

Abstract
Chromosomal domains in Drosophila are marked by the insulator-binding proteins (IBPs) dCTCF/Beaf32 and cofactors that participate in regulating long-range interactions. Chromosomal borders are further enriched in specific histone modifications, yet the role of histone modifiers and nucleosome dynamics in this context remains largely unknown. Here, we show that IBP depletion impairs nucleosome dynamics specifically at the promoters and coding sequence of genes flanked by IBP binding sites. Biochemical purification identifies the H3K36 histone methyltransferase NSD/dMes-4 as a novel IBP cofactor, which specifically co-regulates the chromatin accessibility of hundreds of genes flanked by dCTCF/Beaf32. NSD/dMes-4 presets chromatin before the recruitment of transcriptional activators including DREF that triggers Set2/Hypb-dependent H3K36 trimethylation, nucleosome positioning, and RNA splicing. Our results unveil a model for how IBPs regulate nucleosome dynamics and gene expression through NSD/dMes-4, which may regulate H3K27me3 spreading. Our data uncover how IBPs dynamically regulate chromatin organization depending on distinct cofactors.
Keywords: chromatin barrier, higher-order chromatin organization, nucleosome positioning, physical borders, RNA splicing
Introduction
Recent Chromosome Capture Conformation (3C/Hi-C) data have highlighted that chromosomes are physically partitioned into distinct epigenetically marked chromatin domains bordered by insulators (Ghirlando et al, 2012; Hou et al, 2012; Sexton et al, 2012). The genomic distribution of the thousands of Drosophila insulator protein binding sites tightly correlated with the “physical borders” that restrict long-range interactions between chromosomal domains (Sexton et al, 2012). This may provide with a distinct mechanism of gene regulation through higher-order physical organization of chromatin into topologically associating domains (TADs) that is not strictly linked to the demarcation of chromosomes into epigenetically marked domains (Hou et al, 2012; reviewed in Phillips-Cremins & Corces, 2013).
The function of chromatin insulators in long-range interactions was initially suspected genetically from “enhancer-blocking” assays showing that, when interposed, insulators can block the long-range interactions between distant regulatory elements (Cai & Levine, 1995; reviewed in Maeda & Karch, 2007; Gohl et al, 2011). Enhancer-blocking activity requires the binding of CCCTC-binding factor (CTCF) or one of the additional insulator-binding proteins (IBPs) identified in Drosophila—GAGA Factor (GAF), Boundary Element-Associated Factor (Beaf32), or suppressor of Hairy-wing [Su(Hw)]; (reviewed in Vögelmann et al, 2011). Only a fraction of the identified genomic IBP sites were shown to function in standard enhancer-blocking assays (Negre et al, 2010), maybe as such function may depend on genomic contexts, the presence of nearby insulators (Gohl et al, 2011) or on additional cofactors. Genome-wide analyses of long-range contacts highlighted that IBPs may actually favor long-range interactions between distant sites, thereby contributing to the physical organization of TADs (reviewed in Phillips-Cremins & Corces, 2013). This requires additional cofactors for proper insulation of chromatin into domains, including CP190 (Bushey et al, 2009; Liang et al, 2014), that interact with all types of IBPs, chromator (Hou et al, 2012; Sexton et al, 2012), or key chromatin regulators like cohesin (reviewed in Dorsett, 2011; Phillips-Cremins & Corces, 2013).
IBPs were also proposed to function as “insulator barriers” that could block the spreading of condensed regions toward euchromatin (Gaszner & Felsenfeld, 2006; Maeda & Karch, 2007). Whether barrier and enhancer-blocking activities are conferred by distinct types of factors has been questioned (Vögelmann et al, 2011). Genome-wide, the binding sites of IBPs including CTCF and Beaf32 are largely enriched at the borders of repressed domains epigenetically marked by histone H3 tri-methylated on lysine 27 (H3K27me3) (Guelen et al, 2008; Cuddapah et al, 2009; Sexton et al, 2012). Only a fraction of the IBP sites could actually restrict H3K27me3 spreading (Schwartz et al, 2012), again suggesting that additional cofactors may be required. Remarkably, the barrier and enhancer-blocking activities of the chicken beta-globin locus were shown to be conferred by different types of nearby elements recognized by either USF/VEZF or CTCF, respectively (Ghirlando et al, 2012; Gowher et al, 2012). Nucleosome occupancy is particularly low at the IBP binding sites including those flanking H3K27me3 domains (Emberly et al, 2008; Bartkuhn et al, 2009; Cuddapah et al, 2009), yet it remains unknown whether specific histone modifiers or chromatin remodelers play a role in this context. Whether chromatin barriers involve a division of labor through distinct IBPs, cofactors or distinct chromatin regulators required for barrier activity, remains open questions.
Here, we report that Beaf32 depletion specifically regulates the expression of hundreds of genes flanked by marked Nucleosome Free Regions (NFRs). Biochemical purification of Beaf32 identifies nuclear SET domain-containing proteins (NSD)/Drosophila Maternal-effect sterile gene 4 (dMes-4), an essential HMT needed for dimethylation of histone H3 on lysine 36 (H3K36me2) and involved in recruiting histone acetyltransferases (HATs) (Bell et al, 2007). Strikingly, dMes-4 depletion recapitulates gene expression defects upon Beaf32 depletion, supporting its role as a co-regulator. H3K36me2/3 genome-wide patterns tightly correlate with NFRs/nucleosome positioning, respectively, in complete agreement with recent data showing the role of H3K36 methylation in regulating histone exchange (Venkatesh et al, 2012). Beaf32/dMes-4 depletions impair subsequent recruitment of DREF, a key transcriptional activator required for Set2/Hybp-dependent H3K36me3. As such, H3K36me2 presets chromatin for transcription-coupled, H3K36me3-dependent nucleosome positioning that is further implicated in regulating H3K27me3 spreading. Taken altogether, our results suggest a pivotal role of H3K36 HMTs in regulating the expression of genes flanked by IBPs, through chromatin dynamics.
Results
Beaf32 differentially regulates genes harboring marked NFRs flanking high nucleosome positioning
Drosophila IBP sites including Beaf32 and dCTCF or GAF were shown to be highly enriched within marked Nucleosome Free Regions (NFRs) (Emberly et al, 2008; Gurudatta & Corces, 2009; Jiang et al, 2009; Negre et al, 2010; Mukhopadhyay et al, 2011). Similarly, human CTCF sites were shown to overlap with as much as 28% of the mapped DNase I hypersensitive sites (DHSs) linking transcription programs to such chromatin landscape (Natarajan et al, 2012; Thurman et al, 2012). In the case of Drosophila, the binding sites of IBPs often localize near promoters (reviewed in Raab & Kamakaka, 2010; Vögelmann et al, 2011), as illustrated by ChIP-Seq data of Beaf32 (Fig1A; < 500 bp from TSS; P-value < 1e-300), prompting us to test whether such genomic features reflect a role of IBPs in regulating gene expression through chromatin organization.
Figure 1. Beaf32-mediated regulation of gene expression correlates with high NFR and nucleosome positioning.
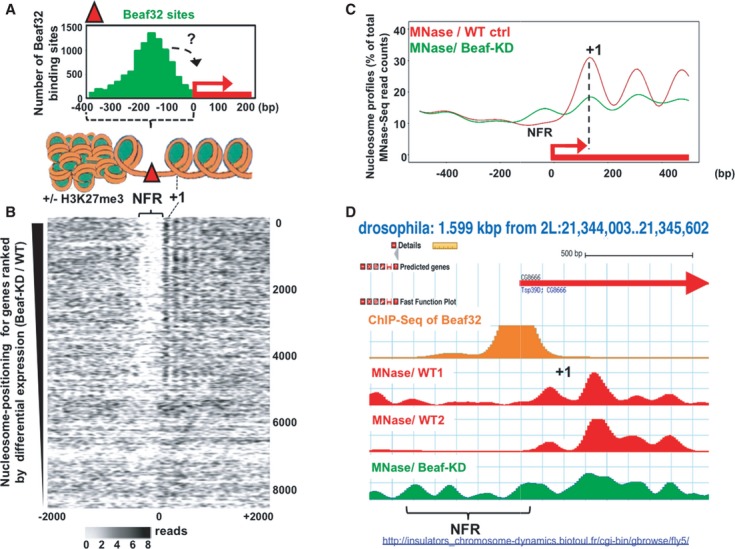
- Histogram representing the mapping of Beaf32 sites identified by ChIP-Seq using specific anti-Beaf32 antibodies (Supplementary Fig S1A) with respect to promoters (TSS = 0) and to chromatin structure (see scheme illustrating the results obtained by MNase-Seq in B). “+/− H3K27me3” indicates that Beaf32 sites are enriched but not necessarily flanking H3K27me3/heterochromatin domains as previously shown (Sexton et al, 2012).
- Heat map of nucleosome positioning as measured by MNase-Seq (see Materials and Methods) along the promoter regions of genes ranked according to their differential expression between wild-type control (“WT ctrl”) or Beaf32-depleted (“Beaf32KD”) cells (top: most differentially expressed; bottom: similarly expressed genes). Nucleosomes were aligned relative to TSS (x-axis; position 0) of genes. Nucleosome positions are indicated by a gradient proportional to the distribution of MNase-Seq reads (see Materials and Methods), as previously done (Schones et al, 2008). NFR, Nucleosome Free Regions; +1, positioning of the first nucleosome after the TSS of genes (see also Supplementary Fig S2A).
- Averaged nucleosome positioning as determined by MNase-Seq in Beaf32-KD (green curve) and control cells (red curve). Nucleosome positions (y-axis, number of MNase-Seq reads) were aligned along the TSS of genes (x-axis, TSS = position 0) where most significant changes in nucleosome positioning were scored (˜2,000 genes; see Materials and Methods). Note that genes harboring most significant changes in nucleosome positioning upon Beaf32-KD are highly enriched among Beaf32 bound genes (see Supplementary Fig S2B-C).
- Example illustrating the variations in nucleosome positioning from our MNase-Seq data accessible through http://insulators_chromosome-dynamics.biotoul.fr/IBPs as observed in Beaf32-KD compared to control cells from this study (“WT1”) or an independent study (“WT2”) (Gilchrist et al, 2010). NFR, Nucleosome Free Regions; +1, positioning of the first nucleosome after the TSS of genes.
In such contexts, Beaf32 may regulate hundreds of genes as shown by genome-wide expression analyses (Supplementary Fig S1). These data were obtained through Digital Gene Expression-sequencing (DGE-Seq) in S2 cells depleted of Beaf32 (Beaf32-KD ˜95% efficiency) as compared to mock-depleted control cells (“WT control”), and the impact of Beaf32 on gene expression could be verified by RT-qPCR and microarray analyses (Supplementary Fig S1F and G). Intersecting expression data with ChIP-Seq data showed that 57% of the differentially expressed (DE) genes harbor a Beaf32 binding site in their promoter (P-value = 1e-291). 28.7% of the promoters harboring a Beaf32 site were differentially regulated as compared to 7.9% without such site. The fold changes in expression as measured by DGE-Seq were limited (WT/Beaf32-KD ˜twofold on average) showing a moderate influence of Beaf32, as expected if it is not a transcription activator (Cuvier et al, 1998). Supporting this interpretation, no effect of Beaf32 depletion was detected using in vitro transcription assays choosing Beaf32 promoters as naked DNA templates (unpublished data).
To investigate whether IBPs such as Beaf32 regulate gene expression through their role in chromatin organization, we performed MNase-Seq (see Materials and Methods) (Barski et al, 2007; Schones et al, 2008; Gilchrist et al, 2010) and ranked genes according to the influence of Beaf32 on their expression (Fig1B). The generated heat maps highlighted a good correlation between DE genes upon Beaf32-KD and the presence of NFRs or of high nucleosome positioning signals in promoters or in gene bodies, respectively (Fig1B, NFR/“+1”), providing Beaf32 was bound to promoters (Supplementary Fig S2A). In agreement, high nucleosome positioning has been observed for highly active, housekeeping genes such as those regulated by Beaf32/DREF or dCTCF (Emberly et al, 2008; Bushey et al, 2009; Gilchrist et al, 2010).
Beaf32-KD significantly affected nucleosome positioning in approximately 2,000 genes (Fig1C, see “+1” for “Beaf32KD”), as evidenced by changes in MNase-Seq reads along their bodies upon Beaf32-KD compared to control cells (see Materials and Methods). By contrast, the averaged nucleosome levels slightly increased in the corresponding NFRs (Fig1C), as illustrated by the tsp39D gene promoter region (Fig1D; NFR). Such variations upon Beaf32-KD were not systematically found in every gene flanked by a Beaf32 binding site (differentially expressed or not). The thousands of genes with marked changes in nucleosome positioning were however specifically enriched among genes flanked by a Beaf32 binding site as well as genes differentially regulated upon Beaf32-KD (Supplementary Fig S2B and C), supporting the functional implication of Beaf32 in regulating nucleosome dynamics.
Nucleosome positioning may function in regulating gene expression by preventing spurious transcription (Carrozza et al, 2005; Gilchrist et al, 2010) or RNA splicing (reviewed in Schwartz & Ast, 2010). In agreement, further RNASeq analysis in Beaf32-KD highlighted increasing levels of spurious/aberrant intronic RNASeq reads compared to WT cells (Supplementary Fig S2D). This phenomenon was specifically encountered for genes bound by Beaf32 or where significant changes in nucleosome positioning were scored upon Beaf32-KD (P-values = 1e-37 and 1e-3, respectively). Aberrant transcripts were found in as much as 48% of the differentially regulated genes harboring nucleosome positioning changes (226/469 genes; P-value = 3e-21; see below). Our data therefore strengthen the view that the function of Beaf32 in regulating gene expression may be linked to nucleosome dynamics, which very likely involves additional cofactors.
Beaf32 interacts with the histone methyltransferase dMes-4
Genome-wide analysis by ranking genes according to the amount of ChIP-Seq reads of Beaf32 in their promoters confirmed that Beaf32 binding in the top quartile (q1) correlated with marked NFRs in promoters as well as high nucleosome positioning over gene bodies (Fig2A). We thus sought to identify its cofactors through high-salt elution of factors biochemically co-purified with the Beaf32 complex (Fig2B; see Materials and Methods). The specificity of the assay was confirmed by the presence of the Beaf32 doublet and of CP190, a key cofactor that interacts with all types of Drosophila IBPs including dCTCF (Bushey et al, 2008; Wood et al, 2011; Liang et al, 2014). In addition, this analysis further identified a unique histone modifier, dMes-4 (Maternal-effect sterile 4; Fig2B; see arrow), an essential HMT that dimethylates lysine 36 of histone H3 (H3K36me2) (Bell et al, 2007) involved in epigenetic mechanisms (Pirrotta, 2002). Co-immunoprecipitation experiments using anti-Beaf32 antibodies confirmed the specific interaction of Beaf32 and dMes-4 (Fig2C) compared to H3 loading control, as confirmed by two-hybrid assays (Supplementary Fig S3A). Ranking of genes according to the ChIP-Seq reads of Beaf32 highlighted a genome-wide correlation between its binding and H3K36me2 levels in promoters (Fig2D) as measured by ChIP-Seq (see Materials and Methods). Beaf32-KD actually led to a reduction of approximately 50% of the chromatin levels of H3K36me2/me3 compared to control cells (Fig2E), unlike what was found for the soluble pool of H3K36 (Supplementary Fig S3B and C). H3K36me2/3 chromatin levels are readily dependent on dMes-4 as shown (Fig2E, left blot) (Bell et al, 2007). ChIP analysis showed that Beaf32-KD impaired the recruitment of dMes-4 specifically for promoters bound by Beaf32 compared to control promoters (Fig2F), in complete agreement with data showing that Beaf32-KD decreased H3K36me2 levels in such promoters as compared to control cells (Supplementary Fig S3E).
Figure 2. Beaf32 interacts with NSD/dMes-4.

- Averaged nucleosome positioning for genes ranked by quartiles defined according to the amount of ChIP-Seq reads of Beaf32 in their promoters (highest to lowest, quartile q1–q4, respectively; see Materials and Methods). Note that each quartile includes approximately 3,650 (± 5) genes (the top quartile q1 is mostly enriched in Beaf32 ChIP-Seq reads). y-axis, number of MNase-Seq reads; x-axis, alignment along the TSS of genes (TSS = position 0).
- Biochemical purification of Beaf32 complex identifies dMes-4 (see arrow) as a novel cofactor. Beaf32 (see doublet at the bottom) and cofactors were purified by affinity chromatography using anti-Beaf32 antibodies (Supplementary Fig S1) in parallel with control IgG (see additional source data; http://insulators_chromosome-dynamics.biotoul.fr/IBPs). Bound proteins were eluted by high-salt extraction (see Materials and Methods) for further mass-spectrometric analysis. 100/33 peptides corresponded to CP190/dMes-4 as the major cofactors eluted (versus 1/0 peptide for IgG control, respectively) with no other histone modifier present.
- Co-immunoprecipitation experiments using affinity-purified anti-Beaf32 in parallel with control IgG followed by Western blotting analysis using anti-Beaf32, -dMes-4, -CP190 or -histone H3 as a control, compared to input nuclear extract (10% “input”) (see additional source data; see also Supplementary Fig S3).
- Box plot showing the enrichment in H3K36 dimethylated levels for genes ranked according to the ChIP-Seq reads of Beaf32 in their promoters as previously (A). y-axis, log ratio for the enrichment in H3K36me2 levels for each quartile.
- Western blotting analysis of the chromatin-bound fractions purified through sucrose cushions (see Materials and Methods) from dMes-4- or Beaf-depleted cells showing the levels of histone H3 di- and tri-methylated on lysine 36 (H3K36me2 or H3K36me3, respectively) compared to total histone H3 levels (see also Supplementary Fig S3).
- Box plot showing the results of ChIP experiments showing dMes-4 recruitment to 16 promoters harboring a Beaf32 site or not (see Materials and Methods for a list) in Beaf32-KD (red boxes) compared to WT control (mock-depleted) cells (green boxes) in percent of input (y-axis) as obtained by performing ChIP with anti-dMes-4 antibodies or IgG control and after normalization to three control loci (see Materials and Methods). Samples were analyzed by qPCR in triplicates and for three independent measures (see also Supplementary Fig S3).
- Venn diagram showing the genome-wide intersection between sites harboring high H4K16 acetylation and Beaf32 sites.
- Box plot representing the ChIP data obtained with specific anti-acetylated H4K16 in control or Beaf32-KD cells for two independent measures for 16 promoters harboring a Beaf32 binding site or not 16 promoters (see Materials and Methods for a list). The P-values (P-value = 0.14 and 1e-7) were obtained by a Wilcoxon test for the difference between WT and Beaf32-KD (see also Supplementary Fig S3).
Source data are available online for this figure.
dMes-4-driven methylation is required for histone acetylation by favoring the recruitment of histone acetyltransferases (HATs) (Bell et al, 2007; Venkatesh et al, 2012). This may account for the observed enrichment of Beaf32 binding sites among genomic sites harboring high acetylated histone levels as shown for H4K16ac (Fig2G). ChIP of H4K16ac in Beaf32-KD showed a significant reduction of H4K16ac levels compared to control cells (Fig2H; P-value = 1e-7), strengthening its implication in chromatin accessibility. In agreement, intersection analyses suggested that genes differentially expressed upon Beaf32-KD were also highly enriched among those harboring high acetylation levels (Supplementary Fig S3D). Altogether, our data supported a functional interplay between Beaf32 and dMes-4 as a key co-regulator of chromatin dynamics, as further addressed below.
dMes-4 is a key co-regulator of Beaf32 in gene expression
The functional interplay of dMes-4 and Beaf32 was addressed by genome-wide expression analyses after efficient depletion of either factor (Fig3A, “dMes-4-KD” and “Beaf32KD”, respectively; Supplementary Fig S3F–H). Strikingly, a large overlap was found between genes whose expression was impaired by Beaf32 and differentially expressed (DE) genes upon dMes-4-KD (Fig3B), strongly supporting a key role of dMes-4 as a co-regulator of Beaf32. Accordingly, DE genes upon dMes-4-KD were largely enriched in the same gene ontologies as found upon Beaf32-KD, including the cell cycle and/or cell death (Supplementary Fig S4A). Such regulations likely implicate DREF as a transcriptional activator enriched within Beaf32 sites (Supplementary Fig S4B; see below) (Emberly et al, 2008) or dCTCF that shares many binding sites with Beaf32 and that regulates similar cellular functions (Bushey et al, 2009; Gurudatta et al, 2013).
Figure 3. dMes-4 depletion recapitulates the gene expression defects upon Beaf32-KD.
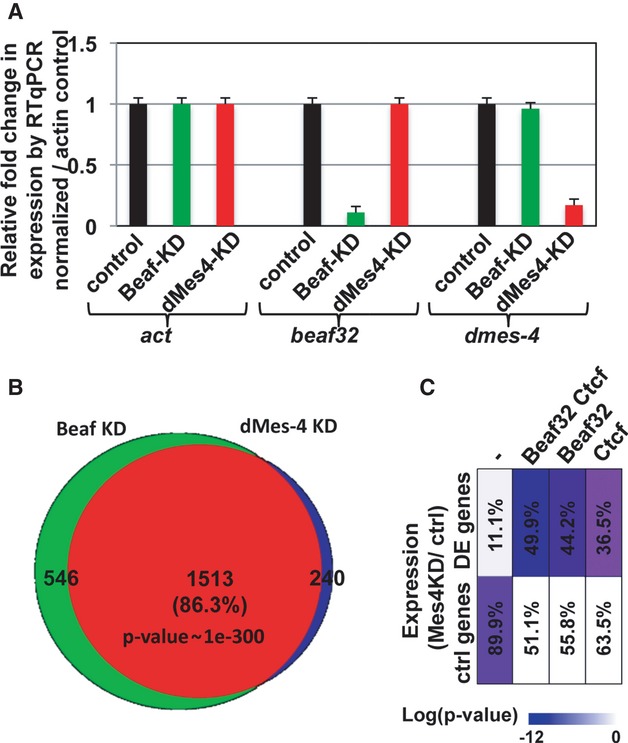
- Graph showing the expression levels of dmes-4 or beaf32 normalized to actin control as measured by three independent RT-qPCR analyses in dMes-4-KD, Beaf32-KD, or control cells (see also Supplementary Fig S3 for confirmation of dMes-4 depletion by Western blotting).
- Venn diagram showing the intersection between differentially expressed genes in Beaf32-KD or dMes-4-KD compared to control cells as obtained by genome-wide expression analyses (see Materials and Methods). Note that differentially expressed genes are enriched in specific gene ontologies (see Supplementary Fig S4; Supplementary Table S1).
- Graphical representation of the enrichment in differentially expressed genes upon dMes-4 depletion as a function of the presence of unique or multiple binding site(s) for the IBPs dCTCF and Beaf32 (see also Supplementary Fig S4E). Note that many gene promoters are flanked by both dCTCF and Beaf32 binding sites as shown (Bushey et al, 2009). The scale bar represents the enrichments as the log of the P-values. The percentages of differentially regulated genes (dMes-4KD/WT) are also indicated for each category of promoters.
Given the overlapping binding sites between Beaf32 and dCTCF, we then tested whether dMes-4 might specifically influence genes that are flanked by dCTCF sites. The percentage of DE genes uniquely bound by dCTCF and under the influence of dMes-4 was lower compared to those bound by Beaf32 (Fig3C; 36.5 and 49.9%, respectively). The influence of dMes-4 on genes flanked by dCTCF binding sites was however specific (207 genes uniquely bound by dCTCF; P-value = 1e-8) as confirmed by the enrichment in dCTCF sites in the promoters of genes uniquely regulated by dMes-4 but not Beaf32 (122 genes; 50.8%; P-value = 1e-70). Such specificity was confirmed by inspecting our data at various threshold settings using receiver operating characteristic (ROC) curve analysis (Supplementary Fig S4E). We therefore conclude that dMes-4 is an important co-regulator of Beaf32 and dCTCF, which may in part account for high expression levels of genes flanked by IBP sites (Bushey et al, 2009; Negre et al, 2010).
H3K36me2/me3 patterns correlate with NFRs and nucleosome positioning
dMes-4-mediated H3K36me2 appears to be a key mechanism for the recruitment of various HATs that would favor nucleosome remodeling/eviction over NFRs (Bell et al, 2007; Venkatesh et al, 2012). We thus sought to test its impact on nucleosome positioning. Heat maps generated by ranking genes according to the ChIP-Seq counts of H3K36me2 (see Materials and Methods) showed a good correlation between H3K36me2 levels and the presence of NFRs in promoter regions (Fig4A), as confirmed by inspection of averaged nucleosome profiles (Fig4C). H3K36me2 levels further correlated with nucleosome positioning (Fig4A and C), yet a more significant correlation was detected by ranking genes with H3K36me3 levels (Fig4, compare panel A and B, C and D). These results are highly consistent with recent findings showing that by preventing interaction of histone H3 with chaperones, H3K36me3 prevents histone exchange along gene bodies (Venkatesh et al, 2012), which may in turn drive nucleosome positioning as shown (Fig4B and D).
Figure 4. dMes-4 prone Set-2/Hypb H3K36me3-mediated nucleosome positioning.
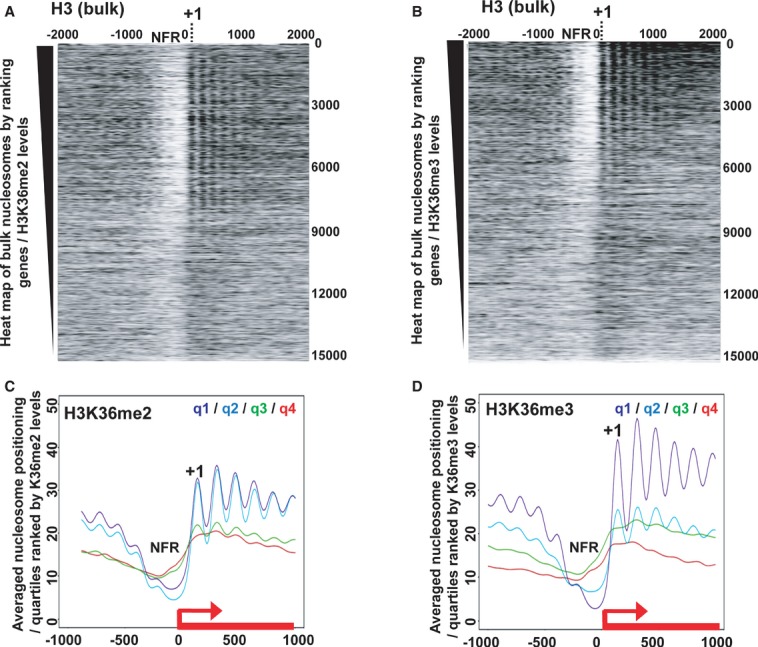
- Heat map of nucleosome positioning as measured by MNase-Seq along all Drosophila genes ranked according to H3K36me2 levels. Top: highest H3K36me2 levels in wild-type cells. Nucleosomes were aligned relative to TSS (x-axis; position 0) of genes. NFR, Nucleosome Free Region; +1, positioning of the first nucleosome.
- Same as in (A) except that genes were ranked according to H3K36me3 levels.
- Averaged nucleosome positioning for genes ranked according to quartiles defined by the amount of ChIP-Seq reads of H3K36me2 (from q1/highest to q4/lowest levels; see Materials and Methods). y-axis, number of MNase-Seq reads; x-axis, alignment along the TSS of genes (TSS = position 0).
- Same as in (C) except that quartiles were defined according to H3K36me3 levels.
Tri-methylation of H3K36 occurs over the bodies of genes, and it requires the HMT Hypb/Set2 family (Bell et al, 2007). Beaf32 binds to promoters and it may not be directly responsible for nucleosome positioning over gene bodies. dMes-4 is however pre-required for subsequent tri-methylation of H3K36 (Bell et al, 2007) that further involves Hypb/Set2-mediated H3K36me3 upon transcription elongation (Joshi & Struhl, 2005; Govind et al, 2010) (see below). As such, Beaf32/dMes-4 may preset chromatin for subsequent H3K36me3-driven nucleosome positioning. Supporting this view, DE genes upon dMes4-KD were enriched among genes harboring high nucleosome positioning in wild-type cells (Supplementary Fig S5A), as well as genes with defects in nucleosome positioning upon Beaf32-KD (Supplementary Fig S5B). These DE genes were mostly enriched in high H3K36me2 levels even in the absence of Beaf32 (Supplementary Fig S5C), supporting a strong linkage between this mark and the impact of dMes-4 on gene expression. Higher H3K36me3 levels were found in DE genes upon dMes-4-KD, providing Beaf32 was bound. In agreement, DE genes by dMes-4-KD harbored high levels of nucleosome positioning when Beaf32 was bound to their promoters, as shown in wild-type cells (Supplementary Fig S5A and D). Taken altogether, these data suggested that Beaf32/dMes-4 are involved in presetting chromatin for subsequent H3K36me3 deposition upon transcriptional activation, as addressed below.
Beaf32/dMes-4 preset chromatin for subsequent regulation of RNA splicing
We next sought to test whether interactions between Beaf32 and dMes-4 are required for subsequent deposition of H3K36me3 and/or for nucleosome positioning. Ranking of genes according to Beaf32 ChIP-Seq reads in promoters showed a tight correlation with H3K36me3 levels in the corresponding gene bodies (Supplementary Fig S6A). ChIP analysis showed no significant variations in H3K36me3 levels upon Beaf32-KD compared to control cells, for promoters bound by Beaf32 or not (Supplementary Fig S6A and B). Genome-wide, little variations of H3K36me3 levels may be found in promoter regions (Supplementary Fig S6C, see arrow), however, which may reflect an equilibrium between dMes-4/Hybp HMTs and demethylases that are recruited to promoter regions (Lin et al, 2012) (see Discussion). By contrast, H3K36 methylation decorates gene bodies (Supplementary Fig S6C) (Bell et al, 2007; Kolasinska-Zwierz et al, 2009) where H3K36me3 levels decreased upon Beaf32-KD providing the gene was flanked by a Beaf32 site (Fig5A; P-value = 1e-4), which would affect nucleosome positioning. Strongly supporting this hypothesis, the nucleosome positioning defects scored upon Beaf32-KD tightly correlated with H3K36me3 levels (Fig5B, red bars) and to a lesser extent with H3K36me2 (Fig5B, grey bars). Altogether, these data therefore strongly supported that Beaf32 binding to promoters is required for subsequent transcriptional elongation, thereby mediating H3K36me3-coupled nucleosome positioning (see below).
Figure 5. Beaf32-KD decreases H3K36 tri-methylation thereby affecting RNA maturation.
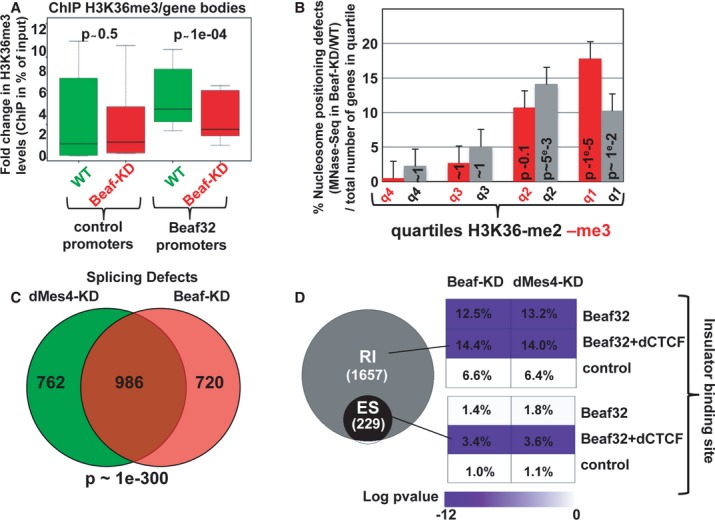
- Box plot showing the results of ChIP experiments showing the H3K36me3 levels in Beaf32-KD (red boxes) compared to control cells (green boxes) in percent of input (y-axis) as obtained by performing ChIP with anti-H3K36me3 antibodies or IgG control and after normalization to three control loci (see Materials and Methods). Samples were analyzed by qPCR analyses in triplicates and for three independent measures for the bodies of genes whose promoters were bound by Beaf32 or not (see Materials and Methods for a list; see also Supplementary Fig S6).
- Histogram showing the percentage of genes with nucleosome positioning defects as measured by MNaseSeq read counts in Beaf32-KD compared to WT cells as previously (see Supplementary Fig S2; Materials and Methods), as a function of the H3K36-me2 or -me3 quartiles. The error bars represent the variations found between two independent measures. The number of genes corresponding to four quartiles is 204/355, 281/213, 101/53, and 44/9 for H3K36-me2/-me3, respectively.
- Venn diagram showing the intersection analysis of genes with splicing defects (see Materials and Methods) upon dMes4-KD (left) or Beaf32-KD (right) as compared to control cells.
- Venn diagram showing the specific RNA splicing defects (expressed in log P-value) measured upon Beaf32-KD compared to WT cells. Splicing defects were scored according to the presence or not of Beaf32 sites alone or together with dCTCF sites as indicated, both for defects identified as “retained introns” (RI) or for defects specifically associated with alternative splicing defects, identified as “exon skipped” (ES). The percentage indicate the number of genes with splicing defects over the total number of genes in each category, that is with Beaf32 and/or dCTCF sites or not, as measured upon Beaf- and dMes4-KD compared to WT control (mock-depleted) cells.
H3K36 methylation may serve to drive nucleosome positioning, thereby preventing spurious transcription and the presence of aberrant RNAs as shown (Supplementary Fig S2D) (Carrozza et al, 2005). Alternatively, such aberrant transcripts (scored by counting RNASeq counts outside exons) may be related to the influence of H3K36me3-coupled transcriptional elongation on RNA splicing (Kolasinska-Zwierz et al, 2009; Schwartz et al, 2009; Luco et al, 2011; Shukla et al, 2011). H3K36 marks may therefore play a pivotal role coupling nucleosome positioning and RNA splicing (reviewed in Schwartz & Ast, 2010). Genome-wide analysis of RNASeq reads using DiffSplice (Hu et al, 2013) showed a significant accumulation of reads from unspliced RNAs upon Beaf32-KD compared to control cells (Fig5C), which was specific for genes bound by IBPs (P-value = 1e-19), as confirmed by RT-qPCR analyses using oligos that span exon–intron junctions (Supplementary Fig S7A). They were tightly correlated with H3K36me3 levels over gene bodies, varying from < 3 to approximately 50% (Supplementary Fig S7B, red bars) and to a lesser extent with H3K36me2 levels (Supplementary Fig S7B, grey bars). In complete agreement, a high proportion of the splicing defects observed in Beaf32-KD were also found upon dMes4-KD compared to WT cells (> 57%; P-value = 1e-300; Fig5C). They were specifically encountered for genes flanked by a Beaf32 together or not with a dCTCF binding site (Fig5D; “RI”, retained introns; 391 and 242 genes, respectively), strengthening the overall implication of IBPs in RNA splicing through dMes4-mediated H3K36 methylation. Specific alternative splicing defects were also scored upon Beaf32-KD giving rise to 229 alternative transcripts with skipped exons (Fig5D, “ES”), similar to what was reported for murine CTCF in alternative splicing (Shukla et al, 2011). The alternative splicing defects detected upon Beaf32-KD were specific of genes harboring both Beaf32 and dCTCF binding sites (Beaf+/− dCTCF; P-value = 1e-6 and 1, respectively), which may be related to the role of murine CTCF in alternative splicing (Shukla et al, 2011). Such defects were also encountered upon dMes4-KD in the presence of dCTCF sites (52 genes; P-value = 1e-6) in complete agreement with our results implicating H3K36 methylation in IBP-mediated regulation of gene expression. Altogether, our data therefore show a role of insulator proteins in RNA splicing involving their interaction with the H3K36 HMTs.
Beaf32/dMes-4 are required for the recruitment of the transcriptional activator DREF
Our data show that the observed defects upon Beaf32-KD involve H3K36me3 deposition. This may likely involve the HMT dHypb/Set2 whose activity has been associated with transcription activation/elongation (Joshi & Struhl, 2005; Govind et al, 2010). The binding sites of Beaf32 largely overlap with that of the transcriptional activator DREF (Emberly et al, 2008; Gurudatta et al, 2013) and we thus sought to characterize the interplay between Beaf32/dMes-4 or DREF and H3K36 methylation.
ChIP analysis showed that Beaf32-KD specifically impaired the recruitment of DREF to Beaf32 bound promoters as compared to control cells (Fig6A), which was not found for control promoters. Of interest, dMes4-KD impaired the recruitment of DREF to Beaf32 bound promoters as compared to control promoters (Fig6B), supporting its role in opening chromatin. The reduction in DREF recruitment was more significant upon Beaf32-KD as compared to dMes-4-KD, which may involve direct interactions of DREF with Beaf32 as suggested by co-immunoprecipitation experiments (Supplementary Fig S8F). In contrast to Beaf32-KD, DREF-KD did not affect the recruitment of dMes-4 (Supplementary Fig S8E). Curiously, its depletion was accompanied by a significant increase in H3K36me2 levels for promoters bound by Beaf32 (Fig6C), similarly to the depletion of HypB (Supplementary Fig S6D). These results raise the possibility that DREF favors the Hypb-mediated H3K36me2 to H3K36me3 transition, upon transcriptional activation. Strongly supporting this view, DREF-KD led to a significant decrease over gene bodies of H3K36me3—but not H3K36me2—levels, as compared to control cells (Fig6D and E). Moreover, ranking of genes according to their relative enrichment in H3K36me3–over H3K36me2 marks showed a tight correlation with the elongation rate of RNA polymerase II (Fig6F; see Materials and Methods), showing that H3K36me3-mediated nucleosome positioning reflects transcriptional elongation.
Figure 6. H3K36me3 deposition requires Beaf32/dMes-4-dependent recruitment of a transcriptional activator such as DREF.
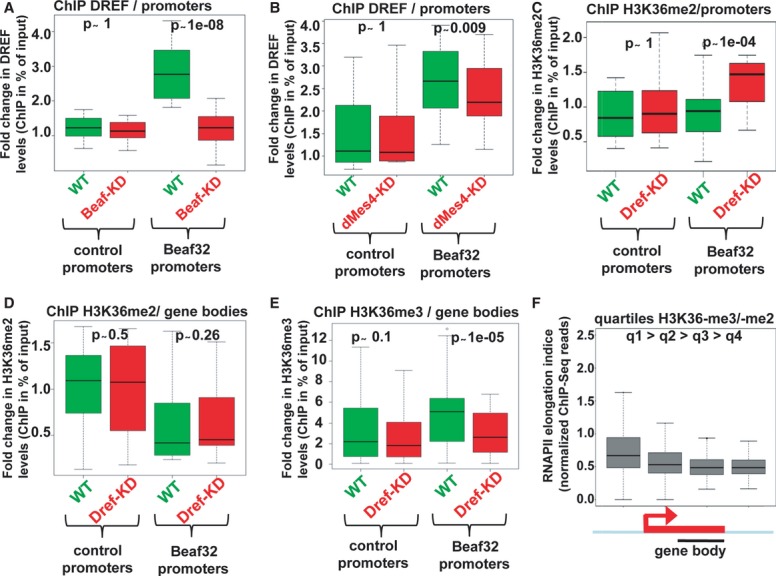
- Box plot showing the results of ChIP experiments showing the DREF levels in Beaf32-KD (red boxes) compared to control cells (green boxes) in percent of input (y-axis) as obtained by performing ChIP with anti-DREF antibodies or IgG control and after normalization to three control loci (see Materials and Methods). Samples were analyzed by qPCR analyses in triplicates and for three independent measures in 16 control promoters or in Beaf32 bound promoters (see Materials and Methods for a list).
- Same as in (A) except that ChIP of DREF was performed in dMes4-KD (red boxes) compared to control cells (green boxes) for the same genes.
- ChIP of H3K36me2 in DREF-KD (red boxes) compared to control cells (green boxes) in percent of input (y-axis) for the same promoters as in (A) and (B).
- Same as in (C) except that levels were measured in the bodies of the same genes bound by Beaf32 or not.
- Same as in (D) except that ChIP was performed using anti-H3K36me3 or IgG antibodies for the same genes bound by Beaf32 or not.
- Box plot showing RNA polymerase II elongation indices as estimated by measuring the log ratio of ChIP-Seq reads found in gene bodies over reads in promoters (see Materials and Methods) as previously (Rahl et al, 2010). Genes were ranked according to the ratio of H3K36me3 over H3K36me2 levels as measured by ChIP-seq (q1/high to q4/low; see Materials and Methods).
Taken altogether, our result therefore shows that IBP/dMes-4 play an important role by coupling the recruitment of transcriptional activators, such as DREF, to the presetting of chromatin through H3K36me2 that is required for subsequent H3K36me3-mediated RNA splicing (see Discussion).
Beaf32 and dMes-4 regulate H3K27me3 spreading
The tight correlation among the various genomic features including IBP binding, H3K36me2/3, and the presence of NFRs was statistically relevant as shown by clustering analyses (Fig7A), in stark contrast with the clear anti-correlation of these features with H3K27me3 levels (Supplementary Fig S9A). IBP binding sites are enriched at the borders of H3K27me3 domains (Sexton et al, 2012), prompting us to test the function of Beaf32/dMes-4 in regulating H3K27me3 spreading. Our MNase-Seq analysis followed by ChIP-Seq of H3K27me3 showed a significant increase in the averaged levels of H3K27me3 upon Beaf32-KD as compared to control mock-depleted cells, for a limited subset of 990 genes (Fig7B). Of the promoters bound by Beaf32, 11.9% harbored higher H3K27me3 levels upon Beaf32-KD as compared to only 4.1% without a Beaf32 site (Fig7C; P-value = 1e-12 and 1, respectively). Beaf32 specifically protected genes from H3K27me3 spreading as 49.5% (480/990) of the genes with higher H3K27me3 levels upon Beaf-KD corresponded to genes bound by Beaf32. Furthermore, a significant proportion of the genes exposed to H3K27me3 spreading intersected with the genes with increasing nucleosome levels within NFRs (326/990 genes; P-value = 1e-6; Fig1) highlighting a good correlation between Beaf32/dMes-4-driven chromatin dynamics and such phenotype. Genes exposed to H3K27me3 spreading were further enriched among the differentially expressed genes (˜44%), as compared to genes with Beaf32 binding yet without no variation in H3K27me3 levels upon Beaf32-KD (˜26%; Fig7D; see also Supplementary Fig S10A-C). Similarly, genes exposed to H3K27me3 spreading upon Beaf32-KD were more enriched in differentially regulated genes upon dMes4-KD as compared to genes with no variations in H3K27me3 (52 versus 33%; Fig7D).
Figure 7. Influence of IBP/dMes-4 on H3K27me3 deposition.

- Dendogram representing the genome-wide correlations among distinct genomic features including H3K36 methylation and nucleosome positioning as a function of Beaf32/DREF binding (right) or not (left). The dendogram reflects the minimal variance (Ward distance) (see Materials and Methods) among all indicated genome-wide data by ChIP-Seq (Beaf32, H3K36me1/me2/me3 H4K16ac) or MNase-Seq (nucleosome positioning, NFR) after running principal component analyses for genes harboring Beaf32 binding sites or not. See also Supplementary Fig S9A.
- Averaged H3K27me3 profiles for 990 promoter regions where most significant variations in H3K27me3 levels were scored between Beaf32-KD and control (WT) cells as measured by ChIP-Seq (see Materials and Methods). Note that such variations were systematically scored over ± 2 Kbp regions surrounding all Drosophila TSSs. See Supplementary Fig S10A for the H3K27me3 profiles of the complementary list of genes showing no variation in H3K27me3 levels upon Beaf32-KD.
- Histogram representing the percentage of genes with increasing H3K27me3 levels in Beaf32-KD/WT (y-axis) depending on the presence or absence of a Beaf32 binding site in their promoters. See Supplementary Fig S10B for a similar analysis of control genes (with no variation in H3K27me3 levels).
- Histogram showing the intersection analysis between the list of genes with variations in H3K27me3 levels (“Beaf32-KD > WT”; panel A) or not (“Beaf32-KD˜WT”; Supplementary Fig S10A) and the list of DE genes upon Beaf32-KD or dMes4-KD. The percentage of DE genes was scored depending on variations or not in H3K27me3 levels, for promoters with or without a Beaf32 binding site. See also Supplementary Fig S10C.
- Box plot showing the fold changes in H3K27me3 levels as measured by qPCR of ChIP with anti-H3K27me3 antibodies or IgG control in dMes4-KD (red boxes), Beaf32-KD (green boxes) compared to control cells (grey boxes) normalized to input (y-axis). Samples were analyzed by qPCR analyses in triplicates and for three independent measures of Beaf32 bound promoters or control promoters (see Materials and Methods for a list). See also Supplementary Fig S10D for a correlation between Beaf32-KD and dMes4-KD.
Our data thus suggested a role of Beaf32 and dMes-4 in controlling the deposition of H3K27me3, in agreement with recent data involving NSD/MES4 in the regulation of H3K27me3 (see Discussion) (Yuan et al, 2011; Gaydos et al, 2012). Accordingly, ChIP analysis of H3K27me3 upon depletion of dMes-4 or of Beaf32 showed a reproducible and significant increase in H3K27me3 levels as compared to control cells (Fig7E). The effect of dMes4-KD on H3K27me3 levels was specific of promoters harboring a Beaf32 binding site as compared to control promoters (P = 1e-4 and 1, respectively), accounting for the good correlation between the impact of dMes4-KD on H3K27me3 levels, compared to that of Beaf32-KD (Supplementary Fig S10D). Taken altogether, our results suggest that the influence of Beaf32/dMes-4 on H3K27me3 deposition may reflect their activity in chromatin dynamics, yet it may not condition their more general influence on gene expression (see Discussion).
Discussion
The identification of dMes-4 as a novel cofactor interacting with IBPs shed new light into how they may impact gene expression through chromatin dynamics. Such regulations involve cycles of NSD/dMes-4-mediated H3K36me2 followed by Set2/Hypb-mediated H3K36me3 (Venkatesh et al, 2012) together with the recruitment and/or activation of various HATs (Bell et al, 2007; Venkatesh et al, 2012) and HDACs (Barski et al, 2007; Schones et al, 2008) including Rpd3 (Joshi & Struhl, 2005; Govind et al, 2010). In turn, such cycles may provide with a highly dynamic regulation of chromatin locally, involving the eviction or fixation of histones/nucleosomes, respectively (Venkatesh et al, 2012), which appears to play a pivotal role in nucleosome positioning. These genomic features are statistically relevant (Fig7A; Supplementary Fig S9A), summarizing strong links between H3K36 di- or tri-methylation, NFRs, and nucleosome positioning, respectively, depending on the presence of insulator protein sites and of DREF (see model, Fig8).
Figure 8. Model for the role of IBP/dMes-4 in chromatin dynamics.
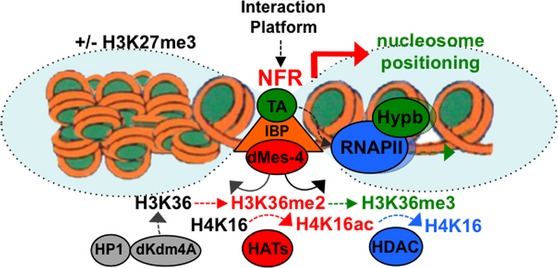
- Model representing how the interactions of the IBP cofactor dMes-4 may regulate the expression of genes through the controlled accessibility of chromatin by H3K36me2-dependent recruitment of HATs for acetylation followed by H3K36me3-coupled nucleosome positioning and HDACs activation, closing back chromatin upon elongation of Pol II. Note that H3K36me3 is most likely coupled with transcription elongation upon activated by transcription activators (“TA”) such as DREF whose binding sites are highly enriched among DE genes regulated by dMes-4, as well as in “active” genes harboring high nucleosome positioning (Gilchrist et al, 2010) (see Discussion). Note that H3K36 methylation levels in promoters further depend on the interaction of HP1 with the H3K36 demethylase dKdm4A as shown (Lin et al, 2012) (see text).
NSD/dMes-4 may account for the enrichment of “active” histone marks, NFRs, or DHSs at IBP sites including CTCF (Natarajan et al, 2012; Thurman et al, 2012). H3K36 methylated marks and IBPs are enriched at the borders of globular H3K27me3 domains (Gurudatta & Corces, 2009; Negre et al, 2010; Schwartz et al, 2012; Sexton et al, 2012). Our data further suggest that the role of IBPs in restricting H3K27me3 spreading involves the recruitment of dMes-4/NSD. H3K36 methylation actually antagonizes PRC2-dependent H3K27me3 as shown in HeLa cells (Yuan et al, 2011). In C. elegans, MES-4 interferes with the spreading of this repressive mark as shown (Gaydos et al, 2012), which may account for the global exclusion of H3K27me3 and H3K36me2/3 marks. Of interest, these two histone marks define bivalent nucleosomes that participate in controlling developmentally regulated genes through PRC2 recruitment (Cai et al, 2013). By interacting with H3K36 HMTs, IBPs like Beaf32 may thus regulate H3K27 deposition through a dynamic interplay with transcription-coupled chromatin dynamics involving H3K36 methylation.
dMes-4/Hypb-driven H3K36 methylation participates in the activation of housekeeping genes flanked by insulator sites that, unlike genes developmentally regulated “paused genes” (Gilchrist et al, 2010), harbor high nucleosome positioning over their bodies. dCTCF/Beaf32 sites flank promoters particularly enriched in genes that regulate specific cell functions including the cell cycle (Emberly et al, 2008; Bushey et al, 2009; Gurudatta et al, 2013). By regulating chromatin accessibility, IBP and dMes-4 may participate in the formation of NFRs, thereby favoring the recruitment of transcriptional activators including DREF. Beaf32/dMes-4-mediated H3K36me2 is pre-required for subsequent Hybp/Set2-driven tri-methylation of H3K36 upon transcription elongation (Bell et al, 2007; Kolasinska-Zwierz et al, 2009). This directionality is evidenced by increasing levels of H3K36me2 upon depletion of—DREF or more directly of—dHypb (Bell et al, 2007). Such mechanisms involving the evolutionary conserved factors, DREF, or dMes-4/NSD also identified as Wolf–Hirschhorn syndrome proteins (e.g. WHSC1), are likely to be essential for the regulation of genes involved in cell cycle and/or proliferation.
The role of IBPs/dMes-4 appears not limited to the regulation of nucleosome occupancy over NFRs as H3K36 methylation functions both in chromatin organization by triggering nucleosome positioning and in RNA splicing (Kolasinska-Zwierz et al, 2009; reviewed in Schwartz & Ast, 2010; Luco et al, 2011). H3K36me3 regulates RNA splicing by favoring the recruitment of splicing factors concomitantly with transcriptional elongation and vice versa, and splicing activity influences nucleosome organization (Keren-Shaul et al, 2013) raising the potent interplay between RNA splicing and nucleosome positioning through H3K36 marks (reviewed in Schwartz & Ast, 2010). The role of IBPs and/or dMes4/H3K36me2 may be to couple the recruitment of transcriptional activators such as DREF with the concomitant presetting of chromatin for Hypb-dependent H3K36me3 deposition, which requires prior H3K36me2 (Bell et al, 2007). Splicing defects may thus be exacerbated when transcriptional elongation is uncoupled from H3K36me3 (and by extension H3K36me2) deposition. Supporting this idea, DREF is required for H3K36me3—but not for H3K36me2—deposition along gene bodies, and its depletion has a lower impact on RNA splicing as compared to Beaf32 (Supplementary Fig S9B). Additional IBPs including CTCF and the insulator barrier protein Vezf1 were shown to affect alternative RNA splicing in other organisms including mouse (Shukla et al, 2011; Gowher et al, 2012), involving their ability to pause RNAPII by binding directly over exon–intron junctions. H3K36 marks are enriched within such genomic locations (Kolasinska-Zwierz et al, 2009) raising the possibility that CTCF-mediated regulation of alternative splicing similarly involves its interaction with dMes4/NSD-driven H3K36 methylation.
Only a sub-fraction of the binding sites of Beaf32 or dCTCF appears to influence H3K27me3 levels (Schwartz et al, 2012) (this work). The large overlap between Beaf32 and dCTCF binding sites may in part account for such limited impact, if these IBPs play a redundant function. Alternatively, the influence of IBPs on H3K27me3 may depend on the recruitment of additional cofactors of IBPs such as CP190, cohesin or chromator and their contribution to the physical organization of chromosomes into topological domains (Wood et al, 2011; Dixon et al, 2012; Hou et al, 2012; Sanyal et al, 2012; Phillips-Cremins & Corces, 2013). By regulating the chromatin accessibility, the interaction of IBPs with HMTs such as dMes-4 may act as a platform for the recruitment of additional IBP cofactors (see Fig8) as illustrated for cohesin loading (reviewed in Dorsett, 2011; Fasulo et al, 2012). A division of labor was suggested in the context of the beta-globin insulator, where USF1/2-VEZF and CTCF distinctly contribute to local chromatin accessibility involving histone acetylation and higher-order chromatin organization (Ghirlando et al, 2012). The division of labor is thus not limited to various sets of DNA–protein interactions, defining which IBP (e.g. dCTCF or Beaf32) interacts with chromosomal borders, but also extends to interactions with multiple cofactors that influence each other. How IBPs participate in restraining H3K27me3 spreading over domain borders may further reflect such a dependence on additional IBP cofactors participating in transcription-coupled chromatin dynamics, such as dMes-4.
dMes-4 is an histone modifier implicated in epigenetic mechanisms of germ line cells involving an interplay with additional HMTs of the Maternal-effect sterile (MES) or set family involved in X-chromosome regulations (Pirrotta, 2002). Such HMTs may dictate specific genomic landscapes contributing to regulate genes, including those associated with dosage compensation (Kharchenko et al, 2011; Regnard et al, 2011; Schwartz et al, 2012). The recruitment of dMes-4 by IBPs may thus provide with key information regarding genomic features specific to cell types (male or female germ cell lines) or to chromatin domains. Following dMes-4/HATs and dSET2/HDACs, the resulting chromatin is deacetylated, yet resetting H3K36 methylation additionally requires the JmjC-domain-containing factor dKdM4A that also demethylates H3K9 (Fig8) (Lin et al, 2012), which may be controlled by IBPs including Beaf32 (Emberly et al, 2008). Of interest, dKdm4A is brought to the borders of repressed domains through its interaction with HP1a, thereby regulating repressive marks (Lin et al, 2012). The regulation of genes at chromosomal borders thus likely involves both a highly dynamic organization of chromatin locally, through multiple interactions of IBPs with NSD/dMes-4 HMTs and additional players including HP1 with histone demethylases, and higher-order chromatin organization through a distinct set of IBP cofactors.
Materials and Methods
RNAi, genome-wide expression data (RT-qPCR, microarray, DGE-Seq, and RNASeq)
Drosophila Schneider SL2 cells were treated with specific interfering RNAs to knock down Beaf32, dMes-4, DREF, or control RNA as a mock control essentially as previously described (Emberly et al, 2008). For RNAi-mediated depletion, T7-driven synthesis (Fermentas TranscriptAid™ T7 High Yield Transcription Kit) of double-stranded RNAs (dsRNA) specific for beaf32, dmes-4, dref, or control dsRNA (against luciferase; for mock-depleted control cells) was checked for potential off-target effects using NCBI primer designing tool and dsCheck (http://dsCheck.RNAi.jp/; Emberly et al, 2008) was used. 400/400/900 μg of dsRNA were added to 30 millions cells in 10 ml media without FBS for beaf32/dref/dmes-4 depletion, respectively. Cells were incubated for 2 h at 25°C, and 20 ml of media with FBS were added. After incubation for 5 days, the cells were harvested, followed by RNA extraction (Qiagen RNeasy; deep-seq and RT-qPCR) or formaldehyde cross-linking (ChIP). Samples were analyzed as replicates by RNA sequencing (DGE/RNA-seq; HiSeq2000; Illumina; INRA platform (GeT-PlaGe; (http://genomique.genotoul.fr/intranet/)) or BGI) using Tophat or the Burrows Wheeler Alignment tool (BWA) software (default parameters) on genome annotations of release 5.41 of Flybase for parsing, HTseq for counting the reads, and DEGseq package to identify differentially expressed genes (P-value < 0.01) from independent replicates. Data were independently verified through analyses by RT-qPCR and microarray analysis for validation (Supplementary Fig S1) using Fisher's exact test by intersecting the groups of differentially expressed genes. Parsing was performed using the TopHat or Burrows Wheeler Alignment tool (BWA) software (default parameters) on genome annotations of release 5.41 of Flybase for parsing, HTseq for counting the reads, and DEGseq package to identify differentially expressed genes (adjusted P-value < 0.01) from four independent replicates for RNASeq. Data were independently verified through analyses by RT-qPCR and microarray analysis for validation (Supplementary Fig S1F and G) using Fisher's exact test by intersecting the groups of differentially expressed genes. Gene expression was measured by real-time PCR analysis using cDNAs prepared from S2 control, Beaf32 KD, and dMes-4-depleted or control cells, for the following (control or Beaf32 bound promoters) genes: control: CG9988, CG13766,CG9520,CG17715,sara,gapdh2; Beaf32 bound: B-tub56D,vha100-2,crc,ter-94,CG5284,tsp39D,spin,ror,kel,CG4210. Data were analyzed using the absolute quantification component of pyQPCR. Quality of mRNA levels was controlled with Experion (Biorad) and quantified in parallel with at least five different concentrations of cDNA and genomic DNA for standard curves (see example in Supplementary Fig S10E) for all oligos used in this study using Biorad iQ SYBR Green Supermix in an Eppendorf Realplex. qPCR or RT-qPCR data were analyzed using the relative quantification component of pyQPCR software developed by M. Hennion and T. Gastine accessible at http://pyqpcr.sourceforge.net/ verifying that the quality of the data obtained with Eppendorf Realplex by confidence interval (95%) or Applied Biosystems Viia7. Further analyses of post-transcriptional defects (Supplementary Figs S2 and S9) were performed using DiffSplice to quantify significant splicing defects (Hu et al, 2013) or by measuring RNASeq reads in introns normalized to the reads in exons. Data were verified by RT-qPCR using Applied biosystem Viia7 with oligos that span exon–intron junctions, and immature RNA levels were normalized to mRNA levels measured with exon-specific oligos for the same list of genes used to measure expression except CG9988 (due to low expression) and the addition of the Ucp4A gene. For intersection analysis between genome-wide expression analyses of Beaf32 or dCTCF and/or dMes-4, receiver operating characteristics (ROC) analysis was performed using R as a convenient classifier tool (Fawcett, 2006) to visualize true positives (e.g. dCTCF or Beaf32 binding) independently of the threshold of differential expression. In the context of genes flanked by IBPs, Fisher's exact was used to measure the relative enrichment in differentially regulated genes as a function of which binding sites were present with respect to the total number of genes of the same category (binding site or other genomic features).
Chromatin immunoprecipitations (ChIP-Seq)
ChIP-Seq data of H3K36me2, H3K36me3, H3K27me3, and RNA polymerase II (“Pol II”) were performed using exponentially growing S2 cells essentially as previously described (Emberly et al, 2008; Schones et al, 2008). Chromatin pellets were analyzed using Qubit fluorometer and Agilent (2100) before library construction and SE- or PE-sequencing at BGI. Data were analyzed using a rMAT package by comparison with read counts obtained by precipitating chromatin with IgG controls (not shown). For pair-end sequencing, data were further processed by mapping positive and negative reads separately (average distance ˜82.5 bp) followed by Gaussian smoothing to generate genome-wide profiles. The average fragment size and variance was calculated using the spatial correlation function between the + and − reads (25 bp reads from either the 5′ (+) or 3′ (−) end of each fragment) by making a histogram of the distances between all + and − reads across the genome. Each read's contribution was obtained by shifting (˜41 bp) +/− by +/− half the average fragment size providing a Gaussian density with a fixed standard deviation (50 bp) for genome-wide profiles (the total score at a given location is the sum of all the read densities). For ranking of genes according to the log ratio of H3K36me3 over H3K36me2 levels, normalized ChIP-Seq reads were counted over gene bodies (+500 to end of gene). RNAPII elongation was estimated by the ratio of ChIP-Seq reads in the same region normalized to the amount of reads in promoters, as previously done (Rahl et al, 2010). For motif search, the MEME program (http://meme.sdsc.edu/meme4_6_1/intro.html) was used.
ChIP of dMes-4, DREF, H4K16ac, H3K36me2/me3, and H3K27me3 were analyzed in depleted (Beaf32, dMes-4, DREF, or mock-deplete control/WT) cells, using equivalent amounts of chromatin prepared from Beaf32-KD or WT cells used for immunoprecipitations in triplicates. DNA was analyzed by real-time qPCR using Applied Biosystems Viia7 and a two-sided Wilcoxon paired test. Statistical analysis was performed using a two-sided Wilcoxon paired test between two conditions for each of the two groups (control or Beaf32 bound promoters): control: CG9988, CG13766, CG9520,CG17715,sara,gapdh2; Beaf32 bound: B-tub56D,tina-1,vha100-2,crc,chico,CG5284,tsp39D,spin,ror,CG4210. Data were analyzed using the absolute quantification component of pyQPCR. For ChIP with anti-H3K27me3 antibodies in dMes4-KD, Beaf32-KD versus control cells, a total of 25 genes were analyzed including the same list of genes or additional genes selected based on variations in H3K27me3 levels as measured by ChIP-Seq in Beaf32-KD compared to control cells (CG3812, CG9422, CG11851, CG3408, sav, CG11975, CG2264, CG14683, Prx). To score such variations in H3K27me3 levels, ChIP-seq reads were counted over a +/− 1 kbp region surrounding every Drosophila TSS genome-wide. A z-score was then defined for each gene/TSS as the number of ChIP-Seq reads in the window in Beaf32-KD minus WT control normalized to the square root of the mean of reads, providing a list of 990 genes increasing H3K27me3 levels as shown (z > 2; Fig7). The significance of such variations and further correlation with dMes4-KD was further tested through three independent ChIP analyses by qPCR measurements of the variations in Beaf32-KD/WT compared to variations in dMes4-KD/WT, providing a determination coefficient (R2) of 0.57 and 0.08 for Beaf32 bound and control (unbound) promoters, respectively (Supplementary Fig S10D).
Nucleosome positioning by MNase-Seq
Genome-wide nucleosome positioning (MNase-seq) was measured from exponentially growing control (WT control) or Beaf32-depleted (“Beaf32KD”) cells after purifying nucleosomes followed by pair-end sequencing essentially as previously described (Schones et al, 2008). Nucleosome profiles upon Beaf32 depletion were generated after normalization to the total number of reads in WT using Gaussian smoothing of the raw read counts at each position in the genome for both plus and minus reads. A shift of 80 bp and a standard deviation of 20 bp were applied to generate smoothened nucleosome profiles. ChIP-Seq of H3K27me3 was performed on chromatin digested with MNase (Schones et al, 2008).
Antibodies, biochemical purification of IBP cofactors
Affinity-purified antibodies specific of dMes-4 or DREF were generated essentially as described (Cuvier & Hirano, 2003) using recombinant DREF protein injected in mouse or using the middle region of dMes-4 injected in rabbit as previously (Bell et al, 2007). Anti-Beaf32 antibodies were generated in the laboratory as described (Liang et al, 2014). Antibody specificity was confirmed by Western blotting of nuclear extracts and by competition with peptides (Supplementary Fig S1) and by RNAi-mediated depletion. Additional antibodies were purchased: anti-H3 (Abcam ab1791), H3K36me3 (Abcam, ab9050), H3K27me3 (Upstate #07-449), H4K16ac (Active motif), anti-RNAPII (Covance), H3K36me2 (Upstate #07-369) anti-actin (Sigma).
Nuclear extracts, Western blotting, chromatin purification, purification of IBP cofactors
Chromatin was prepared from purified nuclei by mild detergent extraction following purification of chromatin-associated proteins through a 30% sucrose cushion essentially as previously described (Cuvier et al, 2008). Nuclear extracts were prepared by lysing purified nuclei in NEB-360 mM KCl (NEB: 10 mM Hepes KOH pH 7.6; 3 mM MgCl2; 0.1 mM EDTA/K; 0.1% Trasylol; 0.2 mM PMSF; 10% glycerol) for 30 min at 4°C and spinning them at 158,000 g for 1 h. nuclear extracts (10 μg/well) were separated on 4–16% SDS–PAGE and passively transferred to nitrocellulose membranes as previously described (Cuvier et al, 2002). Filter strips were then incubated with the indicated antibody ± 0.4 mg/ml of antigen peptide (e.g. Supplementary Fig S1), followed by horseradish peroxidase-conjugated donkey anti-rabbit (1:10,000) (GE Healthcare). Signals were developed using an ECL Plus detection kit (GE Healthcare) and FUJIFILM Luminescent Image Analyzer LAS-4000 for images acquisition. Signal quantification was performed through MultiGauge software following the manufacturer instructions. Identification of dMes-4 was allowed by high-salt elution of cofactors associated with affinity-purified Beaf32 complex using affinity-purified anti-Beaf32 antibodies as compared to IgG control essentially as previously described (Cuvier & Hirano, 2003). Columns were then extensively washed before bound proteins were eluted by adding one bed volume (0.5 ml) of 0.6 M NaCl (“high-salt” extraction) followed by analysis on SDS–PAGE (Fig2B) and mass-spectrometric analyses.
Yeast two-hybrid assays
For yeast two-hybrid, bait constructs and target constructs including beaf32,dmes-4, or cp190 were cloned into pDest22 and pDest32 to express fusion proteins and interactions were evaluated for various combinations as a function of yeast ura3 reporter expression essentially according to the manufacturer's instructions (Invitrogen) by growing the yeast strain MaV203 in replicated cells on nonselective (+Ura) and selective (−Ura) media followed by incubation at 30°C for 2 days on SD media.
Statistical analyses by principal component analysis (PCA) and clustering ascendant hierarchical (CAH)
Statistical analyses by PCA were performed using the package FactoMiner from R (Josse et al, 2011) by taking the total read counts as measured from our ChIP-Seq data of Beaf32, H3K36me2, H3K36me3, from additional ChIP-Seq available at modENCODE (H4K16ac and H3K36me1) (Kharchenko et al, 2011; Alekseyenko et al, 2012), or from our MNase-Seq data in Beaf32-KD or control cells. ChIP-Seq or MNase-Seq data were analyzed by comparison with read counts obtained by precipitating chromatin with IgG controls by counting reads within the indicated windows corresponding to gene bodies and/or promoters defined by Flybase (release 5.41). Reads were counted within windows corresponding to gene bodies (“H3K36me2/3”, gene body from +500 to the end of each gene) or from promoter regions (from −300 to 0 with respect to TSS). Nucleosome positioning was estimated by adding the maximum peak intensity (in read counts) obtained from three separated widows (+100, +150), (+290, +320), (470, 500) and corresponding to +1, +2 and +3 nucleosome windows. Nucleosome Free Regions (NFRs) were calculated by normalizing read counts in the promoter window (−150 to 0 from each TSS) normalized to the average read counts in neighboring regions (−1,000 to −500). The generated averaged values were then provided for each ChIP-Seq or MNase-Seq for each individual gene allowing to perform clustering ascendant hierarchical (CAH) for all centered and normalized data using the package FactoMiner from R. CAH measures Ward distances reflecting the minimal variance among all data sets provided (Josse et al, 2011), which was performed separately for Beaf32 bound or control genes. Analysis of the influence of dMes-4 with respect to Beaf32 or dCTCF peaks was based upon intersection analysis between RNA-Seq data and ChIP-Seq using Fisher's exact test or pairwise Wilcoxon test.
Data access
All genomic sequencing data including ChIP-Seq of H3K36me2 /H3K36me3/Pol II, MNase-Seq of bulk or H3K27me3 nucleosomes in WT or Beaf32-KD cells (“nuc-WT/ nuc-Beaf32-KD”), and RNASeq data in WT, Beaf32-KD or dMes-4-KD cells are available at NCBI (GEO accession number GSE57168) and are accessible through our genome browser links: http://insulators_chromosome-dynamics.biotoul.fr/IBPs
Acknowledgments
We thank Artem Barski and Kairong Cui for performing the Hi-Seq sequencing of MNase data, S. Gadat and G. Fichant for suggestion regarding statistical analyses, L. Lacroix and additional OC's laboratory members for critical reading of the manuscript, C. Carles for help with the web server, P. Martin for technical help. E.E.'s laboratory was supported by NSERC and the Canadian Institute For Advanced Research (CIFAR) and by the University of Toulouse (while in O.C.'s laboratory). K.Z.'s laboratory was supported by the Division of Intramural Research Program of the National Heart, Lung and Blood Institute, NIH. P.L. and M.H. were supported by a fellowship from La Ligue Nationale Contre le Cancer (LNCC). O.C.'s laboratory was supported by grants from the Region Midi-Pyrénées, the Cancer Research funding of the ARC, the ATIP-AVENIR program of the CNRS and Inserm joint program and the ANR “INSULa”.
Author contributions
PL, MH, SC, and OC designed experiments, performed research, and interpreted data. MNase- and ChIP-Seq experiments were performed by MH, SC, and PL under the supervision of OC and KZ. AG conducted all bioinformatic and statistical analyzes with OC and with the help of EE. GM performed RNA splicing analyses. Protein purifications were done and analyzed by SU and OC. Confirmations of interactions were done by PL and JL. PL, MH, DS, PM, and SQ performed the RNAi experiments and expression studies by qPCR, DGE, or RNASeq. OC designed experiments and directed the study, with the help of KZ and EE. PL, MG, JL, AG, and OC prepared the figures. OC wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Alekseyenko AA, Ho JW, Peng S, Gelbart M, Tolstorukov MY, Plachetka A, Kharchenko PV, Jung YL, Gorchakov AA, Larschan E, Gu T, Minoda A, Riddle NC, Schwartz YB, Elgin SC, Karpen GH, Pirrotta V, Kuroda MI, Park PJ. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS Genet. 2012;8:e1002646. doi: 10.1371/journal.pgen.1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bartkuhn M, Straub T, Herold M, Herrmann M, Rathke C, Saumweber H, Gilfillan GD, Becker PB, Renkawitz R. Active promoters and insulators are marked by the centrosomal protein 190. EMBO J. 2009;28:877–888. doi: 10.1038/emboj.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Wirbelauer C, Hild M, Scharf AN, Schwaiger M, MacAlpine DM, Zilbermann F, van Leeuwen F, Bell SP, Imhof A, Garza D, Peters AH, Schubeler D. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007;26:4974–4984. doi: 10.1038/sj.emboj.7601926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushey AM, Dorman ER, Corces VG. Chromatin insulators: regulatory mechanisms and epigenetic inheritance. Mol Cell. 2008;32:1–9. doi: 10.1016/j.molcel.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushey AM, Ramos E, Corces VG. Three subclasses of a Drosophila insulator show distinct and cell type-specific genomic distributions. Genes Dev. 2009;23:1338–1350. doi: 10.1101/gad.1798209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Levine M. Modulation of enhancer-promoter interactions by insulators in the Drosophila embryo. Nature. 1995;376:533–536. doi: 10.1038/376533a0. [DOI] [PubMed] [Google Scholar]
- Cai L, Rothbart SB, Lu R, Xu B, Chen WY, Tripathy A, Rockowitz S, Zheng D, Patel DJ, Allis CD, Strahl BD, Song J, Wang GG. An H3K36 methylation-engaging Tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol Cell. 2013;49:571–582. doi: 10.1016/j.molcel.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvier O, Hart CM, Laemmli UK. Identification of a class of chromatin boundary elements. Mol Cell Biol. 1998;18:7478–7486. doi: 10.1128/mcb.18.12.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvier O, Hart CM, Kas E, Laemmli UK. Identification of a multicopy chromatin boundary element at the borders of silenced chromosomal domains. Chromosoma. 2002;110:519–531. doi: 10.1007/s00412-001-0181-1. [DOI] [PubMed] [Google Scholar]
- Cuvier O, Hirano T. A role of topoisomerase II in linking DNA replication to chromosome condensation. J Cell Biol. 2003;160:645–655. doi: 10.1083/jcb.200209023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvier O, Stanojcic S, Lemaitre JM, Mechali M. A topoisomerase II-dependent mechanism for resetting replicons at the S-M-phase transition. Genes Dev. 2008;22:860–865. doi: 10.1101/gad.445108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D. Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev. 2011;21:199–206. doi: 10.1016/j.gde.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberly E, Blattes R, Schuettengruber B, Hennion M, Jiang N, Hart CM, Kas E, Cuvier O. Beaf32 regulates cell-cycle genes through the controlled deposition of H3K9 methylation marks into its conserved dual-core binding sites. PLoS Biol. 2008;6:2896–2910. doi: 10.1371/journal.pbio.0060327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo B, Deuring R, Murawska M, Gause M, Dorighi KM, Schaaf CA, Dorsett D, Brehm A, Tamkun JW. The Drosophila MI-2 chromatin-remodeling factor regulates higher-order chromatin structure and cohesin dynamics in vivo. PLoS Genet. 2012;8:e1002878. doi: 10.1371/journal.pgen.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. An introduction to ROC analysis. Pattern Recognit Lett. 2006;27:861–874. [Google Scholar]
- Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7:703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- Gaydos LJ, Rechtsteiner A, Egelhofer TA, Carroll CR, Strome S. Antagonism between MES-4 and Polycomb repressive complex 2 promotes appropriate gene expression in C. elegans germ cells. Cell Rep. 2012;2:1169–1177. doi: 10.1016/j.celrep.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R, Giles K, Gowher H, Xiao T, Xu Z, Yao H, Felsenfeld G. Chromatin domains, insulators, and the regulation of gene expression. Biochim Biophys Acta. 2012;1819:644–651. doi: 10.1016/j.bbagrm.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, Li L, Adelman K. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell. 2010;143:540–551. doi: 10.1016/j.cell.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohl D, Aoki T, Blanton J, Shanower G, Kappes G, Schedl P. Mechanism of chromosomal boundary action: roadblock, sink, or loop? Genetics. 2011;187:731–748. doi: 10.1534/genetics.110.123752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, Swaminathan V, Workman JL, Li B, Hinnebusch AG. Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell. 2010;39:234–246. doi: 10.1016/j.molcel.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowher H, Brick K, Camerini-Otero RD, Felsenfeld G. Vezf1 protein binding sites genome-wide are associated with pausing of elongating RNA polymerase II. Proc Natl Acad Sci USA. 2012;109:2370–2375. doi: 10.1073/pnas.1121538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Gurudatta BV, Corces VG. Chromatin insulators: lessons from the fly. Brief Funct Genomic Proteomic. 2009;8:276–282. doi: 10.1093/bfgp/elp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurudatta BV, Yang J, Van Bortle K, Donlin-Asp PG, Corces VG. Dynamic changes in the genomic localization of DNA replication-related element binding factor during the cell cycle. Cell Cycle. 2013;12:1605–1615. doi: 10.4161/cc.24742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Li L, Qin ZS, Corces VG. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell. 2012;48:471–484. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Huang Y, Du Y, Orellana CF, Singh D, Johnson AR, Monroy A, Kuan PF, Hammond SM, Makowski L, Randell SH, Chiang DY, Hayes DN, Jones C, Liu Y, Prins JF, Liu J. DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 2013;41:e39. doi: 10.1093/nar/gks1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Emberly E, Cuvier O, Hart CM. Genome-wide mapping of boundary element-associated factor (BEAF) binding sites in Drosophila melanogaster links BEAF to transcription. Mol Cell Biol. 2009;29:3556–3568. doi: 10.1128/MCB.01748-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Josse J, Pages J, Husson F. Multiple imputation in principal component analysis. Adv Data Anal Classif. 2011;5:231–246. [Google Scholar]
- Keren-Shaul H, Lev-Maor G, Ast G. Pre-mRNA splicing is a determinant of nucleosome organization. PLoS ONE. 2013;8:e53506. doi: 10.1371/journal.pone.0053506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, Linder-Basso D, Plachetka A, Shanower G, Tolstorukov MY, Luquette LJ, Xi R, Jung YL, Park RW, Bishop EP, Canfield TK. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Lacroix L, Gamot A, Cuddapah S, Queille S, Lhoumaud P, Lepetit P, Martin PG, Vogelmann J, Court F, Hennion M, Micas G, Urbach S, Bouchez O, Nollmann M, Zhao K, Emberly E, Cuvier O. Chromatin immunoprecipitation indirect peaks highlight long-range interactions of insulator proteins and pol II pausing. Mol Cell. 2014;53:672–681. doi: 10.1016/j.molcel.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Paulson A, Abmayr SM, Workman JL. HP1a targets the Drosophila KDM4A demethylase to a subset of heterochromatic genes to regulate H3K36me3 levels. PLoS ONE. 2012;7:e39758. doi: 10.1371/journal.pone.0039758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda RK, Karch F. Making connections: boundaries and insulators in Drosophila. Curr Opin Genet Dev. 2007;17:394–399. doi: 10.1016/j.gde.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Schedl P, Studitsky VM, Sengupta AM. Theoretical analysis of the role of chromatin interactions in long-range action of enhancers and insulators. Proc Natl Acad Sci USA. 2011;108:19919–19924. doi: 10.1073/pnas.1103845108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012;22:1711–1722. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, Brown CD, Shah PK, Kheradpour P, Morrison CA, Henikoff JG, Feng X, Ahmad K, Russell S, White RA, Stein L, Henikoff S, Kellis M, White KP. A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet. 2010;6:e1000814. doi: 10.1371/journal.pgen.1000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Corces VG. Chromatin insulators: linking genome organization to cellular function. Mol Cell. 2013;50:461–474. doi: 10.1016/j.molcel.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V. Silence in the germ. Cell. 2002;110:661–664. doi: 10.1016/s0092-8674(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Raab JR, Kamakaka RT. Insulators and promoters: closer than we think. Nat Rev Genet. 2010;11:439–446. doi: 10.1038/nrg2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnard C, Straub T, Mitterweger A, Dahlsveen IK, Fabian V, Becker PB. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS Genet. 2011;7:e1001327. doi: 10.1371/journal.pgen.1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon-intron structure. Nat Struct Mol Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Ast G. Chromatin density and splicing destiny: on the cross-talk between chromatin structure and splicing. EMBO J. 2010;29:1629–1636. doi: 10.1038/emboj.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Linder-Basso D, Kharchenko PV, Tolstorukov MY, Kim M, Li HB, Gorchakov AA, Minoda A, Shanower G, Alekseyenko AA, Riddle NC, Jung YL, Gu T, Plachetka A, Elgin SC, Kuroda MI, Park PJ, Savitsky M, Karpen GH, Pirrotta V. Nature and function of insulator protein binding sites in the Drosophila genome. Genome Res. 2012;22:2188–2198. doi: 10.1101/gr.138156.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- Vögelmann J, Valeri A, Guillou E, Cuvier O, Nollmann M. Roles of chromatin insulator proteins in higher-order chromatin organization and transcription regulation. Nucleus. 2011;2:358–369. doi: 10.4161/nucl.2.5.17860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AM, Van Bortle K, Ramos E, Takenaka N, Rohrbaugh M, Jones BC, Jones KC, Corces VG. Regulation of chromatin organization and inducible gene expression by a Drosophila insulator. Mol Cell. 2011;44:29–38. doi: 10.1016/j.molcel.2011.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.