

Abstract
The pharyngeal pouches are a segmental series of epithelial structures that organize the embryonic vertebrate face. In mice and zebrafish that carry mutations in homologs of the DiGeorge syndrome gene TBX1, a lack of pouches correlates with severe craniofacial defects, yet how Tbx1 controls pouch development remains unclear. Using mutant and transgenic rescue experiments in zebrafish, we show that Tbx1 functions in the mesoderm to promote the morphogenesis of pouch-forming endoderm through wnt11r and fgf8a expression. Consistently, compound losses of wnt11r and fgf8a phenocopy tbx1 mutant pouch defects, and mesoderm-specific restoration of Wnt11r and Fgf8a rescues tbx1 mutant pouches. Time-lapse imaging further reveals that Fgf8a acts as a Wnt11r-dependent guidance cue for migrating pouch cells. We therefore propose a two-step model in which Tbx1 coordinates the Wnt-dependent epithelial destabilization of pouch-forming cells with their collective migration towards Fgf8a-expressing mesodermal guideposts.
Keywords: Epithelial morphogenesis, Fgf8, Pharyngeal pouches, Tbx1, Wnt11r, Zebrafish
INTRODUCTION
From the embryonic endoderm emerge a number of epithelial outpocketings that generate the initial anlagen of diverse organs. In the head, these include a series of pharyngeal pouches that guide the development of neural-crest-derived cells into bone and cartilage and fuse with ectodermal clefts to generate the gills, thymus, parathyroid and other structures (Crump et al., 2004a; Piotrowski et al., 2003; Schwend and Ahlgren, 2009). Although clearly crucial for vertebrate head development, how pouches achieve their stereotyped positions and morphologies remains poorly understood.
Given the importance of pouches in craniofacial development, it is not surprising that mutations in genes that regulate their morphogenesis are implicated in human birth defects. DiGeorge syndrome, commonly associated with heterozygous deletion of a region of chromosome 22 containing the TBX1 gene, is characterized by defects in the facial skeleton, thymus and heart (Lindsay et al., 2001). It is thought that DiGeorge defects can be attributed, at least in part, to earlier malformations of pouches (Epstein, 2001). In support of this, the zebrafish van gogh mutant, which is mutated for tbx1, has severe defects in pouch formation and facial skeletal development (Piotrowski and Nusslein-Volhard, 2000; Piotrowski et al., 2003). Similarly, Tbx1 mutant mice lack pouches and have many of the craniofacial and organ defects of DiGeorge individuals (Vitelli et al., 2002a). Tbx1 is expressed in both the pre-pouch endoderm and surrounding mesoderm; however, the tissue requirements for Tbx1 in pouch development, as well as its crucial downstream targets, remain controversial. In one study, it has been shown that conditional deletion of Tbx1 in the Mesp1-positive mesoderm of mice results in pouch defects, and re-introduction of Tbx1 into Mesp1-positive mesoderm rescues pouches (Zhang et al., 2006). However, another study concludes that deletion of Tbx1 in the Foxg1-positive endoderm likewise results in defective pouch outgrowth (Arnold et al., 2006). In zebrafish, transplantation of wild-type endoderm has been reported to rescue some of the facial cartilage defects of tbx1 mutants, yet the observed rescue is mild and restoration of pouch morphology was not examined (Piotrowski et al., 2003). By using a transgenic strategy to reintroduce Tbx1 and its putative effectors into the mesoderm and/or endoderm of zebrafish, we show here that Tbx1 has crucial functions in the mesoderm for pouch morphogenesis yet is dispensable for the earlier segmentation of the pouch-forming endoderm.
We have previously identified a role for the nkx2.5-expressing subpopulation of mesoderm in guiding pouch formation, with genetic ablation of this mesoderm resulting in pouch defects similar to those of tbx1 mutants (Choe et al., 2013). One common target of Tbx1 in the mesoderm of mice and fish might be Fgf8. In mice, mesoderm-specific deletion of Tbx1 results in loss of Fgf8 expression from the anterior heart field mesoderm (Zhang et al., 2006), and heterozygosity of Fgf8 enhances DiGeorge-like defects in Tbx1 heterozygous animals (Vitelli et al., 2002b). In zebrafish, loss of fgf8a, in particular in combination with reduction of Fgf3 levels, results in severe pouch defects that can be partially rescued by wild-type mesoderm transplants; similar pouch defects are seen upon global inhibition of Fgf signaling with the drug SU5402 (Crump et al., 2004a). However, insertion of Fgf8 into the Tbx1 locus fails to rescue Tbx1 mutant defects (Vitelli et al., 2006). Thus, loss of Fgf8 alone cannot account for the severe pouch defects seen in Tbx1 mutants. Indeed, a number of other potential targets of Tbx1, such as Pitx2 (Nowotschin et al., 2006), Crk1 (Guris et al., 2001), retinoic acid (Roberts et al., 2006; Zhang et al., 2006), Vegf signaling (Stalmans et al., 2003) and Tgfβ signaling (Wurdak et al., 2005) have been identified, yet how they might interact with Tbx1 to control craniofacial morphogenesis remains poorly understood.
Although canonical Wnt signaling (i.e. nuclear β-catenin) has been proposed to negatively regulate Tbx1 expression (Huh and Ornitz, 2010), less is known about potential roles of Wnt signaling downstream of Tbx1. In zebrafish, we have previously reported crucial roles for cytoplasmic (i.e. non-canonical) Wnt signaling in orchestrating distinct steps of pouch morphogenesis (Choe et al., 2013). Pouches develop through a transient multi-layering of endodermal epithelial cells, followed by their lateral collective migration and reorganization into mature bilayers. Mesoderm-derived Wnt11r promotes the loss of columnar morphology that accompanies the multi-layering process, whereas ectodermal Wnt4a promotes the reacquisition of columnar morphology during bilayer maturation. Here, we show that Tbx1 is required for the expression of wnt11r in the developing mesoderm, with Wnt11r acting together with Fgf8a to coordinate pouch morphogenesis. In vivo time-lapse imaging further reveals how Wnt11r-dependent destabilization of the pre-pouch endoderm allows epithelial cells to collectively migrate towards sources of Fgf8a in the adjacent mesoderm. Taken together, the data from this study reveal how two targets of Tbx1 in the mesoderm – Wnt11r and Fgf8a – coordinate the morphogenesis of growing pouches.
RESULTS
Tbx1 is required for the morphogenesis but not the initial segmentation of pouch-forming endoderm
Tbx1 is clearly required for pouch formation across vertebrates, but its roles in specifying pouch-forming regions in the pharyngeal endoderm versus promoting their later morphogenesis has remained unclear. In order to address the requirements for Tbx1, we first performed time-lapse imaging of pouch cell behavior in tbx1 mutant zebrafish harboring a her5:mCherryCAAX transgene that labels endodermal cell membranes (Choe et al., 2013). Compared with wild-type siblings that showed outpocketing of a new pouch every 4 h (Fig. 1A-G; supplementary material Movie 1A), her5:mCherryCAAX-positive endodermal cells were present in tbx1 mutants yet failed to display outpocketing behavior within the 10-h timeframe of the recordings (Fig. 1H-K; supplementary material Movie 1B). As previously reported (Piotrowski and Nusslein-Volhard, 2000), tbx1 mutants also retained expression of the immunoglobulin-domain protein Alcama, a marker of maturing pouches, despite the absence of morphological pouches (Fig. 2G). These data support the notion that Tbx1 is required for the morphogenesis but not generation of pouch-forming endoderm.
Fig. 1.

Requirement of Tbx1 for pouch morphogenesis. (A) A zoomed out view of a 32 hpf her5:mCherryCAAX (red) embryo shows the position of pouches relative to the developing eye and ear. (B,C) The first two pouches (p1, p2) form by 20 hpf, with the remaining pouches (p3-p6) developing at one per 4 h. (D-K) Still images from time-lapse confocal recordings of wild-type and tbx1 mutant pouch development at the times indicated (bottom right of the images) (see supplementary material Movie 1A,B). her5:mCherryCAAX labels endodermal cell membranes. Recordings were started at 26 hpf (T0), a stage when the fourth and fifth pouches (p4 and p5) are beginning to develop in the wild-type example. Compared with the typical pouch outpocketing behavior seen in all three wild-type siblings, no endodermal outpocketing was observed in three out of three tbx1 mutants (H-K). (L,M) Fluorescent in situ hybridization for fgf3 (green) and GFP immunohistochemistry to label the her5:GFP-positive pouch endoderm (red). fgf3 was expressed in a segmental pattern in all 39 wild-type siblings and all 14 tbx1 mutants. (N,O) Confocal sections of pre-pouch endoderm in dusp6:dGFP; her5:mCherryCAAX transgenic embryos. In wild type, dusp6:dGFP (green) was expressed segmentally in already formed pouches (arrowhead) and in clusters of presumptive pouch endoderm before outpocketing (arrows); strong expression was also seen in adjacent mesoderm (asterisks) (n=18). In tbx1 mutants, dusp6:dGFP was expressed at lower levels and in fewer cells, yet a segmental pattern in the endoderm was still detected (n=7). Note that we increased the relative gain of the green channel in tbx1 mutants to reveal the weak segmental dusp6:dGFP expression. See supplementary material Fig. S1 for the unaltered images. L′-O′ show the green channel alone. Scale bars: 40 μm (B,C,L,M); 20 μm (D-K,N,O).
Fig. 2.

Tbx1 is required in the nkx2.5-positive mesoderm for pouch development. (A-E) Fluorescent in situ hybridization for tbx1 (green) and GFP immunohistochemistry to detect her5:GFP-positive endoderm (red) at 34 hpf. In all 58 wild-type siblings, tbx1 expression was observed in her5:GFP-positive endoderm (arrows), adjacent mesoderm (arrowheads) and the ear (asterisk). In all 21 tbx1 mutants, all tbx1 expression was lost. tbx1 expression was selectively restored in the her5:GFP-positive endoderm of all 17 nkx2.3:Tbx1; tbx1−/− embryos and in the mesoderm of all nine nkx2.5:Tbx1; tbx1−/− embryos. Inclusion of both transgenes restored tbx1 expression over a broader area (brackets for presumptive endoderm and arrowheads for presumptive mesoderm) in all four nkx2.3:Tbx1; nkx2.5:Tbx1; tbx1−/− embryos, although for technical reasons her5:GFP was not included. (F-J) In wild-type zebrafish at 34 hpf, immunohistochemistry for Alcama (green) labeled five pouches (p1-p5). nkx2.5:Tbx1; tbx1−/− and nkx2.3:Tbx1; nkx2.5:Tbx1; tbx1−/− embryos displayed partial rescue of pouches compared with tbx1 mutants, whereas nkx2.3:Tbx1; tbx1−/− embryos did not. Sensory ganglia are indicated with red asterisks. (K-O) Fluorescent in situ hybridization for dlx2a (green) and GFP immunohistochemistry for her5:GFP (red) at 30 hpf. In wild-type zebrafish, dlx2a was expressed in the neural-crest-derived mesenchyme of each arch (numbered), with higher expression in posterior arches. In all tbx1−/− (n=9), nkx2.3:Tbx1; tbx1−/− (n=6), nkx2.5:Tbx1; tbx1−/− (n=4) and nkx2.3:Tbx1; nkx2.5:Tbx1; tbx1−/− (n=4) embryos, dlx2a expression was reduced in the second arch and lost from the more posterior arches. For technical reasons, her5:GFP was not included in O. (P-T) Ventral whole-mount views of dissected facial cartilages. Wild-type zebrafish (P) invariantly formed five ceratobranchials (CBs) on each side. CBs were missing in all tbx1 mutants (Q, n=41) and not recovered in nkx2.3:Tbx1; tbx1−/− (R, n=23), nkx2.5:Tbx1; tbx1−/− (S, n=26) or nkx2.3:Tbx1; nkx2.5:Tbx1; tbx1−/− (T, n=24) embryos. (U) Quantification of pouch defects as assessed by Alcama staining in wild-type zebrafish (n=342), tbx1−/− (n=63), nkx2.3:Tbx1; tbx1−/− (n=54), nkx2.5:Tbx1; tbx1−/− (n=22) and nkx2.3:Tbx1; nkx2.5:Tbx1; tbx1−/− (n=23). Data represent mean±s.e.m., P values are shown. n.s., not significant. Scale bars: 40 μm (A-O).
We next investigated whether the failure to initiate pouch outpocketing in tbx1 mutants reflects a loss of segmental character in the pre-pouch endoderm, or instead defects in the morphogenesis of pre-specified segmental units. In wild-type zebrafish, an early marker of developing pouches is fgf3 expression (Fig. 1L) (David et al., 2002; Nissen et al., 2003). Remarkably, despite the near complete loss of morphological pouches, we still observed segmental expression of fgf3 in the presumptive pouch-forming endoderm of tbx1 mutants (Fig. 1M). To further explore dynamic Fgf signaling in tbx1 mutants, we also examined a dusp6:dGFP transgenic line in which a destabilized GFP protein is expressed under control of the Fgf target gene dusp6 (Molina et al., 2007). As with fgf3 itself, we found that dusp6:dGFP marks nascent pouch-forming segments in wild type, with fluorescence apparent in epithelial cell clusters up to two segmental units preceding pouch formation, as well as in adjacent mesoderm (Fig. 1N). dusp6:dGFP intensity was diminished in tbx1 mutants (supplementary material Fig. S1), but enhancement of the fluorescent signal revealed segmental expression within the presumptive pouch-forming endoderm (Fig. 1O). Taken together, the retention of segmental fgf3 expression and Fgf reporter activity in tbx1 mutant endoderm indicates that Tbx1 is dispensable for at least some aspects of pre-pouch segmentation.
Mesodermal Tbx1 is sufficient for pouch morphogenesis but not cartilage formation
As tbx1 was expressed in both the pharyngeal endoderm and mesoderm (Fig. 2A; supplementary material Fig. S2A-C) (Piotrowski et al., 2003), we investigated the requirements of Tbx1 in each tissue for pouch morphogenesis. To do so, we used an endodermal nkx2.3 (Choe et al., 2013) or mesodermal nkx2.5 (Witzel et al., 2012) promoter to restore Tbx1 function in each tissue or both. In situ hybridization in combination with her5:GFP labeling of endoderm confirmed that stably integrated nkx2.3:Tbx1 and nkx2.5:Tbx1 transgenes selectively restored tbx1 expression to the endoderm and mesoderm, respectively, of tbx1 mutants – although nkx2.3:Tbx1 restored tbx1 expression to lower levels than those in wild type and in only the posterior pouch-forming endoderm (Fig. 2B-E; supplementary material Fig. S2I). nkx2.3:Tbx1 and nkx2.5:Tbx1 animals were healthy and viable with no pouch defects on their own (supplementary material Fig. S2D-F). Supporting a role for Tbx1 in the mesoderm, nkx2.5:Tbx1 was able to partially restore pouch formation in tbx1 mutants. Although nkx2.3:Tbx1 alone did not restore pouches to a significant extent, both transgenes together increased the average number of pouches over nkx2.5:Tbx1 alone, indicating that Tbx1 functions in both the mesoderm and endoderm for pouch formation (Fig. 2J).
Surprisingly, despite the robust rescue of pouches by combined nkx2.5:Tbx1 and nkx2.3:Tbx1 transgenes, we failed to observe any recovery of the ceratobranchial cartilages that depend on pouches for their development (Fig. 2P-T). This inability to rescue cartilage formation might be due to earlier requirements for Tbx1 in neural crest development. In tbx1 mutants at 36 h post-fertilization (hpf), we observed severe reductions in pharyngeal arch expression of dlx2a, a marker of neural-crest-derived skeletogenic ectomesenchyme, particularly in the branchial arches that generate the ceratobranchial cartilages (Fig. 2L). Branchial arch reduction of dlx2a was seen as early as 16.5 hpf, and more so at 18 hpf, stages that are well in advance of formation of the third through sixth pouches (supplementary material Fig. S2L-O). Consistent with the lack of cartilage rescue, dlx2a expression was also not restored by nkx2.5:Tbx1 and nkx2.3:Tbx1 transgenes (Fig. 2M-O). Our findings differ somewhat from those of Piotrowski and Nusslein-Volhard (Piotrowski and Nusslein-Volhard, 2000), which found no change in dlx2a expression in 18 hpf tbx1 mutants (although reduced dlx2a was reported at 33 hpf). However, we note that the skeletal defects observed in our tbx1 mutant line are more severe than those presented in this earlier study. Furthermore, the reduction in dlx2a expression did not appear to be due to earlier defects in neural crest specification or migration. Cranial neural crest cells expressing sox10 were present in normal numbers in tbx1 mutant embryos at 12 hpf (supplementary material Fig. S2J,K), and sox10:GFP-positive neural-crest-derived cells still populated the arches of tbx1 mutants at 30 hpf (supplementary material Fig. S2Q,S). We did detect a modest increase in cell death (as determined using LysoTracker Red staining), both in the arches and throughout the head of tbx1 mutants, yet proliferation was unaffected (as determined using BrdU staining; supplementary material Fig. S2Q,S,T). Our data indicate an important function of Tbx1 in the nkx2.5-positive mesoderm for pouch morphogenesis; however, it is likely that Tbx1 has other requirements for the identity and/or survival of skeletogenic ectomesenchyme.
Tbx1 is required for mesodermal expression of wnt11r and fgf8a
Given the crucial role for mesodermal Tbx1 in pouch formation, we next investigated its potential targets in this tissue. As the pouch defects of tbx1 mutants are reminiscent of those seen upon inhibition of Wnt (Choe et al., 2013) or Fgf (Crump et al., 2004a) signaling, we explored whether the expression of individual Wnt and Fgf ligands might require Tbx1 function. In particular, fgf8a and wnt11r are expressed in the mesoderm during pouch initiation (Choe et al., 2013; Nechiporuk et al., 2007; Reifers et al., 2000), and we have previously reported requirements for both these genes in pouch morphogenesis (Choe et al., 2013; Crump et al., 2004a). In wild type, we observed expression of fgf8a and wnt11r in distinct subsets of the nkx2.5:GFP-positive mesoderm adjacent to pouch-forming endoderm (Fig. 3A,F,V). wnt11r appeared in the anterior arch mesoderm slightly before fgf8a, with expression of both shifting to more posterior mesoderm over time – consistent with the anterior to posterior progression of pouch formation (supplementary material Fig. S3A,B,E,F). This mesodermal expression of fgf8a and wnt11r was lost in tbx1 mutants, and reintroduction of Tbx1 in nkx2.5-positive mesoderm (but not nkx2.3-positive endoderm) restored fgf8a and wnt11r expression (Fig. 3A-D,F-I). Also consistent with Wnt11r and Fgf8a acting downstream of Tbx1, we found that tbx1 expression was unaltered in wnt11r or fgf8a mutants (supplementary material Fig. S3D,H). By contrast, expression of wnt4a in the overlying ectoderm, where it regulates the later maturation of pouch epithelia (Choe et al., 2013), as well as its endodermal receptor fzd8a, was largely unaffected in tbx1 mutants (supplementary material Fig. S3L,N). The abnormal shape of the wnt4a ectodermal expression domain in tbx1 mutants might reflect a requirement of endodermal contacts for proper expression, as wnt4a expression was lost in sox32 mutants that lack endoderm (supplementary material Fig. S3M). Hence, abnormal wnt4a expression in tbx1 mutants could be an indirect consequence of defective pouch outpocketing and altered endoderm-ectoderm contacts. In summary, we find that Tbx1 has a mesoderm-autonomous requirement for fgf8a and wnt11r expression, with the related fgf3 and wnt4a ligands, as well as the fzd8a receptor, being regulated independently.
Fig. 3.

Rescue of tbx1−/−pouch defects with mesodermal Fgf8a and Wnt11r. (A-J) Fluorescent in situ hybridization for fgf8a or wnt11r (green) and GFP immunohistochemistry to detect nkx2.5:GFP-positive mesoderm (red) at 30 hpf. In wild type, fgf8a was expressed in ventral nkx2.5:GFP-positive mesodermal cores of arches 3 and 4 (m3 and m4), whereas wnt11r was in more dorsal subsets of these mesodermal cores. In tbx1 mutants, mesodermal expression of fgf8a (n=12 of 12) and wnt11r (n=10 of 11) was lost, although fgf8a and wnt11r expression in the anterior (arrow) and posterior (arrowheads) portions of the otic vesicle was unaffected. An nkx2.3:Tbx1 transgene did not restore mesodermal fgf8a (n=0 of 7) and wnt11r expression (n=0 of 5), whereas an nkx2.5:Tbx1 transgene restored mesodermal fgf8a (n=5 of 6) and wnt11r expression (n=9 of 9). Also, nkx2.5:Fgf8-GFP and nkx2.5:Wnt11r transgenes restored fgf8a (n=27 of 27) and wnt11r (n=21 of 21) expression, respectively. (K-O) Alcama immunohistochemistry (green) showed five pouches (p1-p5) in wild-type fish at 34 hpf. tbx1−/− mutants lost all pouches except for the first (p1), whereas individual nkx2.5:Wnt11r or nkx2.5:Fgf8a-GFP transgenes modestly rescued, and combined nkx2.5:Wnt11r and nkx2.5:Fgf8a-GFP transgenes strongly rescued posterior pouches (p2-p5). Sensory ganglia are indicated with red asterisks. (P-T) Ventral views of dissected facial cartilages. A bilateral set of five CBs formed in wild-type zebrafish, and no CBs formed in tbx1 mutants. No rescue of CB cartilage was seen in nkx2.5:Wnt11r; tbx1−/− (n=39), nkx2.5:Fgf8a-GFP; tbx1−/− (n=34), or nkx2.5:Wnt11r; nkx2.5:Fgf8a-GFP; tbx1−/− larvae (n=31). (U) Quantification of pouch defects based on Alcama staining in wild type (n=51), nkx2.5:Wnt11r (n=49), nkx2.5:Fgf8a-GFP (n=36), tbx1−/− (n=62), nkx2.5:Wnt11r; tbx1−/− (n=44), nkx2.5:Fgf8a-GFP; tbx1−/− (n=37), and nkx2.5:Wnt11r; nkx2.5:Fgf8a-GFP; tbx1−/− (n=24). Data represent mean±s.e.m., P values are shown. (V) Low magnification view of an embryo at 32 hpf showing nkx2.5:GFP-positive mesoderm (green) relative to her5:mCherryCAAX-positive pouch endoderm (red) and the developing eye and ear. The schematic shows expression of fgf8a (green) and wnt11r (yellow) within distinct subsets of nkx2.5-positive mesoderm (red) during the formation of endodermal pouches (blue). Scale bars: 40 μm (A-O).
Restoration of Wnt11r and Fgf8a in mesoderm rescues tbx1−/− pouch defects
As we found wnt11r and fgf8a to be positively regulated by Tbx1, we next tested whether restoring these proteins in mesoderm would rescue tbx1 mutant pouches. To do so, we used the nkx2.5 promoter to stably reintroduce Wnt11r and/or a green fluorescent protein (GFP)-tagged functional Fgf8a protein (Yu et al., 2009) into the tbx1 mutant mesoderm, which we confirmed using in situ hybridization (Fig. 3E,J). In contrast to stably transgenic nkx2.5:Wnt11r embryos, which never displayed pouch or cartilage defects on their own, a few nkx2.5:Fgf8a-GFP embryos did display subtle pouch and ceratobranchial cartilage defects, consistent with the local guidance function of Fgf8a that is discussed below (supplementary material Fig. S3O-T). Compared with non-transgenic tbx1−/− siblings, mutants expressing nkx2.5:Wnt11r or nkx2.5:Fgf8a-GFP alone formed a slightly greater number of pouches, with co-expression of both transgenes rescuing pouch number to a greater extent (Fig. 3K-O,U). As with mesodermal restoration of Tbx1, mesodermal restoration of Wnt11r, Fgf8a, or both failed to rescue ceratobranchial cartilage defects despite the rescue of pouches (Fig. 3P-T). Taken together, these findings support Wnt11r and Fgf8a as being major mesodermal effectors of Tbx1 for pouch morphogenesis.
Synergistic effects of wnt11r and fgf8a loss on pouch formation
If Wnt11r and Fgf8a are parallel effectors of Tbx1 for pouch morphogenesis, then their combined loss should produce severe pouch defects that are similar to loss of tbx1. Individual wnt11r and fgf8a mutants form on average four pouches by 34 hpf and four ceratobranchial cartilages, compared with the normal complement of five each in wild-type zebrafish (Fig. 4). By contrast, wnt11r; fgf8a double mutants formed on average only two pouches and two to three ceratobranchials (Fig. 4D,H,I), with loss of just one copy of wnt11r enhancing the pouch and ceratobranchial defects of fgf8a mutants (supplementary material Fig. S4D,J,M). By contrast, loss of wnt4a, whose expression does not depend on Tbx1, failed to enhance the pouch and cartilage defects of fgf8a mutants (supplementary material Fig. S4F,L,M). Also consistent with parallel roles of Wnt11r and Fgf8a, we found that the mesodermal expression of each does not depend on the other (supplementary material Fig. S4N-Q).
Fig. 4.
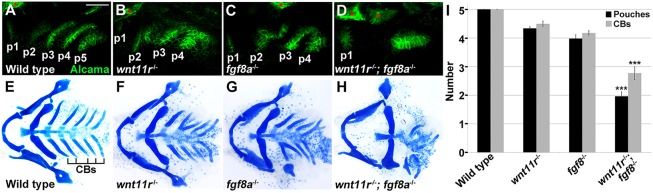
Synergistic pouch and cartilage defects in compound wnt11r; fgf8a mutants. (A-D) Alcama immunohistochemistry (green) labeled five pouches (p1-p5) in wild-type embryos at 34 hpf. Compared with the mild reductions of pouches in wnt11r or fgf8a single mutants, compound wnt11r−/−; fgf8a−/− mutants had a near complete loss of pouches. Sensory ganglia are indicated with red asterisks. (E-H) Dissections of facial cartilage at 5 dpf. Wild type had five CBs on each side, wnt11r and fgf8a single mutants had variable fusions and losses of on average one CB and compound wnt11r−/−; fgf8a−/− mutants lost multiple CBs. (I) Quantification of defects. Number of embryos examined (pouches, CBs): wild type (106, 102), wnt11r−/− (66, 57), fgf8a−/− (68, 98), and wnt11r−/−; fgf8a−/− (24, 20). Data represent mean±s.e.m. ***P<0.001 relative to fgf8a mutants alone. Scale bar: 40 μm (A-D).
Reduced directional persistence of endodermal epithelial cells in fgf8a mutants
We have previously reported that Wnt11r functions to initially destabilize the pouch-forming endoderm so that these epithelial cells can initiate outpocketing (Choe et al., 2013). However, in contrast to wnt11r mutants in which the multi-layering of pouch-forming cells is delayed, we found that fgf8a mutant epithelial cells did become multi-layered at the initial stages (supplementary material Fig. S5G-I). Hence, we performed time-lapse recordings of pouch development, using her5:mCherryCAAX to track individual epithelial cells and dusp6:dGFP to monitor dynamic Fgf activity, to assess the potential later requirements for Fgf8a in epithelial cell behavior. In all five wild-type embryos (Fig. 5A-E; supplementary material Movies 2, 3A), we observed segmental clusters of dusp6:dGFP-positive cells in both forming pouches and presumptive pre-pouch zones, as well as in the adjacent mesoderm. By tracking individual epithelial cells, we found that those that initially expressed dusp6:dGFP or turned it on during the recording selectively contribute to growing pouches compared with dusp6:dGFP-negative cells, consistent with Fgf activity prefiguring the later contributions of cells to pouches (supplementary material Fig. 5). For fgf8a mutants, several differences were consistently observed in three separate embryos (Fig. 5F-J; supplementary material Movie 3B). First, fewer her5:mCherryCAAX-positive epithelial cells expressed dusp6:dGFP in the pre-pouch endoderm and did so at lower levels; mesodermal dusp6:dGFP expression was affected to a lesser extent. Second, nascent mutant pouches comprised mosaics of cells with high, low, or no dusp6:dGFP, compared with the more uniform high expression levels of dusp6:dGFP in wild-type pouches. By tracking individual pouch-forming cells in wild-type and fgf8a mutant zebrafish, we quantified how loss of Fgf8a affects the migratory behavior of these epithelial cells. Although wild-type and fgf8a−/− cells had similar velocities, we found that fgf8a−/− cells had less-directed migration paths, as measured by both the persistence of migration and the distribution of the angles of migration (Fig. 5K-M). One interpretation of this is that the mosaic reduction of endodermal Fgf activity results in reduced directional coherence of migrating pouch cells, which in turn might explain the wide variation of pouch defects that has been observed in fgf8a mutants (Crump et al., 2004a).
Fig. 5.

Requirement for Fgf8a in the directional persistence of pouch cells. (A-J) Representative confocal sections from time-lapse recordings show the development of pouches p4-p6 in a wild type (n=5) and fgf8a mutant (n=3) (see supplementary material Movie 3A,B). her5:mCherryCAAX labels endodermal cell membranes (red) and dusp6:dGFP shows dynamic Fgf activity (green). T0 is 26 hpf. Merged images are shown in A-J and dusp6:dGFP alone in A″-J″. Schematics in A′-J′ show the graded intensity of dusp6:dGFP (green) relative to endodermal cells (red). Individual cell tracks used for the quantification are shown in A‴-J‴. (K-M) The velocity, persistence of migration and distribution of angles of tracked cells over a 7-h period. For distribution of angles, each bin represents the number of cells moving in a particular direction relative to the last cell position. We tracked the cells of two embryos for each wild type and fgf8a mutant. Mean±s.e.m. and P values are shown. n.s., not significant. Scale bar: 20 µm (A-J).
Deviation of growing pouches by ectopic Fgf8a requires Wnt11r function
The analysis of dusp6:dGFP in fgf8a mutants indicates a transcriptional response to Fgf signaling in the endoderm that, at least in part, requires mesodermal Fgf8a. In order to investigate whether Fgf8a might also have more immediate effects on directed pouch outgrowth, we used time-lapse imaging to assess the effects of ectopic Fgf8a-GFP mesodermal clones on neighboring pouch epithelial cells. To do so, we took advantage of our observation that expression of the endogenous fgf8a gene was restricted to a ventral subset of the nkx2.5-positive mesoderm (Fig. 3A), and that injection of DNA into one-cell-stage zebrafish embryos results in mosaic transgene expression during later development. We therefore injected the nkx2.5:Fgf8a-GFP transgene into her5:mCherryCAAX embryos and selected those pouch-stage embryos with clones of Fgf8a-GFP in more dorsal mesoderm. In four out of six wild-type embryos, time-lapse imaging revealed that ectopic expression of Fgf8a-GFP in dorsal mesodermal cells caused adjacent pouch-forming epithelial cells to diverge from their normal lateral migration path (two examples are shown in Fig. 6A-H; supplementary material Movie 4A,B). Eventually, diverted pouch-forming cells resumed their normal lateral migration path, perhaps in response to the endogenous fgf8a-expressing mesodermal cells that were located more ventrally. As a control, we observed no effects on pouch outgrowth in all three wild-type embryos that expressed a nkx2.5:GFP transgene in similar domains of dorsal mesoderm (Fig. 6I-L; supplementary material Movie 4C).
Fig. 6.

Ectopic Fgf8a redirects pouch outgrowth. (A-P) Still images from time-lapse recordings monitoring the development of the third pouch in wild-type embryos with mosaic mesodermal mis-expression of a nkx2.5:Fgf8a-GFP transgene (two examples are shown: A-D and E-H) or a control nkx2.5:GFP transgene (I-L), as well as wnt11r mutants with nkx2.5:Fgf8a-GFP mis-expression (M-P) (see supplementary material Movie 4). her5:mCherryCAAX labels endodermal cell membranes in red. Recordings started at 25 hpf (T0), and subsequent stills were taken every 2 h. nkx2.5:Fgf8a-GFP-expressing clones transiently diverted developing third pouch cells (arrows) in four out of six embryos, whereas nkx2.5:GFP-expressing clones had no effect on adjacent third pouch cells (arrowheads) in all three of the embryos examined. nkx2.5:Fgf8a-GFP-expressing clones failed to attract wnt11r−/− endodermal cells (arrowheads) in all embryos (n=3). (Q) A two-step model of Tbx1 function in pouch morphogenesis. Tbx1 promotes wnt11r and fgf8a expression in distinct domains of the mesoderm, with Wnt11r initiating pouch morphogenesis through epithelial destabilization and Fgf8a guiding subsequent pouch outgrowth. Scale bar: 20 μm (A-P).
Next, we investigated what role the Wnt11r-dependent destabilization of the pre-pouch epithelium plays in the ability of cells to respond to ectopic Fgf8a. Consistent with Wnt11r being required for the Fgf8a response, ectopic nkx2.5:Fgf8a-GFP mesodermal clones failed to attract adjacent presumptive pouch epithelial cells in all three wnt11r mutants examined (Fig. 6M-P; supplementary material Movie 4D). This inability of wnt11r−/− epithelial cells to efficiently respond to Fgf8a was not due to a requirement for Wnt11r in the Fgf transcriptional response, as dusp6:dGFP expression was unaffected in wnt11r mutants (supplementary material Fig. S6). Instead, our data are consistent with the notion that Wnt11r-mediated destabilization of the pre-pouch epithelium is required for cells to be guided by local sources of Fgf8a in the ventral nkx2.5-positive mesoderm (Fig. 6Q).
DISCUSSION
Our study reveals that Tbx1 functions in the facial mesoderm to coordinate multiple steps in the outpocketing of the endodermal epithelium into pouches. Through high-resolution recordings of pouch development and transgenic rescue experiments, we provide evidence that Tbx1 initiates epithelial morphogenesis through Wnt11r and then guides directional pouch outgrowth through Fgf8a. This previously underappreciated morphogenetic role of mesodermal Tbx1 in pouch development helps to clarify the tissue-specific functions and molecular targets of this crucial DiGeorge syndrome gene during craniofacial development.
Morphogenetic role of mesodermal Tbx1 for endodermal pouch development
Our rescue experiments using the nkx2.5:Tbx1 transgene establish important roles of Tbx1 within the facial mesoderm for the outpocketing of the endoderm into pouch epithelia. Restoration of Tbx1 to the nkx2.3-positive endoderm failed to rescue pouch development on its own, but it did improve the extent of pouch rescue upon expression of nkx2.5:Tbx1. Thus, Tbx1 probably functions in both the mesoderm and endoderm for pouch development. The observed enhanced rescue by the nkx2.5:Tbx1 transgene alone, compared with the nkx2.3:Tbx1 transgene, might reflect a more prominent role of mesodermal, compared with endodermal, Tbx1 for pouch formation. Alternatively, the nkx2.3:Tbx1 transgene might not restore Tbx1 early enough or to sufficient levels in the endoderm to strongly rescue pouches. Indeed, nkx2.3:Tbx1 restored tbx1 expression primarily to just the posterior pouch-forming endoderm.
A mesodermal role for Tbx1 in zebrafish is consistent with mouse studies showing that tissue-specific deletion of Tbx1 in the Mesp1-positive mesoderm results in severe pouch defects and that restoration of Tbx1 in Mesp1-positive mesoderm largely rescues pouch defects in Tbx1 null embryos (Zhang et al., 2006). We used a mesoderm-specific nkx2.5 promoter to restore tbx1 and rescue pouches, but deletion of Tbx1 with an Nkx2.5:Cre driver in mouse has no effect on pouch development (Xu et al., 2004). This discrepancy might be explained by the inefficient and hence mosaic activity of this Nkx2.5:Cre line (Zhang et al., 2005), or by different timings of Nkx2.5 gene expression between mouse and fish. In mouse, deletion of Tbx1 using an endoderm-specific isolate of the Foxg1:Cre construct also resulted in pouch defects, and endoderm-specific Tbx1 deletion with Fgf15:Cre resulted in loss of the fourth pharyngeal artery, which might be interpreted as secondary to altered development of the fourth pouch (Arnold et al., 2006; Zhang et al., 2005). In mouse, endodermal Tbx1 has been proposed to promote the proliferation of pouch endodermal progenitors (Arnold et al., 2006; Xu et al., 2005). One explanation then for the pouch defects that are seen upon endoderm-specific Tbx1 deletion in mouse is that there are simply less cells available to make normal pouches. Indeed, we also observed extensive proliferation in the pre-pouch endoderm of zebrafish, although only limited proliferation was apparent during pouch outpocketing (data not shown). Hence, a parsimonious view is that Tbx1 acts primarily in the mesoderm for pouch morphogenesis yet has additional roles in the endoderm for other aspects of pouch cell biology, such as their proliferative expansion.
Segmental patterning of the pre-pouch endoderm in the absence of Tbx1
Tbx1 is essential for the morphological outpocketing of the endoderm into pouches; however, we find that the pre-pouch endoderm still retains some segmental characteristics even in the absence of Tbx1. In particular, expression of the fgf3 ligand and Fgf activity itself remain apparent in segmental clusters of tbx1 mutant endodermal cells despite their failure to undergo morphogenesis. Consistent with Tbx1 being dispensable for the initial segmentation of pharyngeal endoderm, hemichordates segment an apparently homologous population of pharyngeal endoderm despite lacking Tbx1 expression in their pharynx (Gillis et al., 2012). One explanation is that pharyngeal segmentation evolutionarily pre-dates Tbx1 expression, with mesodermal Tbx1 in vertebrates driving more complex morphologies of pouches, coinciding with the newfound investment of pharyngeal arches with neural crest cells. Although the nature of this early Tbx1-independent segmentation of the endoderm remains unclear, it is interesting that the Fgf reporter dusp6:dGFP is an early marker of epithelial cells that are destined to generate pouches. In the pre-somitic mesoderm, iterative Fgf activity plays an important role in the segmentation of this tissue into somites (reviewed by Pourquié, 2011). Hence, it will be interesting to explore whether the pharyngeal endoderm utilizes a similarly constructed segmentation clock.
Wnt11r is a novel morphogenetic effector of Tbx1 in pouch development
Several lines of evidence indicate that wnt11r is a crucial effector of Tbx1 for pouch development. First, time-lapse imaging of tbx1 mutants revealed a failure to initiate outpocketing of the endodermal epithelium, a phenotype similar to that of wnt11r mutants and embryos in which the cytoplasmic Wnt effectors Dishevelled and Rac1 have been inhibited (Choe et al., 2013). Second, wnt11r expression was lost in the nkx2.5-positive mesoderm of tbx1 mutants. Third, restoration of Wnt11r in this mesoderm partially rescued tbx1−/− pouch defects, and did so more effectively when combined with Fgf8a. Recently, a role for Wnt11r downstream of Tbx1 has been reported for the looping of the heart in zebrafish (Choudhry and Trede, 2013). This dual requirement of Tbx1 in the outpocketing of pouch endoderm and looping of heart mesoderm might reflect a common regulation of wnt11r in the nkx2.5-positive mesoderm, with this mesodermal subpopulation not only contributing to the myocardium but also organizing the adjacent pouch endoderm. In mice, the wnt11r homolog Wnt11 has a similar requirement to Tbx1 for development of the second heart field, although this is only revealed when Wnt5a is also disrupted (Cohen et al., 2012). Wnt11−/− mutants are viable and presumably lack the pouch defects seen in wnt11r mutant fish, but pouch development in Wnt11; Wnt5a double mutants, which die by embryonic day (E)10.5, has not been analyzed. Indeed, the relatively weaker pouch defects of wnt11r mutants versus embryos expressing dominant-negative Wnt signaling constructs indicates the presence of other functionally redundant Wnt proteins downstream of Tbx1 in pouch development (Choe et al., 2013).
Guidance role of Fgf signaling in pouch epithelial outpocketing
The ability of mesodermal Fgf8a plus Wnt11r to rescue tbx1 mutant pouches indicates an important role for mesoderm-derived Fgf ligands in Tbx1-dependent pouch morphogenesis. Our results are consistent with those in mouse that show loss of mesodermal Fgf8a expression upon deletion of Tbx1 in the Mesp1-positive mesoderm (Zhang et al., 2006). Although it has been reported that insertion of Fgf8 into the Tbx1 locus failed to rescue Tbx1 mutant defects (Vitelli et al., 2006), this could be due to ectopic expression of Fgf8 in non-mesodermal Tbx1-positive tissues or the need to also restore Wnt ligands. It has been proposed that Fgf8 in mouse functions in a tissue-autonomous manner for mesodermal cell proliferation (Zhang et al., 2006). By contrast, our finding that expression of the Fgf reporter dusp6:dGFP is reduced in the endoderm but not mesoderm of zebrafish fgf8a mutants suggests that there is a non-autonomous requirement for mesodermal Fgf8a in endoderm development.
Cellular resolution time-lapse imaging also provides direct evidence for a role of Fgf8a in guiding collective cell migration during pouch outpocketing. Guidance roles for Fgf signaling have been well characterized in the Drosophila tracheal system (Ghabrial and Krasnow, 2006; Sutherland et al., 1996) and recently in the migration of Drosophila caudal visceral mesoderm cells (Kadam et al., 2012). In vertebrates, Fgf10 promotes the collective migration of posterior lateral line neuroblasts in zebrafish (Breau et al., 2012), whereas Fgf4 and Fgf8 appear to oppositely attract and repel mesenchymal cells of the chick primitive streak (Yang et al., 2002). Although Fgf10 is essential for branching of the endodermal epithelium to form the mammalian lung, a direct role in guiding collective epithelial cell migration has not been provided (Bellusci et al., 1997; Sekine et al., 1999). Our finding that ectopic sources of Fgf8a can redirect pouch epithelial migration therefore raises the possibility that Fgf ligands might guide the collective migration of branching epithelia along the length of the endoderm.
The observation that Fgf8a is required for a transcriptional response in the endoderm (i.e. expression of dusp6:dGFP) might seem at odds with the chemotactic role that we have revealed using ectopic Fgf8a-GFP mis-expression, with chemotaxis presumably involving rapid cytoskeletal changes in the cytoplasm. By contrast, work in the lateral line system has shown that Fgf signaling can influence collective cell migration by controlling chemokine receptor expression (Aman and Piotrowski, 2008; Nechiporuk and Raible, 2008). Previous studies have also shown that Fgf signaling can elicit multiple responses within a cell through distinct downstream pathways (Baron et al., 2000; Maher, 1999; Raucci et al., 2004), and hence Fgf signaling in the growing pouch might also serve to coordinate chemotactic cytoskeletal changes with nuclear transcription responses that promote the maturation, proliferation and/or survival of pouch cells. Further work is also needed to elucidate how Wnt11r induces competency of the endodermal epithelium to respond to Fgf8a. One possibility is that Wnt11r-dependent destabilization of adherens junctions allows pre-pouch epithelial cells to more easily extend membrane extensions that promote migration towards Fgf8a sources. The cytoplasmic effectors of Fgf8a signaling that promote chemotaxis remain unknown, but it is possible that Wnt11r and Fgf8a signaling might converge on common targets in the cytoplasm that promote collective cell migration. By contrast, Wnt11r does not appear to be required for either fgf8a expression or the Fgf transcriptional response, supporting our earlier work that cytoplasmic and not nuclear Wnt signaling mediates pouch morphogenesis (Choe et al., 2013).
Pouch-independent roles for Tbx1 in craniofacial skeletal development
A surprising finding from our study was the lack of craniofacial skeletal rescue despite the robust rescue of pouch morphogenesis in tbx1 mutants. This was true when Tbx1 was restored to both the mesoderm and endoderm of tbx1 mutants, or when combined Wnt11r and Fgf8a were restored to the mesoderm. It is well established that defective pouch development results in decreased cartilage and bone formation from the hyoid and more posterior branchial arches, with the latter generating the ceratobranchial cartilages that are most affected by the loss of tbx1 in zebrafish. For example, mutations in either integrinα5 or sox32 in zebrafish result in both pouch defects and losses of the posterior facial skeleton, with skeletal rescue by wild-type endoderm transplants demonstrating that skeletal defects are due to non-autonomous roles of these genes in the endoderm (Crump et al., 2004b; David et al., 2002). Our data indicate that, beyond its role in inducing pouch formation, Tbx1 also has an earlier role in the development of the skeletogenic neural crest. In particular, we observed reduced expression of the ectomesenchyme marker dlx2a in the posterior arches of tbx1 mutants, even at stages before those at which the pouches normally form. At least in mouse, Tbx1 does not appear to function cell-autonomously within cranial neural crest cells to promote their ectomesenchyme potential, as neural-crest-specific deletion of Tbx1 using the Wnt1:Cre driver does not result in any craniofacial defects (Aggarwal et al., 2010). Instead, it is likely that Tbx1 has non-autonomous requirements for early cranial neural crest development, for example within mesoderm and/or endoderm populations that are not labeled by the nkx2.5 and nkx2.3 promoters used in this study. Indeed, the dissociation of pouch and craniofacial skeletal defects in these ‘rescued’ tbx1 mutants further highlights the multiplicity of functions of this transcription factor in the assembly of the vertebrate head.
MATERIALS AND METHODS
Zebrafish lines
Zebrafish (Danio rerio) were raised in strict accordance with good animal practice as defined by the relevant national and/or local animal welfare bodies, and all animal work was approved by the University of Southern California Institutional Animal Care and Use Committee. Published lines used include tbx1tu285 (van gogh) (Piotrowski et al., 2003), wnt11rfh224 (Banerjee et al., 2011), fgf8ati282a (acerebellar) (Reifers et al., 1998), Tg(∼3.4her5:EGFP)ne1911 (Tallafuss and Bally-Cuif, 2003), Tg(her5:mCherryCAAX)el72 (Choe et al., 2013), Tg(nkx2.5:GFP)el83 (Witzel et al., 2012), Tg(sox10:LOX-GFP-LOX-hDLX3)el8 (Das and Crump, 2012), and Tg(dusp6:dGFP) (Molina et al., 2007). For genotyping of tbx1tu285, primers TBX-09 and TBX-10 converted tbx1tu285 into a co-dominant polymorphism, with a wild-type product of 438 bp and mutant products of 276 and 162 bp after PacI digestion. Genotyping of wnt11rfh224 was as described previously (Banerjee et al., 2011). fgf8ati282a mutant embryos were scored by loss of cerebellum (Reifers et al., 1998). nkx2.3:Tbx1, nkx2.5:Tbx1, nkx2.5:Wnt11r and nkx2.5:Fgf8a-GFP transgenic constructs were generated using the Gateway (Invitrogen) Tol2kit (Kwan et al., 2007). p5E-nkx2.3, p5E-nkx2.5 and pME-Wnt11r plasmids have been published previously (Choe et al., 2013). For pME-Tbx1, the coding sequence of tbx1 was amplified using primers Tbx1-B1F and Tbx1-B2R from multi-stage zebrafish cDNA according to the manufacturer’s instructions (Phusion, NEB) and cloned into pDONR221. The Fgf8a-GFP fusion protein was generated through fusion PCR (Szewczyk et al., 2007) to insert GFP between amino acid residues 22 and 23 of Fgf8a. This fusion has been previously verified to be functional (Yu et al., 2009). The Fgf8a-GFP cDNA was then cloned into pDONR221 to generate pME-Fgf8a-GFP. After LR cloning with p3E-pA and pDestTol2CG2 (cmlc2:GFP) or pDestTol2AB (α-crystallin:Cerulean), plasmids were injected at 30 ng/μl with 35 ng/μl Tol2 transposase RNA into one-cell stage embryos. Four independent transgenic lines were isolated for Tg(nkx2.3:Tbx1:pA) and Tg(nkx2.5:Wnt11r:pA) based on cmlc2:GFP heart fluorescence. Two independent transgenic lines were isolated for Tg(nkx2.5:Tbx1:pA) based on α-crystallin:Cerulean lens fluorescence. Four independent transgenic lines were isolated for Tg(nkx2.5:Fgf8a-GFP:pA) based on mesodermal GFP fluorescence. We used stable lines Tg(nkx2.3:Tbx1:pA)el513, Tg(nkx2.5:Tbx1:pA)el567, Tg(nkx2.5:Wnt11r:pA)el493 and Tg(nkx2.5:Fgf8a-GFP:pA)el562 for this study. In order to generate nkx2.5:Fgf8a-GFP mosaics, 30 ng/μl of the nkx2.5:Fgf8a-GFP construct with 35 ng/μl Tol2 RNA was injected into one-cell stage wild-type or wnt11r mutant her5:mCherryCAAX embryos. We then used a Leica M165 FC fluorescence dissecting stereoscope with 3.2× magnification to select mosaic animals displaying desired GFP fluorescence at 22 hpf, use of confocal microscopy allowed further selection for time-lapse imaging. See supplementary Materials and Methods for primers.
Staining
Immunohistochemistry for Alcama/ZN8 (Zebrafish International Resource Center, 1:400) was performed as described previously (Crump et al., 2004a). Alcian Blue staining, fluorescent in situ hybridizations and GFP immunohistochemistry (Torrey Pines Biolabs, 1:1000) protocols have been published previously (Zuniga et al., 2011). The in situ probes included wnt11r, wnt4a and fzd8a (Choe et al., 2013). For tbx1, fgf8a and fgf3, probes were generated by using PCR and cloned into pGEM®-T Easy Vector Systems (Promega), and digoxigenin-labeled RNAs were synthesized using T7 or SP6 RNA polymerase. See supplementary Materials and Methods for primers. Direct visualization of LysoTracker Red staining was used to monitor cell death in embryos and antibody-mediated staining of BrdU incorporation was used to follow cell division. Further details are given in the supplementary Materials and Methods.
Imaging
Craniofacial cartilages were dissected with fine insect pins and were then flat-mounted for imaging on a Leica DM 2500 upright microscope using Leica software. Fluorescence images of antibody-stained or in situ hybridization embryos were taken on a Zeiss LSM5 confocal microscope using ZEN software. For time-lapse imaging, live embryos were mounted as previously described (Crump et al., 2004a), and approximately 80-μm z-stacks at 1.5-μm intervals were captured by using a Zeiss 40× LD-Plan Neofluar objective lens every 10 min. For single section movies, time-lapse imaging datasets were manually assembled using ZEN software, and then the brightness and contrast of movies were adjusted using Fiji. For cell tracking, we used Fiji freeware to manually annotate the centroid of each pouch cell in each frame of the time-lapse recording. Only cells that could be reliably tracked throughout the whole 6 h window were used for analysis. Cell tracks were then exported to MATLAB (MathWorks) and processed as described previously (Matthews et al., 2008). Velocity was calculated as the total distance traveled divided by time, and persistence equals the ratio between the linear distance from the initial to the final point and the total length of the migratory path. For the angle of migration, lines were first generated between the first and second track position and then compared with the lines generated between the second and third track position. The angle represents the deviation between these lines. The process was then performed iteratively for each subsequent set of time points (i.e. the line between third and fourth track compared with the line between second and third track) and for each cell in the analysis.
Scoring and statistics
Pouches missing or reduced >50% compared with those of wild type were scored as 0 and mis-shapen or normal pouches as 1 (examples shown for each are shown in supplementary material Fig. S7). Wild-type zebrafish invariantly had five pouches per side at 34 hpf. We scored fusions of two ceratobranchials as 1.5, normal ceratobranchials as 1.0, reduced ceratobranchials as 0.5 and absent ceratobranchials as 0. We utilized the multiple comparison test of Tukey–Kramer for pouch and ceratobranchial defects, one-tailed Student's t-test with unequal variance for cell velocity and persistence, chi-square test of independence for distribution of angles and Fisher's exact test for contributions of dusp6:dGFP-positive cells to pouches.
Supplementary Material
Acknowledgements
We thank Megan Matsutani and Jennifer DeKoeyer Crump for fish care, Seth Ruffins for image analysis, and Henry Sucov for comments.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
C.P.C. and J.G.C. designed the experiments, analyzed the data and wrote the paper. C.P.C. performed the experiments.
Funding
Work was funded by a National Institute of Dental and Craniofacial Research (NIDCR) grant [R01DE022572 to J.G.C.] and a California Institute for Regenerative Medicine (CIRM) training fellowship to C.P.C. Deposited in PMC for immediate release.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.111740/-/DC1
References
- Aggarwal, V. S., Carpenter, C., Freyer, L., Liao, J., Petti, M. and Morrow, B. E. (2010). Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev. Biol. 344, 669-681 10.1016/j.ydbio.2010.05.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman, A. and Piotrowski, T. (2008). Wnt/beta-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev. Cell 15, 749-761 10.1016/j.devcel.2008.10.002 [DOI] [PubMed] [Google Scholar]
- Arnold, J. S., Werling, U., Braunstein, E. M., Liao, J., Nowotschin, S., Edelmann, W., Hebert, J. M. and Morrow, B. E. (2006). Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development 133, 977-987 10.1242/dev.02264 [DOI] [PubMed] [Google Scholar]
- Banerjee, S., Gordon, L., Donn, T. M., Berti, C., Moens, C. B., Burden, S. J. and Granato, M. (2011). A novel role for MuSK and non-canonical Wnt signaling during segmental neural crest cell migration. Development 138, 3287-3296 10.1242/dev.067306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron, W., Metz, B., Bansal, R., Hoekstra, D. and de Vries, H. (2000). PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol. Cell. Neurosci. 15, 314-329 10.1006/mcne.1999.0827 [DOI] [PubMed] [Google Scholar]
- Bellusci, S., Grindley, J., Emoto, H., Itoh, N. and Hogan, B. L. (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867-4878. [DOI] [PubMed] [Google Scholar]
- Breau, M. A., Wilson, D., Wilkinson, D. G. and Xu, Q. (2012). Chemokine and Fgf signalling act as opposing guidance cues in formation of the lateral line primordium. Development 139, 2246-2253 10.1242/dev.080275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe, C. P., Collazo, A., Trinh, L. A., Pan, L., Moens, C. B. and Crump, J. G. (2013). Wnt-dependent epithelial transitions drive pharyngeal pouch formation. Dev. Cell 24, 296-309 10.1016/j.devcel.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhry, P. and Trede, N. S. (2013). DiGeorge syndrome gene tbx1 functions through wnt11r to regulate heart looping and differentiation. PLoS ONE 8, e58145 10.1371/journal.pone.0058145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, E. D., Miller, M. F., Wang, Z., Moon, R. T. and Morrisey, E. E. (2012). Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 139, 1931-1940 10.1242/dev.069377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump, J. G., Maves, L., Lawson, N. D., Weinstein, B. M. and Kimmel, C. B. (2004a). An essential role for Fgfs in endodermal pouch formation influences later craniofacial skeletal patterning. Development 131, 5703-5716 10.1242/dev.01444 [DOI] [PubMed] [Google Scholar]
- Crump, J. G., Swartz, M. E. and Kimmel, C. B. (2004b). An integrin-dependent role of pouch endoderm in hyoid cartilage development. PLoS Biol. 2, e244 10.1371/journal.pbio.0020244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, A. and Crump, J. G. (2012). Bmps and Id2a act upstream of Twist1 to restrict ectomesenchyme potential of the cranial neural crest. PLoS Genet. 8, e1002710 10.1371/journal.pgen.1002710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, N. B., Saint-Etienne, L., Tsang, M., Schilling, T. F. and Rosa, F. M. (2002). Requirement for endoderm and FGF3 in ventral head skeleton formation. Development 129, 4457-4468. [DOI] [PubMed] [Google Scholar]
- Epstein, J. A. (2001). Developing models of DiGeorge syndrome. Trends Genet. 17, S13-S17 10.1016/S0168-9525(01)02450-7 [DOI] [PubMed] [Google Scholar]
- Ghabrial, A. S. and Krasnow, M. A. (2006). Social interactions among epithelial cells during tracheal branching morphogenesis. Nature 441, 746-749 10.1038/nature04829 [DOI] [PubMed] [Google Scholar]
- Gillis, J. A., Fritzenwanker, J. H. and Lowe, C. J. (2012). A stem-deuterostome origin of the vertebrate pharyngeal transcriptional network. Proc. R. Soc. B Biol. Sci. 279, 237-246 10.1098/rspb.2011.0599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris, D. L., Fantes, J., Tara, D., Druker, B. J. and Imamoto, A. (2001). Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 27, 293-298 10.1038/85855 [DOI] [PubMed] [Google Scholar]
- Huh, S.-H. and Ornitz, D. M. (2010). β-catenin deficiency causes DiGeorge syndrome-like phenotypes through regulation of Tbx1. Development 137, 1137-1147 10.1242/dev.045534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam, S., Ghosh, S. and Stathopoulos, A. (2012). Synchronous and symmetric migration of Drosophila caudal visceral mesoderm cells requires dual input by two FGF ligands. Development 139, 699-708 10.1242/dev.068791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan, K. M., Fujimoto, E., Grabher, C., Mangum, B. D., Hardy, M. E., Campbell, D. S., Parant, J. M., Yost, H. J., Kanki, J. P. and Chien, C.-B. (2007). The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088-3099 10.1002/dvdy.21343 [DOI] [PubMed] [Google Scholar]
- Lindsay, E. A., Vitelli, F., Su, H., Morishima, M., Huynh, T., Pramparo, T., Jurecic, V., Ogunrinu, G., Sutherland, H. F. and Scambler, P. J.et al. (2001). Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410, 97-101 10.1038/35065105 [DOI] [PubMed] [Google Scholar]
- Maher, P. (1999). p38 mitogen-activated protein kinase activation is required for fibroblast growth factor-2-stimulated cell proliferation but not differentiation. J. Biol. Chem. 274, 17491-17498 10.1074/jbc.274.25.17491 [DOI] [PubMed] [Google Scholar]
- Matthews, H. K., Marchant, L., Carmona-Fontaine, C., Kuriyama, S., Larraín, J., Holt, M. R., Parsons, M. and Mayor, R. (2008). Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development 135, 1771-1780 10.1242/dev.017350 [DOI] [PubMed] [Google Scholar]
- Molina, G. A., Watkins, S. C. and Tsang, M. (2007). Generation of FGF reporter transgenic zebrafish and their utility in chemical screens. BMC Dev. Biol. 7, 62 10.1186/1471-213X-7-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechiporuk, A. and Raible, D. W. (2008). FGF-dependent mechanosensory organ patterning in zebrafish. Science 320, 1774-1777 10.1126/science.1156547 [DOI] [PubMed] [Google Scholar]
- Nechiporuk, A., Linbo, T., Poss, K. D. and Raible, D. W. (2007). Specification of epibranchial placodes in zebrafish. Development 134, 611-623 10.1242/dev.02749 [DOI] [PubMed] [Google Scholar]
- Nissen, R. M., Yan, J., Amsterdam, A., Hopkins, N. and Burgess, S. M. (2003). Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development 130, 2543-2554 10.1242/dev.00455 [DOI] [PubMed] [Google Scholar]
- Nowotschin, S., Liao, J., Gage, P. J., Epstein, J. A., Campione, M. and Morrow, B. E. (2006). Tbx1 affects asymmetric cardiac morphogenesis by regulating Pitx2 in the secondary heart field. Development 133, 1565-1573 10.1242/dev.02309 [DOI] [PubMed] [Google Scholar]
- Piotrowski, T. and Nüsslein-Volhard, C. (2000). The endoderm plays an important role in patterning the segmented pharyngeal region in zebrafish (Danio rerio). Dev. Biol. 225, 339-356 10.1006/dbio.2000.9842 [DOI] [PubMed] [Google Scholar]
- Piotrowski, T., Ahn, D.-G., Schilling, T. F., Nair, S., Ruvinsky, I., Geisler, R., Rauch, G.-J., Haffter, P., Zon, L. I. and Zhou, Y.et al. (2003). The zebrafish van gogh mutation disrupts tbx1, which is involved in the DiGeorge deletion syndrome in humans. Development 130, 5043-5052 10.1242/dev.00704 [DOI] [PubMed] [Google Scholar]
- Pourquiè, O. (2011). Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell 145, 650-663 10.1016/j.cell.2011.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci, A., Laplantine, E., Mansukhani, A. and Basilico, C. (2004). Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J. Biol. Chem. 279, 1747-1756 10.1074/jbc.M310384200 [DOI] [PubMed] [Google Scholar]
- Reifers, F., Bohli, H., Walsh, E. C., Crossley, P. H., Stainier, D. Y. and Brand, M. (1998). Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain-hindbrain boundary development and somitogenesis. Development 125, 2381-2395. [DOI] [PubMed] [Google Scholar]
- Reifers, F., Walsh, E. C., Leger, S., Stainier, D. Y. and Brand, M. (2000). Induction and differentiation of the zebrafish heart requires fibroblast growth factor 8 (fgf8/acerebellar). Development 127, 225-235. [DOI] [PubMed] [Google Scholar]
- Roberts, C., Ivins, S., Cook, A. C., Baldini, A. and Scambler, P. J. (2006). Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Hum. Mol. Genet. 15, 3394-3410 10.1093/hmg/ddl416 [DOI] [PubMed] [Google Scholar]
- Schwend, T. and Ahlgren, S. C. (2009). Zebrafish con/disp1 reveals multiple spatiotemporal requirements for Hedgehog-signaling in craniofacial development. BMC Dev. Biol. 9, 59 10.1186/1471-213X-9-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine, K., Ohuchi, H., Fujiwara, M., Yamasaki, M., Yoshizawa, T., Sato, T., Yagishita, N., Matsui, D., Koga, Y. and Itoh, N.et al. (1999). Fgf10 is essential for limb and lung formation. Nat. Genet. 21, 138-141 10.1038/5096 [DOI] [PubMed] [Google Scholar]
- Stalmans, I., Lambrechts, D., De Smet, F., Jansen, S., Wang, J., Maity, S., Kneer, P., von der Ohe, M., Swillen, A. and Maes, C.et al. (2003). VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat. Med. 9, 173-182 10.1038/nm819 [DOI] [PubMed] [Google Scholar]
- Sutherland, D., Samakovlis, C. and Krasnow, M. A. (1996). branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell 87, 1091-1101 10.1016/S0092-8674(00)81803-6 [DOI] [PubMed] [Google Scholar]
- Szewczyk, E., Nayak, T., Oakley, C. E., Edgerton, H., Xiong, Y., Taheri-Talesh, N., Osmani, S. A. and Oakley, B. R. (2007). Fusion PCR and gene targeting in Aspergillus nidulans. Nat. Protoc. 1, 3111-3120 10.1038/nprot.2006.405 [DOI] [PubMed] [Google Scholar]
- Tallafuss, A. and Bally-Cuif, L. (2003). Tracing of her5 progeny in zebrafish transgenics reveals the dynamics of midbrain-hindbrain neurogenesis and maintenance. Development 130, 4307-4323 10.1242/dev.00662 [DOI] [PubMed] [Google Scholar]
- Vitelli, F., Morishima, M., Taddei, I., Lindsay, E. A. and Baldini, A. (2002a). Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 11, 915-922 10.1093/hmg/11.8.915 [DOI] [PubMed] [Google Scholar]
- Vitelli, F., Taddei, I., Morishima, M., Meyers, E. N., Lindsay, E. A. and Baldini, A. (2002b). A genetic link between Tbx1 and fibroblast growth factor signaling. Development 129, 4605-4611. [DOI] [PubMed] [Google Scholar]
- Vitelli, F., Zhang, Z., Huynh, T., Sobotka, A., Mupo, A. and Baldini, A. (2006). Fgf8 expression in the Tbx1 domain causes skeletal abnormalities and modifies the aortic arch but not the outflow tract phenotype of Tbx1 mutants. Dev. Biol. 295, 559-570 10.1016/j.ydbio.2006.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzel, H. R., Jungblut, B., Choe, C. P., Crump, J. G., Braun, T. and Dobreva, G. (2012). The LIM protein Ajuba restricts the second heart field progenitor pool by regulating Isl1 activity. Dev. Cell 23, 58-70 10.1016/j.devcel.2012.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak, H., Ittner, L. M., Lang, K. S., Leveen, P., Suter, U., Fischer, J. A., Karlsson, S., Born, W. and Sommer, L. (2005). Inactivation of TGFβ signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 19, 530-535 10.1101/gad.317405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H., Morishima, M., Wylie, J. N., Schwartz, R. J., Bruneau, B. G., Lindsay, E. A. and Baldini, A. (2004). Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131, 3217-3227 10.1242/dev.01174 [DOI] [PubMed] [Google Scholar]
- Xu, H., Cerrato, F. and Baldini, A. (2005). Timed mutation and cell-fate mapping reveal reiterated roles of Tbx1 during embryogenesis, and a crucial function during segmentation of the pharyngeal system via regulation of endoderm expansion. Development 132, 4387-4395 10.1242/dev.02018 [DOI] [PubMed] [Google Scholar]
- Yang, X., Dormann, D., Münsterberg, A. E. and Weijer, C. J. (2002). Cell movement patterns during gastrulation in the chick are controlled by positive and negative chemotaxis mediated by FGF4 and FGF8. Dev. Cell 3, 425-437 10.1016/S1534-5807(02)00256-3 [DOI] [PubMed] [Google Scholar]
- Yu, S. R., Burkhardt, M., Nowak, M., Ries, J., Petrásek, Z., Scholpp, S., Schwille, P. and Brand, M. (2009). Fgf8 morphogen gradient forms by a source-sink mechanism with freely diffusing molecules. Nature 461, 533-536 10.1038/nature08391 [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Cerrato, F., Xu, H., Vitelli, F., Morishima, M., Vincentz, J., Furuta, Y., Ma, L., Martin, J. F. and Baldini, A.et al. (2005). Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 132, 5307-5315 10.1242/dev.02086 [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Huynh, T. and Baldini, A. (2006). Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 133, 3587-3595 10.1242/dev.02539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga, E., Rippen, M., Alexander, C., Schilling, T. F. and Crump, J. G. (2011). Gremlin 2 regulates distinct roles of BMP and Endothelin 1 signaling in dorsoventral patterning of the facial skeleton. Development 138, 5147-5156 10.1242/dev.067785 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.