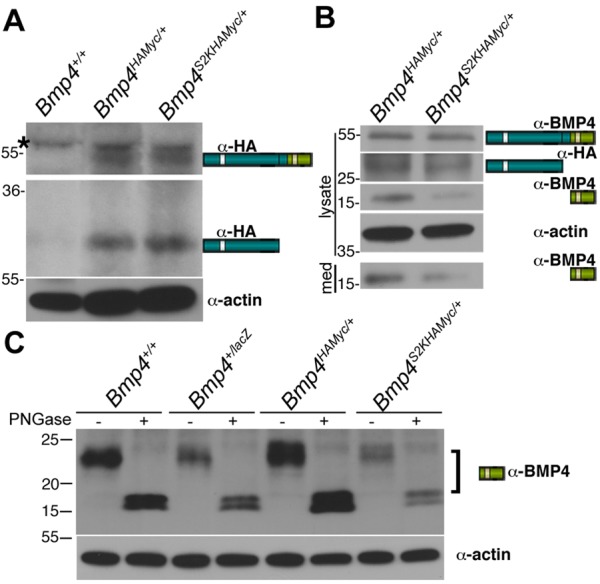
The S2K mutation leads to a reduction in steady state levels of mature BMP4. (A) The S2K mutation does not lead to reduction in steady state levels of BMP4 precursor or cleaved prodomain. Western blot of equivalent amounts of protein from adult lungs of mice probed with anti-HA or anti-actin antibodies. Bands corresponding to precursor and cleaved prodomain are illustrated to the right of the gel. Black asterisk denotes a nonspecific background band. (B) The S2K mutation leads to reduction in steady state levels of cleaved mature BMP4. Western blot of equivalent levels of protein from cell lysates or culture medium (med) from adult lung endothelial cells. Bands corresponding to precursor and cleavage products and antibodies used to detect each band are indicated to the right of the gel. (C) Steady state levels of mature BMP4 are reduced in mice heterozygous for a null mutation or the S2K mutation relative to wild-type controls. Equivalent levels of protein from endothelial cells purified from adult lungs were treated with or without PNGase prior to western blot analysis under reducing conditions. The doublet in PNGase-treated samples most probably reflects incomplete deglycosylation in this experiment.
Because we were unable to reproducibly detect cleaved BMP4 ligand in total lung lysates using antibodies directed against either the mature domain or against the myc tag, we also analyzed levels of BMP4 precursor and cleavage products in cultures of purified lung endothelial cells, which provide a highly enriched source of endogenous BMP4 (Frank et al., 2005). Notably, cells were not acid washed to remove surface-bound proteins prior to lysis and thus cleaved prodomain and mature BMP4 associated with the cell surface, as well as that contained in intracellular compartments, were present in lysates. Western blot analysis revealed equivalent levels of BMP4 precursor in endothelial cells from Bmp4S2KHAMyc/+ and Bmp4HAMyc/+ mice (Fig. 4B). Levels of cleaved prodomain were slightly reduced in cells isolated from Bmp4S2KHAMyc/+ mice and were not detectable in the medium of cells from either line. By contrast, in three independent experiments, steady state levels of mature BMP4 were reduced by 40-60% in the media and lysates of cells isolated from Bmp4S2KHAMyc/+ mice relative to Bmp4HAMyc/+ mice (Fig. 4B).
As a control to demonstrate that our antibody specifically recognizes mature BMP4, we compared ligand levels in lung endothelial cells isolated from wild-type mice with those isolated from mice heterozygous for a null mutation, the S2K mutation or the HAMyc tag in the absence of the S2K mutation (Fig. 4C). Extracts were treated with or without peptide N-glycosidase F (PNGase) to prove that the band we detected is a glycosylated protein, as expected. Relatively equivalent levels of mature BMP4 were detected in cells from wild-type and Bmp4HAMyc/+ mice, whereas significantly lower levels of mature BMP4 were detected in cells from Bmp4 null heterozygotes (Bmp4+/lacZ) and Bmp4S2KHAMyc/+ mice (Fig. 4C). Thus, the S2K mutation leads to a severe reduction in steady state levels of mature BMP4 with little or no loss of precursor or cleaved prodomain.
We were unable to detect endogenous BMP4 cleavage products in early embryos, and thus it has not been possible to compare levels of prodomain and mature ligand in Bmp4S2KHAMyc homozygotes and wild-type littermates. This leaves open the possibility that the remaining wild-type BMP4 protein in Bmp4S2KHAMyc heterozygotes in some way confounds the results. This is unlikely, however, given that we saw an identical selective reduction in steady state levels of mature BMP4, with little or no reduction in levels of precursor protein or cleaved prodomain, in HEK cells transfected with the S2K mutant form of BMP4 (supplementary material Fig. S3).
ProBMP4 traffics through the TGN but is cleaved in more distal subcellular compartments
Our previous analysis supports a model in which the S1 site of BMP4 is cleaved in the trans-Golgi network (TGN), and the prodomain/mature ligand complex then traffics to a more acidic, post-TGN compartment where the S2 site is cleaved (Degin et al., 2004). If this model is correct, then one mechanism by which the S2K mutation could lead to a loss of cleaved ligand is by promoting premature S2 cleavage and release of the mature ligand from the prodomain within the TGN, potentially interfering with ligand stabilization and trafficking to the cell surface. To test this possibility, we used pulse chase analysis to examine accumulation of BMP4 cleavage products in intra- and extracellular compartments over time. HEK cells were transiently transfected with DNA encoding BMP4HAMyc or BMP4S2KHAMyc and then pulse labeled with [35S]methionine-cysteine for one hour. Cells were incubated in unlabeled chase medium for different times, after which cells were acid washed to remove cleavage products bound to the outside of cells. BMP4 precursor and cleavage products were then immunoprecipitated from cell lysates and medium. In cells transfected with wild-type or S2K mutant BMP4, precursor protein was present within cell lysates at the onset of the chase period and its levels decreased over time (Fig. 5A, lysates, top panels). Signal corresponding to cleaved prodomain or mature BMP4 was not detected in cell lysates (Fig. 5A, lysates, middle and bottom panels). By contrast, the intact precursor was detected in conditioned medium from cells transfected with wild-type or S2K mutant BMP4 beginning at 20-60 min postchase, and cleavage fragments became increasingly abundant after that time point (Fig. 5A, medium). The presence of intact proBMP4 in the cell medium and the presence of cleavage products in extracellular, but not intracellular compartments, strongly suggest that both sites of proBMP4 are cleaved coincident with, or following, secretion.
Fig. 5.

ProBMP4 is trafficked through the TGN and cleaved in an intracellular compartment. (A) Pulse chase analysis of HEK293 cells transfected with DNA encoding BMP4HAmyc, BMP4S2KHAmyc or no DNA (−) suggests that BMP4 is cleaved coincident with or after secretion. Precursor protein and cleavage products were immunoprecipitated from cell lysates and medium at various times postchase as indicated above each lane, deglycosylated by incubation in PNGase and analyzed by SDS-PAGE. Arrows designate nonspecific background bands that are detected in untransfected cells. (B) Endo H-resistant, PNGase-sensitive forms of BMP4 precursor and cleavage products (arrowheads) were detected in the medium but not in cell lysates. Samples were not treated (none) or were treated with Endo H or PNGase as indicated above each lane. Arrows designate nonspecific background bands that are detected in untransfected cells. (C) Blocking TGN export does not lead to accumulation of cleavage products in cell lysates, suggesting that BMP4 is cleaved in a post-TGN compartment. Cells were incubated at either 37°C or 19°C throughout the chase period. Samples were treated with PNGase. Arrows designate nonspecific background bands that are detected in untransfected cells. (D,E) Cell-permeable (RVKR-cmk), but not cell-impermeable (D6R) PC inhibitors, block processing of BMP4, whereas the opposite is observed for Xnr2. Cell lysates and medium from transiently transfected HEK293 cells were collected after treatment with vehicle (DMSO), RVKR-cmk or D6R. Western blots were probed with antibodies specific for the HA tag in the prodomain of BMP4 or Xnr2, or with antibodies specific for the mature domain of BMP4. The two prodomain bands present in the medium of Xnr2-transfected cells correspond to two alternative cleavage sites. Arrow designates a nonspecific background band that is detected in untransfected cells.
To determine whether the BMP4 precursor protein present in cell lysates and medium has transited the TGN, we examined its sensitivity to digestion by Endoglycosidase H (Endo H) or PNGase. Carbohydrates that are transferred onto proteins in the ER are sensitive to Endo H digestion. When further modified in the Golgi, these moieties become Endo H resistant, but remain sensitive to PNGase. BMP4 precursor protein present inside of cells was fully Endo H sensitive (Fig. 5B, lysates), suggesting that it had not exited the ER. By contrast, the cleaved prodomain secreted into the culture medium was fully resistant to digestion by Endo H and sensitive to digestion by PNGase, indicating that it had transited the Golgi on the way to the cell surface. As previously noted (Degnin et al., 2004), BMP4 precursor and mature ligand remain partially Endo H sensitive following secretion (Fig. 5B, medium; note shift in mobility following Endo H treatment), suggesting that a subset of carbohydrates present in the mature domain of BMP4 is inaccessible to modifying enzymes in the Golgi once these proteins adopt their fully folded conformation. However, Endo H-resistant/PNGase-sensitive forms of proBMP4, cleaved prodomain and mature BMP4 were detected in the cell medium (Fig. 5B, arrowheads; note the additional shift in mobility following PNGase treatment relative to Endo H-treated samples). These data, showing that the intracellular pool of BMP4 is restricted to the ER, whereas precursor protein present in the medium has trafficked through the TGN, demonstrate that the latter pool is not a contaminant derived from cell lysis. They further suggest that once BMP4 exits the ER, it rapidly traffics through the TGN to the cell surface, without being stably retained in the TGN or in post-TGN compartments.
To more stringently test the possibility that one or both sites of wild-type proBMP4 are cleaved in the TGN, but that cleavage products are not detected in cell lysates because they are rapidly secreted, we repeated pulse chase analysis of BMP4 cleavage in cells cultured at 19°C, which inhibits trafficking out of the TGN (Griffiths et al., 1985). The 19°C temperature block caused an increase in the accumulation of precursor proteins in cell lysates and efficiently inhibited their secretion, but did not lead to accumulation of cleaved prodomain or mature ligand within cells (Fig. 5C). Collectively, these data are most consistent with a model in which proBMP4 traffics through the TGN but is not cleaved until it reaches the cell surface.
To ask whether the BMP4 precursor protein is cleaved by PCs that are active in intracellular compartments or by PCs that are active on the cell surface or in the extracellular space, we examined processing of proBMP4 in HEK cells in the presence of the cell-permeable pan-PC inhibitor decanoyl-RVKR-chloromethyl ketone (RVKR-cmk) (Hallenberger et al., 1992), or the non-cell permeable peptide inhibitor hexapeptide D-Arg (D6R) (Cameron et al., 2000). RVKR-cmk completely abolished cleavage of proBMP4, leading to secretion of intact precursor protein into the cell medium, concurrent with the loss of cleaved prodomain and mature ligand fragments (Fig. 5D). By contrast, D6R did not inhibit cleavage of proBMP4 and cleavage products were found in the medium (Fig. 5D). As a control, we showed that D6R effectively inhibited cleavage of Xenopus nodal-related protein 2 (Xnr2) (Fig. 5E), consistent with previous reports that nodals are cleaved extracellularly (Beck et al., 2002). Collectively, these results suggest that proBMP4 and proBMP4S2K are cleaved in a post-TGN intracellular compartment, coincident with secretion from the cell.
Defects caused by simultaneous cleavage of the S1 and S2 site are not rescued in Bmp4S2KHAMyc/S2G compound mutants
ProBMP4S2K generates the same end cleavage products as does the wild-type precursor, and yet steady state levels of the mature ligand are reduced. These findings suggest that formation of the S1-only cleaved prodomain/mature ligand complex that arises during sequential, but not simultaneous, cleavage (illustrated in Fig. 1A) is essential for stabilization, and thus for full in vivo activity of mature BMP4. As an initial test of whether failure of the prodomain to associate with the mature domain contributes to the loss of function in Bmp4S2KHAMyc mutants, we intercrossed Bmp4S2G/+ mice, which carry a point mutation that prevents cleavage at the S2 site, with Bmp4S2KHAMyc/+ mice to generate compound heterozygotes for S2K and S2G alleles. When proBMP4S2G is cleaved at the S1 site alone, this generates a stable prodomain/mature domain complex that is targeted for lysosomal degradation when overexpressed (Degnin et al., 2004). This leads to slightly lower levels of mature BMP4 protein when expressed from the endogenous locus, albeit sufficient to support development, as Bmp4S2G/S2G mice are adult viable (Goldman et al., 2006). If defects in Bmp4S2KHAmyc mutants are caused by loss of association of the prodomain with the mature domain, then these defects might be rescued in Bmp4S2KHAMyc/S2G compound mutants via the formation of BMP4S2G/S2K heterodimers, in which one prodomain piece remains stably associated with the mature domains (illustrated in Fig. 6A). Alternatively, because the stably associated prodomain/mature domain complex is targeted for more rapid degradation, Bmp4S2KHAMyc/S2G compound heterozygotes might show a greater loss of function than that seen in heterozygotes for either allele. Bmp4S2KHAMyc/S2G mice were present at only 50% of the predicted frequency at weaning (Table 4), demonstrating that the S2G allele enhances the overall loss of BMP4 function associated with the S2K mutation, although less so than the S2K or null allele. Consistent with this interpretation, Bmp4S2G/S2KHAMyc compound heterozygotes showed a more severe loss of PGCs than did embryos heterozyogous for either the S2K or the S2G allele alone (Fig. 6B).
Fig. 6.
Loss of PGCs and defects in skeletal patterning in Bmp4S2KHAmyc/S2G compound mutants. (A) Schematic illustration of heterodimeric BMP4 precursor protein and cleavage products predicted to form in Bmp4S2KHAmyc/S2G compound mutants. (B) PGC number versus somite pairs at E9 demonstrates a more severe reduction in PGC number in Bmp4S2KHAmyc/S2G compound mutants than in either single mutant. (C) Dorsal view of representative skeletal preparations showing normal (left panel), or examples of mild (center panel) or severe (right panel) defects in vertebral fusion (white arrows).
Table 4.
Progeny from Bmp4S2KHAMyc/+ and Bmp4S2G/+ intercrosses
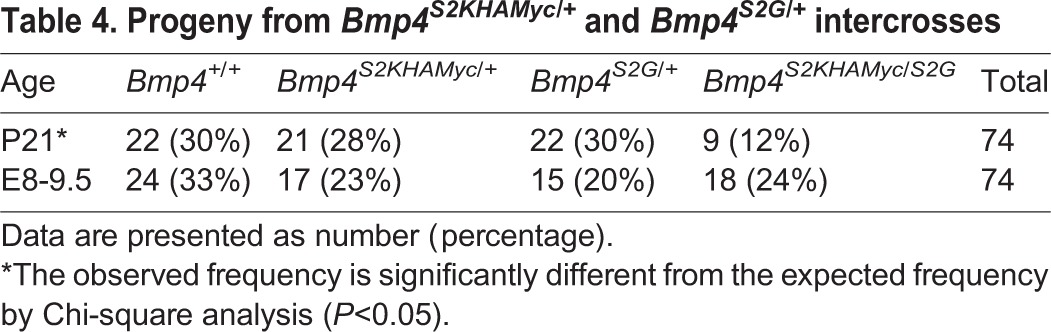
Table 5.
Defects in dorsal vertebral fusion in Bmp4S2KHAMyc/+ and Bmp4S2KHAMyc/S2G/+ mice
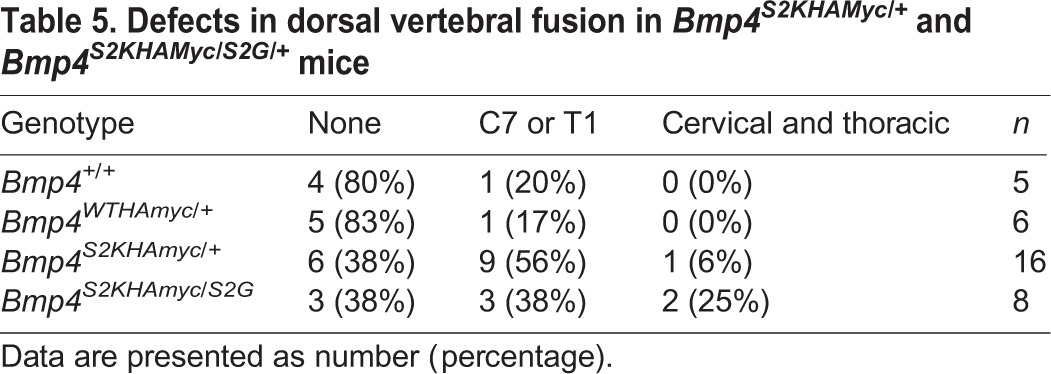
We also examined adult mice for skeletal defects. Mice heterozygous for a null allele of Bmp4 show defects in dorsal fusion of one or more cervical and/or thoracic vertebrae whereas Bmp4S2G/S2G mice show no skeletal defects (Goldman et al., 2006). Defects in dorsal vertebral fusion (Fig. 6C) were observed with similar frequency in Bmp4S2KHAMyc heterozygotes and in Bmp4S2G/S2KHAMyc compound heterozygotes (Table 5). Thus, the S2G allele cannot rescue skeletal defects caused by simultaneous cleavage of proBMP4S2K, and it may increase their severity, although analysis of additional mice would be required to verify this. These data suggest that stable association of the prodomain with the mature domain (Bmp4S2G) cannot substitute for the transient interaction between these two fragments that normally occurs during sequential cleavage of the wild-type precursor. Instead, the phenotype of Bmp4S2G/S2KHAMyc compound heterozygotes is intermediate between that of Bmp4S2KHAMyc/+ and Bmp4S2KHAMyc/lacZ mice, consistent with predicted phenotypes in an allelic series.
DISCUSSION
Previous work in our lab has demonstrated that proBMP4 is sequentially cleaved at two sites within the prodomain, and that cleavage of the upstream (S2) site is essential for normal development in vivo (Goldman et al., 2006; Sopory et al., 2010). The current study extends this work by showing that the presence of a minimal, rather than an optimal, consensus motif at the S2 site is a more crucial determinant of BMP activity than is the ability to cleave this site per se. Specifically, whereas our previous studies have shown that mice carrying a mutation that prevents cleavage of the S2 site (Bmp4S2G/S2G mice) are adult viable (Goldman et al., 2006), in this study we show that mice carrying a mutation that accelerates cleavage of this site die early in embryogenesis due to reduced ligand levels. These results provide the first in vivo evidence that optimal and minimal furin motifs are differentially recognized and functionally important in an endogenous substrate.
Why does simultaneous, rather than sequential, cleavage of proBMP4 lead to a loss of function, despite the fact that both cleavage patterns generate the same end products? The most probable possibility is that the transient formation of a prodomain/mature ligand complex, which occurs during sequential but not simultaneous cleavage, is essential for stabilizing the ligand after its release. One possible function of this complex is to target the mature ligand to the extracellular matrix (ECM) to regulate ligand accessibility, stability and signaling. By analogy, the structurally related ligand TGF-β remains non-covalently associated with its prodomain following cleavage and is targeted to the ECM via covalent interactions between the prodomain and the ECM proteins latent TGF-β binding protein (LTBP) and fibrillin (Ramirez and Rifkin, 2009). The ligand/prodomain/LTBP complex forms intracellularly (Todorovic and Rifkin, 2012) and then binds to fibrillin following secretion. Mature TGF-β remains inactive until it is released from the fibrillin complex by proteolysis or mechanical stress (Robertson and Rifkin, 2013). Fibrillin also binds directly to the prodomain of multiple BMPs (Sengle et al., 2008) and might regulate ECM association of the mature ligand. Genetic and cell biology studies suggest that fibrillins function in a context- and tissue-dependent fashion to either facilitate signaling (Arteaga-Solis et al., 2001; Nistala et al., 2010a) or to sequester BMPs in the matrix in an inactive form (Nistala et al., 2010b).
Our findings lead us to propose the following testable model for how sequential cleavage of BMP4 maximizes ligand bioavailability in the context of a cellular environment (illustrated in Fig. 7): first, our data suggest that the S1 and the S2 sites are cleaved in a membrane-proximal intracellular compartment, closely coupled to the process of secretion. We suggest that prodomain binding to fibrillin and/or to other ECM components occurs intracellularly, prior to cleavage at the S1 site, thereby generating a fibrillin-bound prodomain/mature ligand complex (Fig. 7A). This transient complex serves to deposit mature BMP4 in the ECM coincident with S2 cleavage, thereby facilitating association with heparin sulfate proteoglycans (HSPGs), collagens or other binding proteins crucial for ligand stability and/or signaling. In tissues where the S2 site is not normally cleaved, we propose that the ECM-bound prodomain/mature domain complex is able to signal locally but is then targeted for re-uptake and lysosomal degradation (Fig. 7B). This model is consistent with studies showing that HSPGs can promote endocytosis and lysosomal degradation of BMPs (Jiao et al., 2007) and with our previous studies showing that deletion of the HSPG-binding domain on mature BMP4 partially rescues lysosomal degradation of BMP4S2G (Degnin et al., 2004). Finally, this model offers an explanation for the severe loss of function observed in Bmp4S2KHAmyc mutant mice. Specifically, we propose that introduction of an optimal motif at the S2 site prevents formation of the transient fibrillin/prodomain/mature domain complex such that mature BMP4 is released from the fibrillin-bound prodomain prematurely. Thus, efficient association with HSPGs or other ECM proteins that are required to stabilize the free ligand would be precluded (Fig. 7C). This would account for the specific reduction in levels of cleaved ligand but not cleaved prodomain in Bmp4S2KHAmyc mutant mice, as the prodomain remains anchored in the ECM regardless of the order of cleavages.
Fig. 7.
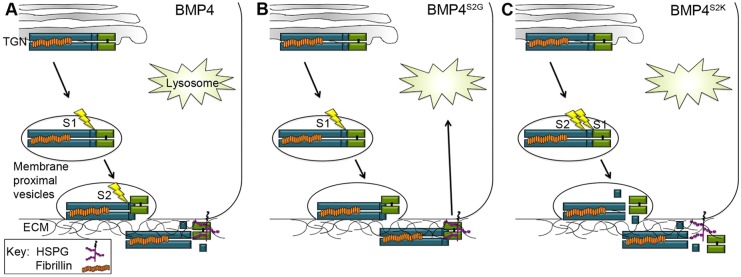
Hypothetical model for regulation of BMP4 ligand activity by cleavages in the prodomain. (A) Fibrillin-bound proBMP4 is sequentially cleaved at the S1 and S2 sites (lightning bolts) in a membrane-proximal vesicle, and cleavage is closely coupled to the process of secretion. The transient fibrillin/prodomain/mature ligand complex that forms after S1 cleavage ensures that mature BMP4 is deposited in the ECM coincident with S2 cleavage, thereby facilitating association of the ligand with HSPGs or other ECM proteins required for ligand stability. (B) In tissues where the S2 site is not cleaved, the ECM-bound prodomain/mature domain complex is able to signal locally but is then targeted for re-uptake and lysosomal degradation. (C) Simultaneous cleavage of the S1 and S2 sites leads to premature release of mature BMP4 from the fibrillin/prodomain complex, precluding efficient association with HSPGs or other binding partners that are required for ligand stability.
Previous studies have identified multiple roles for ECM-associated binding partners in promoting BMP signaling. For example, members of the glypican family of HSPGS have been shown to stabilize BMPs by preventing lysosomal targeting (Akiyama et al., 2008), to facilitate movement of BMP4 across sheets of cells (Belenkaya et al., 2004) and/or to function as obligatory co-receptors (Kuo et al., 2010). Other ECM components or associated proteins, such as fibronectin, collagen and members of the crossveinless family, promote BMP signaling through diverse and context-dependent mechanisms (Harris and Ashe, 2011; Ikeya et al., 2006; Kelley et al., 2009; Kenny et al., 2012; Serpe et al., 2008). Some BMP-binding proteins function as either pro- or anti-BMP factors, contingent on the relative concentrations of mature BMP4 and its binding partner that are present in a given tissue (Kelley et al., 2009). This provides a possible explanation for the ability of the S2K mutation to enhance BMP activity in overexpression studies (Cui et al., 2001), but to inhibit it under more physiological conditions. An alternative explanation is that ECM-binding components are saturated when their substrates are overexpressed, thus bypassing this regulatory step.
Our data showing that proBMP4 transits through the TGN intact raises the question of how it avoids cleavage in this subcellular compartment. The endogenous BMP4 convertases furin and PC5/6 are first active in the TGN, although they also function in more distal vesicles, the cell surface and the extracellular space (Seidah et al., 2008). Precedents exist for post-TGN processing of other TGF-β family members. For example, the Nodal precursor is secreted intact and then cleaved in the extracellular space. However, nodal traffics to the cell surface via a TGN-independent route, thereby avoiding intracellular PCs (Blanchet et al., 2008), whereas our data show that BMP4 traffics through the TGN. Interestingly, LTBP2 and 3 bind to the zymogen form of proPC5/6, allowing it to be sequestered as an inactive protein in the ECM and leading to impaired processing of growth and differentiation factor 11 (Sun et al., 2011). Thus, ECM components can affect both the proteolytic maturation and the signaling activity of TGF-β family members.
Primary and upstream furin cleavage motifs are conserved in vertebrate BMP2 and BMP4 precursor proteins and in the Drosophila ortholog DPP, but there are clear differences in how these sites are used. Whereas all vertebrate BMP2/4 precursors contain an optimal furin motif at the S1 site and a minimal consensus motif at the S2 site, the order is reversed in DPP, thus leading to the suggestion that in Drosophila the upstream prodomain (S2) site is cleaved initially, followed by cleavage of the site adjacent to the mature domain (S1) (Kunnapuu et al., 2009). Furthermore, cleavage of the S2 rather than the S1 site of DPP appears to be the more crucial determinant of ligand activity in Drosophila, unlike in vertebrates. Specifically, whereas mice homozygous for a knock-in mutation (Bmp4S2G) that prevents cleavage at the S2 site have substantial residual BMP4 activity in most tissues (Goldman et al., 2006), a comparable mutation in Dpp completely ablates DPP activity in the fly wing disc (Kunnapuu et al., 2009; Sopory et al., 2010). Conversely, whereas mutations that prevent cleavage of the S1 site, but allow for cleavage at the S2 site alone lead to a nearly complete loss of vertebrate BMP4 activity (Sopory et al., 2006), cleavage at the S2 site alone is sufficient for most DPP activity in the fly wing disc (Kunnapuu et al., 2009). Further analysis and comparison of cleavage site usage across evolution may shed light on mechanisms by which proteolytic maturation regulates the activity of these crucial signaling molecules.
MATERIALS AND METHODS
Generation and genotyping of mice
Animal procedures followed protocols approved by the Oregon Health & Sciences University and the University of Utah Institutional Animal Care and Use Committees. Bmp4S2KHAMyc and Bmp4HAMyc mice were generated using standard gene targeting procedures that are described in detail in the supplementary Materials and Methods. Bmp4lacZ/+ and BRE-lacZ mice were obtained from Dr B. Hogan (Duke University, USA) and Dr C. Mummery (Leiden University, The Netherlands), respectively.
Xenopus embryo assays
RNA (5 pg) was injected near the dorsal midline of four-cell Xenopus embryos. The dorsal quadrant was dissected from ten embryos in each group at stage 10 and proteins extracted as described (Moon and Christian, 1989), with the addition of complete Mini protease inhibitors (Roche), 1 mM NaF and 1 mM Na3VO4. Two embryo equivalents per sample were resolved by SDS-PAGE, followed by western blotting with antibodies specific for phospho- and total Smad1/5/8 (Cell Signaling, 9511 and 9512, respectively).
Transient transfection and pulse chase analysis
HEK293 cells were transfected with 100 ng of DNA encoding BMP4HAMyc or BMP4S2KHAMyc together with 1.9 µg of DNA encoding GFP using Lipofectamine 2000 (Invitrogen). Cells were cultured for three days, rinsed twice with 1× PBS, then pulse labeled with 100 µCi/ml [35S]Met/Cys in DMEM without Met/Cys for 1 h. Cells were again rinsed twice with 1× PBS, then incubated in chase medium (OPTIMEM) for various time points. For temperature block experiments, cells were incubated at either 37°C or 19°C throughout the chase. Medium was removed and cells were acid washed three times (2 min each in 0.1 M glycine, pH 2.5, 150 mM NaCl). Cells were lysed with RIPA (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 50 mM Tris, pH 8.0) and radiolabeled precursor and prodomain proteins were immunoprecipitated by incubation with anti-HA antibody (12CA5, 16 μg/ml) and protein A-Sepharose 4B beads as described (Degnin et al., 2004). Radiolabeled mature proteins were then immunoprecipitated from HA-depleted medium and lysates by incubation with anti-Myc antibody (9E10, 16 μg/ml) and protein G-Sepharose beads. Where indicated, proteins were deglycosylated using PNGase F or Endo H (New England BioLabs). Immunoprecipitated proteins were analyzed by 8-10% SDS-PAGE and imaged by autoradiography. Radiolabeled bands were visualized with a GE Healthcare Phosphorimager.
Inhibitor studies
HEK293 cells were transiently transfected with 200 ng of DNA encoding BMP4HAMyc or Xnr2HA together with 1.8 µg of DNA encoding pCS2+ as described above. After 24 h, the culture medium was replaced with serum-free medium containing vehicle alone (DMSO), or vehicle plus either 25 μM RVKR-cmk (Calbiochem) or 10 μM D6R (Calbiochem), and cells were cultured for an additional 20 h in the presence of inhibitors. Cells were lysed in 150 mM NaCl, 20 mM Tris-Cl pH 7.5, 1 mM EDTA, 1% sodium deoxycholate, 1% NP40, and culture medium was TCA-precipitated. Proteins were separated on a 4-15% gradient gel and transferred onto a PVDF membrane. Membranes were probed with anti-HA (Roche, 3F10; 1:1000), anti-BMP4 (Santa Cruz, sc-12721; 1:1000) or anti-beta-actin antibodies follow by HRP-conjugated anti-mouse, rat or rabbit IgG (Jackson ImmunoResearch, 115-035-03, 112-035-03, 111-035-003, respectively; 1:10,000). Immunoreactive proteins were detected using ECL Plus Western Blotting Detection Reagents (GE Healthcare).
Western blot analysis of mouse tissue and endothelial cells
Whole lungs were homogenized in 1 ml 150 mM NaCl, 1% NP-40, 50 mM Tris pH 8.0+1× complete Mini protease inhibitors (Roche), incubated on ice for 1 h and spun down for 15 min. Lysates were pre-cleared with 100 µl protein G-Sepharose beads on a rocker for 1 h. Lung endothelial cells were prepared as described (Sobczak et al., 2010). Per whole lung sample, 450 µg of protein, 5-30 µg of endothelial cell lysates or 40 µl of endothelial cell medium were resolved by SDS-PAGE and transferred onto a PVDF membrane. Membranes were probed with anti-HA, anti-BMP4 or anti-actin antibodies and immunoreactive proteins were detected as described above.
Detection and counting of PGCs
Primordial germ cells were visualized and counted according to Lawson et al. (1999).
Skeletal preparations
Skeletal staining was performed as described (Hogan et al., 1994).
Immunostaining and β-galactosidase staining
β-galactosidase staining of Bmp4lacZ/+ and BRE-lacZ embryos was performed as described (Lawson et al., 1999), using Red-Gal (Research Organics) or X-Gal (Promega) as a substrate.
Supplementary Material
Acknowledgements
We thank A. Pratt and R. Essalmani for suggesting the PC inhibitor studies and S. Mansour, G. Schoenwolf and members of the Christian laboratory for helpful comments on the manuscript.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
S.M.N. generated targeting constructs, helped analyze mouse phenotypes, conducted pulse chase studies and performed western analysis of HEK cells, mouse lung and Xenopus embryo explants; H.-S.K. analyzed BMP expression in lung endothelial cultures, conducted inhibitor studies and helped with phenotype analysis and manuscript preparation; A.T. analyzed PGC number and BRE-lacZ and BMP4null/S2K mice; N.D. helped generate and maintain mouse lines; A.M. conducted skeletal analysis; H.L. genotyped and maintained mouse lines; J.L.C. conceived the study, analyzed phenotypes, helped interpret data and prepared the manuscript.
Funding
This work was supported by grants from the National Institutes of Health [RO1 HD042598 and RO1HD37976] and the Shriners Hospital for Crippled Children Research Foundation to J.L.C., and by a predoctoral fellowship from the American Heart Association (AHA) to S.M.N. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.110130/-/DC1
References
- Akiyama T., Kamimura K., Firkus C., Takeo S., Shimmi O., Nakato H. (2008). Dally regulates Dpp morphogen gradient formation by stabilizing Dpp on the cell surface. Dev. Biol. 313, 408-419 10.1016/j.ydbio.2007.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga-Solis E., Gayraud B., Lee S. Y., Shum L., Sakai L., Ramirez F. (2001). Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 154, 275-282 10.1083/jcb.200105046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P., Efthymiou M., Klein J. C., Salt A., Bunyan D. J., Wyatt A., Ponting C. P., Martin A., Williams S., Lindley V., et al. (2008). Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet. 82, 304-319 10.1016/j.ajhg.2007.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S., Le Good J. A., Guzman M., Ben Haim N., Roy K., Beermann F., Constam D. B. (2002). Extraembryonic proteases regulate Nodal signalling during gastrulation. Nat. Cell Biol. 4, 981-985 10.1038/ncb890 [DOI] [PubMed] [Google Scholar]
- Belenkaya T. Y., Han C., Yan D., Opoka R. J., Khodoun M., Liu H., Lin X. (2004). Drosophila Dpp morphogen movement is independent of dynamin-mediated endocytosis but regulated by the glypican members of heparan sulfate proteoglycans. Cell 119, 231-244 10.1016/j.cell.2004.09.031 [DOI] [PubMed] [Google Scholar]
- Blanchet M.-H., Le Good J. A., Mesnard D., Oorschot V., Baflast S., Minchiotti G., Klumperman J., Constam D. B. (2008). Cripto recruits Furin and PACE4 and controls Nodal trafficking during proteolytic maturation. EMBO J. 27, 2580-2591 10.1038/emboj.2008.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragdon B., Moseychuk O., Saldanha S., King D., Julian J., Nohe A. (2011). Bone morphogenetic proteins: a critical review. Cell. Signal. 23, 609-620 10.1016/j.cellsig.2010.10.003 [DOI] [PubMed] [Google Scholar]
- Cameron A., Appel J., Houghten R. A., Lindberg I. (2000). Polyarginines are potent furin inhibitors. J. Biol. Chem. 275, 36741-36749 10.1074/jbc.M003848200 [DOI] [PubMed] [Google Scholar]
- Constam D. B., Robertson E. J. (1999). Regulation of bone morphogenetic protein activity by pro domains and proprotein convertases. J. Cell Biol. 144, 139-149 10.1083/jcb.144.1.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Jean F., Thomas G., Christian J. L. (1998). BMP-4 is proteolytically activated by furin and/or PC6 during vertebrate embryonic development. EMBO J. 17, 4735-4743 10.1093/emboj/17.16.4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Hackenmiller R., Berg L., Jean F., Nakayama T., Thomas G., Christian J. L. (2001). The activity and signaling range of mature BMP-4 is regulated by sequential cleavage at two sites within the prodomain of the precursor. Genes Dev. 15, 2797-2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesh S. M., Villasenor A., Chong D., Soukup C., Cleaver O. (2009). BMP and BMP receptor expression during murine organogenesis. Gene Expr. Patterns 9, 255-265 10.1016/j.gep.2009.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnin C., Jean F., Thomas G., Christian J. L. (2004). Cleavages within the prodomain direct intracellular trafficking and degradation of mature bone morphogenetic protein-4. Mol. Biol. Cell 15, 5012-5020 10.1091/mbc.E04-08-0673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn N. R., Winnier G. E., Hargett L. K., Schrick J. J., Fogo A. B., Hogan B. L. M. (1997). Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev. Biol. 188, 235-247 10.1006/dbio.1997.8664 [DOI] [PubMed] [Google Scholar]
- Frank D. B., Abtahi A., Yamaguchi D. J., Manning S., Shyr Y., Pozzi A., Baldwin H. S., Johnson J. E., de Caestecker M. P. (2005). Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ. Res. 97, 496-504 10.1161/01.RES.0000181152.65534.07 [DOI] [PubMed] [Google Scholar]
- Goldman D. C., Hackenmiller R., Nakayama T., Sopory S., Wong C., Kulessa H., Christian J. L. (2006). Mutation of an upstream cleavage site in the BMP4 prodomain leads to tissue-specific loss of activity. Development 133, 1933-1942 10.1242/dev.02368 [DOI] [PubMed] [Google Scholar]
- Griffiths G., Pfeiffer S., Simons K., Matlin K. (1985). Exit of newly synthesized membrane proteins from the trans cisterna of the Golgi complex to the plasma membrane. J. Cell Biol. 101, 949-964 10.1083/jcb.101.3.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallenberger S., Bosch V., Angliker H., Shaw E., Klenk H.-D., Garten W. (1992). Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360, 358-361 10.1038/360358a0 [DOI] [PubMed] [Google Scholar]
- Harris R. E., Ashe H. L. (2011). Cease and desist: modulating short-range Dpp signalling in the stem-cell niche. EMBO Rep. 12, 519-526 10.1038/embor.2011.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B. M., Beddington R. S. P., Constantine F., Lacy E. (1994). Manipulating the Mouse Embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Ikeya M., Kawada M., Kiyonari H., Sasai N., Nakao K., Furuta Y., Sasai Y. (2006). Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development 133, 4463-4473 10.1242/dev.02647 [DOI] [PubMed] [Google Scholar]
- Jiao X., Billings P. C., O'Connell M. P., Kaplan F. S., Shore E. M., Glaser D. L. (2007). Heparan sulfate proteoglycans (HSPGs) modulate BMP2 osteogenic bioactivity in C2C12 cells. J. Biol. Chem. 282, 1080-1086 10.1074/jbc.M513414200 [DOI] [PubMed] [Google Scholar]
- Kelley R., Ren R., Pi X., Wu Y., Moreno I., Willis M., Moser M., Ross M., Podkowa M., Attisano L., et al. (2009). A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 184, 597-609 10.1083/jcb.200808064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny A. P., Rankin S. A., Allbee A. W., Prewitt A. R., Zhang Z., Tabangin M. E., Shifley E. T., Louza M. P., Zorn A. M. (2012). Sizzled-tolloid interactions maintain foregut progenitors by regulating fibronectin-dependent BMP signaling. Dev. Cell 23, 292-304 10.1016/j.devcel.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W., Essalmani R., Szumska D., Creemers J. W., Roebroek A. J. M., D'Orleans-Juste P., Bhattacharya S., Seidah N. G., Prat A. (2012). Loss of endothelial furin leads to cardiac malformation and early postnatal death. Mol. Cell. Biol. 32, 3382-3391 10.1128/MCB.06331-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunnapuu J., Bjorkgren I., Shimmi O. (2009). The Drosophila DPP signal is produced by cleavage of its proprotein at evolutionary diversified furin-recognition sites. Proc. Natl. Acad. Sci. USA 106, 8501-8506 10.1073/pnas.0809885106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W.-J., Digman M. A., Lander A. D. (2010). Heparan sulfate acts as a bone morphogenetic protein coreceptor by facilitating ligand-induced receptor hetero-oligomerization. Mol. Biol. Cell 21, 4028-4041 10.1091/mbc.E10-04-0348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson K. A., Dunn N. R., Roelen B. A. J., Zeinstra L. M., Davis A. M., Wright C. V. E., Korving J. P. W. F. M., Hogan B. L. M. (1999). Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 13, 424-436 10.1101/gad.13.4.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy S. S., Bresnahan P. A., Leppla S. H., Klimpel K. R., Thomas G. (1992). Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267, 16396-16402. [PubMed] [Google Scholar]
- Monteiro R. M., de Sousa Lopes S. M. C., Korchynskyi O., ten Dijke P., Mummery C. L. (2004). Spatio-temporal activation of Smad1 and Smad5 in vivo: monitoring transcriptional activity of Smad proteins. J. Cell Sci. 117, 4653-4663 10.1242/jcs.01337 [DOI] [PubMed] [Google Scholar]
- Moon R. T., Christian J. L. (1989). Microinjection and expression of synthetic mRNAs in Xenopus embryos. Technique 1, 76-89. [Google Scholar]
- Nakayama K. (1997). Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 327, 625-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelsen S. M., Christian J. L. (2009). Site-specific cleavage of BMP4 by furin, PC6 and PC7. J. Biol. Chem. 284, 27157-27166 10.1074/jbc.M109.028506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistala H., Lee-Arteaga S., Siciliano G., Smaldone S., Ramirez F. (2010a). Extracellular regulation of transforming growth factor beta and bone morphogenetic protein signaling in bone. Ann. N. Y. Acad. Sci. 1192, 253-256 10.1111/j.1749-6632.2009.05350.x [DOI] [PubMed] [Google Scholar]
- Nistala H., Lee-Arteaga S., Smaldone S., Siciliano G., Carta L., Ono R. N., Sengle G., Arteaga-Solis E., Levasseur R., Ducy P., et al. (2010b). Fibrillin-1 and -2 differentially modulate endogenous TGF-beta and BMP bioavailability during bone formation. J. Cell Biol. 190, 1107-1121 10.1083/jcb.201003089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F., Rifkin D. B. (2009). Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr. Opin. Cell Biol. 21, 616-622 10.1016/j.ceb.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis L. M., Tyler R. C., Schilter K. F., Abdul-Rahman O., Innis J. W., Kozel B. A., Schneider A. S., Bardakjian T. M., Lose E. J., Martin D. M., et al. (2011). BMP4 loss-of-function mutations in developmental eye disorders including SHORT syndrome. Hum. Genet. 130, 495-504 10.1007/s00439-011-0968-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson I. B., Rifkin D. B. (2013). Unchaining the beast; insights from structural and evolutionary studies on TGFbeta secretion, sequestration, and activation. Cytokine Growth Factor Rev. 24, 355-372 10.1016/j.cytogfr.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008). The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 40, 1111-1125 10.1016/j.biocel.2008.01.030 [DOI] [PubMed] [Google Scholar]
- Sengle G., Charbonneau N. L., Ono R. N., Sasaki T., Alvarez J., Keene D. R., Bachinger H. P., Sakai L. Y. (2008). Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J. Biol. Chem. 283, 13874-13888 10.1074/jbc.M707820200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe M., Umulis D., Ralston A., Chen J., Olson D. J., Avanesov A., Othmer H., O'Connor M. B., Blair S. S. (2008). The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev. Cell 14, 940-953 10.1016/j.devcel.2008.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore E. M., Xu M., Feldman G. J., Fenstermacher D. A., Brown M. A., Kaplan F. S. (2006). A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 38, 525-527 10.1038/ng1783 [DOI] [PubMed] [Google Scholar]
- Sobczak M., Dargatz J., Chrzanowska-Wodnicka M. (2010). Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 14, 2316 10.3791/2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopory S., Nelsen S. M., Degnin C., Wong C., Christian J. L. (2006). Regulation of bone morphogenetic protein-4 activity by sequence elements within the prodomain. J. Biol. Chem. 281, 34021-34031 10.1074/jbc.M605330200 [DOI] [PubMed] [Google Scholar]
- Sopory S., Kwon S., Wehrli M., Christian J. L. (2010). Regulation of Dpp activity by tissue-specific cleavage of an upstream site within the prodomain. Dev. Biol. 346, 102-112 10.1016/j.ydbio.2010.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Essalmani R., Susan-Resiga D., Prat A., Seidah N. G. (2011). Latent transforming growth factor beta-binding proteins-2 and -3 inhibit the proprotein convertase 5/6A. J. Biol. Chem. 286, 29063-29073 10.1074/jbc.M111.242479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S., Marazita M. L., Cooper M. E., Miwa N., Hing A., Jugessur A., Natsume N., Shimozato K., Ohbayashi N., Suzuki Y., et al. (2009). Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am. J. Hum. Genet. 84, 406-411 10.1016/j.ajhg.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabatabaeifar M., Schlingmann K.-P., Litwin M., Emre S., Bakkaloglu A., Mehls O., Antignac C., Schaefer F., Weber S. (2009). Functional analysis of BMP4 mutations identified in pediatric CAKUT patients. Pediatr. Nephrol. 24, 2361-2368 10.1007/s00467-009-1287-6 [DOI] [PubMed] [Google Scholar]
- Todorovic V., Rifkin D. B. (2012). LTBPs, more than just an escort service. J. Cell. Biochem. 113, 410-418 10.1002/jcb.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. W., Godson C., Brazil D. P., Martin F. (2010). Extracellular BMP-antagonist regulation in development and disease: tied up in knots. Trends Cell Biol. 20, 244-256 10.1016/j.tcb.2010.01.008 [DOI] [PubMed] [Google Scholar]
- Weber S., Taylor J. C., Winyard P., Baker K. F., Sullivan-Brown J., Schild R., Knuppel T., Zurowska A. M., Caldas-Alfonso A., Litwin M., et al. (2008). SIX2 and BMP4 mutations associate with anomalous kidney development. J. Am. Soc. Nephrol. 19, 891-903 10.1681/ASN.2006111282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnier G., Blessing M., Labosky P. A., Hogan B. L. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 9, 2105-2116 10.1101/gad.9.17.2105 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.