

Abstract
The epithelial complement inhibitory proteins (CIPs) cluster of differentiation 46 and 55 (CD46 and CD55) regulate circulating immune complex–mediated complement activation in idiopathic pulmonary fibrosis (IPF). Our previous studies demonstrated that IL-17A mediates epithelial injury via transforming growth factor β1 (TGF-β1) and down-regulates CIPs. In the current study, we examined the mechanistic role of TGF-β1 in complement activation–mediated airway epithelial injury in IPF pathogenesis. We observed lower epithelial CIP expression in IPF lungs compared to normal lungs, associated with elevated levels of complement component 3a and 5a (C3a and C5a), locally and systemically. In normal primary human small airway epithelial cells (SAECs) treated with TGF-β1 (10 ng/ml), C3a, or C5a (100 nM), we observed loss of CIPs and increased poly(ADP-ribose) polymerase (PARP) activation [also observed with RNA interference (RNAi) of CD46/CD55]. TGF-β1-mediated loss of CIPs and Snail induction [SNAI1; a transcriptional repressor of E-cadherin (E-CAD)] was blocked by inhibiting mitogen-activated protein kinase (p38MAPK; SB203580) and RNAi silencing of SNAI1. C3a- and C5a-mediated loss of CIPs was also blocked by p38MAPK inhibition. While C3a upregulated TGFb transcripts, both C3a and C5a down-regulated SMAD7 (negative regulator of TGF-β), and whereas TGF-β1 induced C3a/C5a receptor (C3aR/C5aR) expression, pharmacologic C3aR/C5aR inhibition protected against C3a-/C5a-mediated loss of CIPs. Taken together, our results suggest that epithelial injury in IPF can be collectively amplified as a result of TGF-β1-induced loss of CIPs leading to complement activation that down-regulates CIPs and induces TGF-β1 expression.—Gu, H., Mickler, E. A., Cummings, O. W, Sandusky, G. E., Weber, D. J., Gracon, A., Woodruff, T., Wilkes, D. S., Vittal, R. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis.
Keywords: C3a, C5a, CD46, CD55, IPF
Idiopathic pulmonary fibrosis (IPF) is a disease of high mortality for which lung transplantation is considered the only definitive therapy. Its pathogenesis remains largely unknown (1), but emerging concepts point to repeated injury to bronchiole-like epithelial cells and hyperplastic type II alveolar epithelial cells lining areas of honeycomb fibrosis (1, 2). These injured epithelial cells produce key profibrotic factors, including transforming growth factor β (TGF-β), which is implicated in epithelial injury (3–5) and epithelial-to-mesenchymal transition (EMT; refs. 6, 7).
The complement system is an integral arm of innate and adaptive immunity. Early studies demonstrated evidence of circulating immune complexes (8) and complement activation (9) in patients with IPF. In experimental models of IPF, antifibrotic effects due to deletion of complete downstream complement factors (10), specifically complement component 5 (C5; ref. 11), were reported. C3a and C5a are implicated in autoimmune diseases (12), chronic lung transplant rejection (13), experimental allergic asthma (14), and EMT in renal pathology (15, 16). Although the deleterious profibrotic effect of C5 (which generates C5a and C5b) is reportedly dependent on TGF-β1 (11), the specifics of TGF-β1 crosstalk with the anaphylatoxins C3a and C5a are unclear.
The membrane-bound complement inhibitory proteins (CIPs) cluster of differentiation 46 and 55 (CD46 and CD55) are the early regulators of complement activation and are ubiquitously expressed in the upper and lower respiratory tract of normal humans (17). Early studies have demonstrated protective effects of CD46 and CD55 against complement-mediated injury on nasal (18) and glomerular (19) epithelia. However, the specific mechanisms by which the CIPs are down-regulated on epithelial cells in IPF and the role of complement activation products in ongoing epithelial injury are unknown.
In IPF, elevated expression of the transcription factor, SNAI1 [Snail, a known E-cadherin (E-CAD) repressor], has been linked to epithelial injury and repair, including EMT (20). TGF-β1 induces Snail expression via activation of mitogen-activated protein kinase (p38MAPK) signaling (21), and we have previously reported that interleukin-17A (IL-17A) mediates EMT via TGF-β1 and associated signaling pathways, such as p38MAPK (22), and down-regulates CIP expression in lung epithelial cells (13). Recent reports have provided indirect evidence of complement activation in IPF pathogenesis, whereas C3a and C5a have been shown to induce EMT in kidney tubular epithelium. Since the activity of C3a and C5a has been linked to fibrogenesis in nonpulmonary pathologies (15, 16), and TGF-β1 has a key role in fibrogenesis that is characteristic of IPF, we hypothesized that there is crosstalk between complement, TGF-β1, and p38MAPK/Snail-associated signaling pathways in regulating CIP expression in airway epithelium. In addition, we sought to determine whether altered local CIP expression may account for upregulated complement activation in IPF. Our findings suggest that CIPs are down-regulated in the IPF lung. In addition, TGF-β1 may crosstalk with C3a and C5a via shared signaling pathways and down-regulate CIPs via a feed-forward loop and thus link complement activation to epithelial injury in IPF.
MATERIALS AND METHODS
Human studies
As described previously, frozen tissues and plasma from patients with IPF were obtained through the Lung Tissue Research Consortium, sponsored by the U.S. National Institutes of Health/National Heart, Lung, and Blood Institute (http://www.ltrcpublic.com/docs/LTRC_Consent_Jul_2010.pdf), and paraffin-embedded IPF and normal specimens were procured from the Department of Pathology, Indiana University School of Medicine (IUSM). Demographics of the donor patients are presented in Table 1. All protocols were approved by the Institutional Review Board, IUSM.
Table 1.
Demographics and pulmonary function characteristics of patients with IPF
| Characteristic | Statistic |
|---|---|
| n | 25 |
| Age [yr (range)] | 59.2 ± 7.7 (42–75) |
| Male [n (%)] | 14 (56) |
| Race [n (%)] | |
| Caucasian | 20 (80) |
| African American | 2 (8) |
| Hispanic | 1 (4) |
| Asian | 2 (8) |
| Current/former smoker [n (%)]a | 15 (60) |
| Smoking (pack yr) | 29.9 ± 37.6 |
| Pulmonary function test | |
| FEV1, predicted [% (range)] | 58.1 ± 14 (25–86) |
| FVC, predicted [% (range)] | 53.4 ± 14.4 (24–80) |
| DLCO, predicted [% (range)]b | 32.18 ± 13.0 (9–69) |
Values are means ± sd or as indicated.
Data missing from 1 patient.
Data missing from 3 patients.
Cell culture conditions and reagents
Normal primary human small airway epithelial cells (SAECs; Clonetics; Cambrex Biosciences, Walkersville, MD, USA) were cultured in small airway basal medium (SABM; Clonetics) supplemented with growth factors (SAGM; Clonetics) to 70% confluence, followed by culture in SABM containing 1:100 growth factors for 16 h before the specific treatments. In these studies, we used recombinant human C3a and C5a (100 nM each; the concentration presented in this report is equivalent to 90.91 ng/ml of C3a and 83.3 ng/ml of C5a) from R&D Systems (Minneapolis, MN, USA), platelet-derived TGF-β1 (10 ng/ml) from Roche Diagnostics (Mannheim, Germany), and pharmacologic inhibitors: PD98059 (ERK1/2; 20 μM), SB203580 (p38MAPK; 6 μM), SP600125 (Jun N-terminal kinase; 100 nM), and SB290157 (C3aRA; 50, 100 μM) from Calbiochem-Millipore (Billerica, MA, USA), and PMX205 (C5aRA; 1, 5 μM), a generous donation from Dr. Trent Woodruff (University of Queensland, St. Lucia, QLD, Australia). All other reagents were from Sigma-Aldrich (St. Louis, MO, USA).
RNA interference (RNAi)
For in vitro RNAi delivery to the SAECs, single-duplex small interference RNA (siRNA) sequences targeting SNAI1 (GCGAGCUGCAGGACUCUAA; Dharmacon Technologies, Pittsburgh, PA, USA), CD46, CD55 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), or nontargeting control siRNA (100 nM; Dharmacon Technologies) were transfected with Oligofectamine (Invitrogen, Grand Island, NY, USA) for 24 h, per the manufacturer's instructions. Subsequently, the transfected cells were cultured in SABM with 1:100 growth factors for 16 h before treatment.
Western blot analysis of cell lysates and tissue homogenates
SAECs were lysed by in CellLytic M cell lysis buffer (Sigma-Aldrich) containing 2 mM sodium orthovanadate (Sigma-Aldrich) and 1:100 dilution of protease inhibitor cocktail III (Calbiochem). Lung tissue was homogenized in the presence of PBS and lysed in the cell lysis buffer. Subsequently, the samples were centrifuged at 10,000 rpm for 10 min at 4°C, to extract the supernatants for further analyses. Total protein concentrations in both the samples were measured with a nanospectrophotometer (Nanodrop 1000; Fisher Scientific, Wilmington, DE, USA). Equal protein concentrations were subjected to immunoblot analysis, according to published methods (23–26). Primary antibodies were incubated overnight at 4°C at 1000-fold dilution against CD46 and CD55 (sc-166159 and sc-9156, respectively; Santa Cruz Biotechnology); E-CAD (CP1921; ECM Biosciences, Versailles, KY, USA); glyceraldehyde-3-phosphate dehydrogenase (GAPDH; H86504M), Ser423/425 p38MAPK (4511), and poly(ADP-ribose) polymerase (PARP; 9542S) (Cell Signaling Technology, Beverly, MA, USA); Snail (NBT1-19529), C3a receptor (C3aR; NBP2-15649), and C5a receptor (C5aR; NBT1-61567) (Novus Biologicals, Littleton, CO, USA); and β-actin (A2228-200; Sigma-Aldrich). The membranes were washed with TBS-T and incubated with HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) for 1 h, followed by probing with Pierce West Pico chemiluminescent signal (Fisher Scientific). Densitometric analyses were performed with ImageJ 1.32j (U.S. National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
Staining was performed on 4 μm tissue sections, according to a published description (13). In brief, sections obtained from paraffin-embedded, formalin-fixed lungs underwent antigen retrieval treatment, followed by 5 min peroxide and protein blocks (1× Power Block; Biogenex, San Ramon, CA, USA). The sections were then incubated with the following primary Abs: rabbit anti-mouse Crry (1:200; sc-9098), rabbit anti-mouse CD55 (1:200; sc-9156), rabbit anti-human CD46 (1:150), and rabbit anti-human CD55 (1:150) (Santa Cruz Biotechnology) and the corresponding rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA). The sections were washed and incubated with MACH 2 Rabbit HRP-Polymer (RHRP520; Biocare, Concord, CA, USA) for 30 min and then stained with ImmPACT DAB (Vector Laboratories, Burlingame, CA, USA) for 2 min. Nuclei were counterstained with hematoxylin. Images were scanned with the Aperio Scanscope Imaging System (Leica Biosystems, Buffalo Grove, IL, USA).
Digital imaging with positive-pixel algorithm
Using Aperio Scanscope 11.2.0.780 imaging software (Leica Biosystems), we quantified the amount of a specific stain present in a scanned slide image. Default input parameters were preconfigured for brown color intensity fraction of positive pixels for quantification in the 3 intensity ranges (220–175, 175–100, and 100–0). Pixels that were stained but did not fall into the positive color specification were considered negative-stained pixels. These pixels were counted as well, and thus the fraction of positive to total stained pixels was determined. We selected the entire image region for analyses, and saved the algorithm results. Specific algorithms were used for the positive-pixel analysis (25). Staining intensity was analyzed under the supervision of the pathologist.
ELISA
C3a (with or without desArg) and C5a (with or without desArg) were measured in the clinical tissues with the MicroVue C3a Plus EIA Kit (Quidel Corp., San Diego, CA, USA) for serum and plasma and the MicroVue C5a EIA Kit (Quidel) for serum, plasma, and biological materials, respectively, per the manufacturer's instructions. Lung tissue homogenates were processed as described for Western blot analysis. The samples were measured for total protein concentrations and then subjected to ELISA, per the manufacturer's instructions.
Real-time polymerase chain reaction (PCR)
Total RNA was isolated from cells with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and reverse transcribed with qScript cDNA SuperMix (Quanta BioSciences Inc., Foster City, CA, USA). Real-time PCR was performed for each cDNA with SYBRGreen PCR Master Mix (Quanta BioSciences), with gene-specific primer pairs (Table 2). The semiquantitative real-time PCR data for each target gene are expressed as 2−ΔΔCt relative quantitation vs. endogenous control, with error bars representing the standard error for epithelial lines derived from multiple donors.
Table 2.
Real-time primer sequences
| Gene | Forward | Reverse |
|---|---|---|
| SNAIL | GAGGCGGTGGCAGACTAG | GACACATCGGTCAGACCAG |
| TGFb | GTGGAAACCCACAACGAAA | TAAGGCGAAAGCCCTCAAT |
| SMAD7 | CCTTAGCCGACTCTGCGAACTA | CCAGATAATTCGTTCCCCCTGT |
Statistical analyses
Statistical analysis was performed with Student's t test and 1-way ANOVA followed by the Bonferroni post hoc test using GraphPad Prism 4.03 for Windows (GraphPad Software, San Diego, CA, USA), unless otherwise stated. Statistical significance was set at P < 0.05.
RESULTS
CIPs CD46 and CD55 and anaphylatoxins C3a and C5a in clinical IPF
To investigate the clinical relevance of CIPs in IPF, we examined lung tissue explants from patients with IPF or normal lung tissues. Protein extracts from histologically normal lung or IPF lung explants were immunoblotted with antibodies against CD46 and CD55, with GAPDH used as the loading control (Fig. 1). Whereas we observed strong expression of CD46 and CD55 in normal lung tissues, the levels were either significantly lower or absent in the IPF lung homogenates (Fig. 1A). Densitometric analysis of the immunoblots (normalized to GAPDH) comparing normal (n=3) and IPF (n=11) lungs showed significantly lower expression of CD46 (Fig. 1B) and CD55 (Fig. 1C). As expected, immunohistochemical analysis of pathologically normal lungs demonstrated higher levels of CD46 and CD55, specifically in the alveolar and airway epithelia, compared with levels in biopsy tissue harvested from patients with IPF (Fig. 2A). Interestingly, in the IPF tissues, the distribution of CIP expression was dependent on the epithelial injury in that region. In addition, some of the mesenchymal cells in the fibrotic foci expressed these proteins strongly, with most of the interstitium staining negative. The staining specificity was confirmed by using the corresponding secondary IgG (Fig. 2B). The cumulative intensity showed a lower expression of CD46 (P<0.0017) and CD55 (P<0.0002) in the IPF lung tissue (Fig. 2C). Taken together, these data demonstrate the loss of expression of CIPs in the IPF lungs.
Figure 1.

Down-regulation of CIPs CD46 and CD55 in IPF vs. non-IPF human lung biopsy tissues. A) Pathologically normal (non-IPF) and IPF lung biopsies were homogenized separately and then subjected to immunoblot analysis with antibodies against CD46, CD55, and GAPDH (loading control). B, C) Densitometry analyses of individual band intensities showed lower expression of CD46 (B) and CD55 (C) in IPF tissues when normalized to GAPDH. Data represent means ± sem (normal lungs: n=3; IPF lungs, n=11); unpaired t test. *P < 0.05.
Figure 2.

Loss of CD46 and CD55 in IPF lungs. Comparative immunohistochemical analysis of paraffin-embedded human IPF lung biopsy explants obtained at lung transplant and tissue resected from normal (non-IPF) lungs was conducted with CD46 and CD55 staining. A) Top panels: normal lungs at the time of resection for other diseases. Normal lung architecture with significant CD46 and CD55 expression in the airway (2 arrows) and the alveolar epithelium (1 arrow). Bottom panels: IPF lung tissue biopsy. Disrupted airway epithelium (2 arrows) and alveolar epithelium (1 arrow), with loss of CD46 and CD55 staining (DAB, brown) appearing in the mesenchymal cells at the fibroblastic foci (FF). Nuclei were counterstained with hematoxylin (blue). Insets: airway (left panels); alveoli and interstitium (right panels). Representative lesions observed from 5 different patients are presented. AW, airway; AL, alveoli. Scale bars = 100 μm (×20). B) Normal lungs immunostained with corresponding rabbit IgG. Scale bars = 100 μm (×20). C) Intensity analysis of CD46 and CD55 staining in 5 normal and 5 IPF tissue sections. Values represent means ± sem; unpaired t test. *P < 0.05.
Although the conversion products of the complement pathway, C3a and C5a, are reportedly higher in patients with IPF (27), loss of CIPs, as shown in Fig. 1, suggested complement activation in IPF tissues. Furthermore, C3a and C5a levels in local and systemic IPF compartments are not clear. Accordingly, we examined the levels of C3a (with or without desArg) and C5a (with or without desArg), in lung tissue homogenates and plasma derived from patients with IPF. Compared to pathologically normal tissues, we observed >2-fold higher C3a (with or without desArg) levels in plasma (normal lungs, n=6; IPF lungs, n=20; P<0.0001; Fig. 3A) and >4-fold higher C3a (with or without desArg) levels in lung tissue homogenates (normal lungs, n=3; IPF lungs, n=10; P<0.024; Fig. 3C) derived from patients diagnosed with IPF. Interestingly, C5a (with or without desArg) levels in both compartments were significantly less than C3a (with or without desArg) levels. Compared to pathologically normal tissues, we observed >2-fold higher C5a (with or without desArg) levels in plasma (P<0.02; Fig. 3B) and lung homogenates (P<0.0087; Fig. 3D) derived from patients diagnosed with IPF. Together, these data on the systemic and local levels of the anaphylatoxins C3a (with or without desArg) and C5a (with or without desArg) indicate complement activation, which likely influence epithelial injury in the pathogenesis of IPF.
Figure 3.

The anaphylatoxins C3a and C5a are up-regulated systemically and locally in IPF. C3a and C5a (with or without desArg) levels were measured in plasma from normal volunteers (n=6) and patients with IPF (n=20) and in whole-lung homogenates from tissue explants derived from pathologically normal lungs (n=3) and from IPF lungs (n=10). We observed higher levels of plasma C3a (A) and C5a (B) and lung tissue C3a (C) and C5a (D). Values represent means ± sem, analyzed by unpaired t test (A, B) or unpaired t test with Welch's correction (C, D). *P < 0.05.
TGF-β1, C3a, and C5a mediate down-regulation of CD46 and CD55 in primary normal human small-airway epithelium
Although TGF-β1 is widely implicated in epithelial injury and IPF (3–5), our studies in Figs. 1 and 2 suggest a potential link between CIPs and C3a (with or without desArg) and C5a (with or without desArg) levels in IPF tissues. Initially, we determined the optimal dose that might mimic down-regulation of the CIPs, as observed in clinical IPF tissues. Toward this end, we employed normal primary human SAECs for in vitro studies. Although SAECs are not alveolar type II (ATII) epithelial cells, a key target of injury in IPF, they are isolated from the distal portion of normal human lung tissue in the 1 mm bronchiole area and have been proven to replicate many ATII characteristics (22, 28). SAECs were treated with various doses of TGF-β1 for up to 72 h. As expected, we observed a dose-dependent decrease in the expression of E-CAD (an epithelial marker) with complete loss at 10 ng/ml of TGF-β1 that was maximal at 72 h (Fig. 4A). Accordingly, we also observed dose-dependent decreases in CD46 and CD55, with almost near or complete loss at 10 ng/ml of TGF-β1 during the same period (Fig. 4A). We then treated the SAECs with various doses of recombinant human C3a (Fig. 4B) or C5a (Fig. 4C). We observed dose-dependent decreases in CD46 and CD55, with almost near or complete loss at 100 nM of C3a or C5a that was maximal at 24 h. Since down-regulation of CIPs causes stress to these epithelial cells, we focused on PARP, which is one of the main cleavage targets of caspase 3 and is activated in response to stress and DNA repair (29–31). Whereas total PARP helps cells to maintain their viability, cleavage of PARP yields an 89 kDa product, the carboxy-terminal catalytic domain, which facilitates cellular disassembly and serves as a marker of injured cells undergoing apoptosis. At the specific doses of TGF-β1, C3a, and C5a that caused down-regulation of the CIPs, we observed increased cleaved PARP expression (Fig. 4D). To confirm our findings, we subjected SAECs to RNAi-mediated gene silencing of CD46 and CD55 (Fig. 4E). With gene silencing of CD46, we observed marked down-regulation of CD46 expression with the absence of CD55 expression. With gene silencing of CD55, we observed near absence of CD55 expression with no appreciable effect on CD46 levels. Cleaved PARP was higher in cells with lower expression of CD46 or CD55 due to RNAi than in the epithelial cells transfected with the nontargeting siRNA sequence, indicating epithelial injury with loss of CIPs. Together, these data demonstrate that TGF-β1, similar to the effects of the anaphylatoxins C3a and C5a, can cause significant down-regulation of the CIPs with epithelial injury to normal primary human SAECs.
Figure 4.

TGF-β1, C3a, and C5a mediate down-regulation of the CIPs CD46 and CD55 in SAECs. SAECs were cultured in growth medium to 70% confluence and then cultured overnight in medium with 1:100 growth factors. A) SAECs were treated with various doses of TGF-β1 (0.01, 0.1, 1, 2.5, 5, or 10 ng/ml) for 72 h or were not treated. Cell lysates were immunoblotted with antibodies recognizing E-CAD, CD46, and CD55 (β-actin, loading control). B, C) SAECs were treated with various doses (10, 50, or 100 nM) of C3a (B) or C5a (C) for 24 h or were not treated. Cell lysates were immunoblotted with antibodies recognizing E-CAD, CD46, and CD55 (β-actin, loading control). D) Cell lysates from A–C were immunoblotted for total/cleaved PARP (β-actin, loading control). E) SAECs were transfected with nontargeting or CD46- or CD55-specific siRNA sequences for 24 h, followed by overnight culture in basal medium with 1:100 growth factors and then 24 h culture in growth medium. Cell lysates were immunoblotted for CD46, CD55, and total/cleaved PARP (β-actin, loading control). Data are representative of analyses of cells from 3 independent donors.
p38MAPK inhibition blocks TGF-β1-mediated down-regulation of the CIPs CD46 and CD55 in SAECs
Our previous report demonstrated that IL-17A-mediated EMT occurs via TGF-β1-dependent signaling, particularly p38MAPK activation (22), and p38MAPK has been implicated in epithelial injury (32). However, the correlation between TGF-β1-mediated p38MAPK activation and complement activation–mediated epithelial injury is unknown. Therefore, to dissect the mechanism by which TGF-β1 mediates the down-regulation of the CIPs, we used pharmacologic inhibitors against the canonical MAPK pathways (33). Of these, pharmacologic inhibition of p38MAPK blocked TGF-β1-mediated down-regulation of the CIPs and loss of E-CAD (Fig. 5A). Densitometric analyses of data obtained from 3 independent donors demonstrated the protective effect of CIPs due to the blockade of p38MAPK activation (Fig. 5B, C). Notably, we did not observe any effects caused by inhibition of ERK or JNK (data not shown). These findings suggest that p38MAPK activation plays a critical role in the regulation of complement activation.
Figure 5.
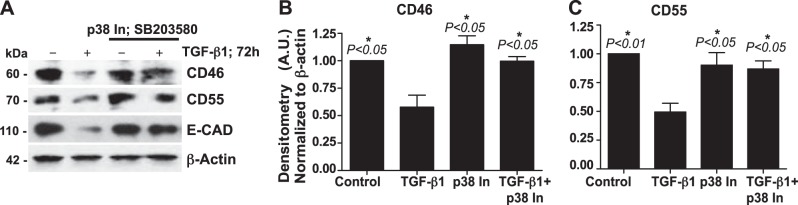
Blockade of p38MAPK activation protects against TGF-β1-mediated loss of the CIPs CD46 and CD55 in SAECs. A) SAECs were pretreated with p38MAPK inhibitor (p38 In; SB203580; 6 μM) for 1 h and then were treated with TGF-β1 (10 ng/ml) for 72 h or were not treated. The cell lysates were immunoblotted with antibodies recognizing E-CAD, CD46, and CD55 (β-actin, loading control). Data are representative of analyses of cells from 3 independent donors. B, C)) Densitometry of CD46 (A) and CD55 (A) from panel A. Data represent means ± sem (n=3). *P < 0.05 vs. TGF-β1; 1-way ANOVA with Bonferroni post hoc test.
RNAi-mediated gene silencing of Snail expression protects against TGF-β1-mediated down-regulation of the CIPs CD46 and CD55 in SAECs
The canonical EMT transcription factor Snail has been implicated in epithelial injury and the pathogenesis of IPF, particularly in the repression of epithelial targets (5, 20) such as E-CAD, which is essential for maintaining the functional integrity of the epithelial cells. To evaluate the role of Snail in the regulation of CIP expression, we first investigated the temporal expression pattern of SNAI1 in cultured SAECs. SAECs were cultured with or without TGF-β1 for specific times. We observed a robust (∼5-fold) increase in SNAI1 expression at 6 h (P<0.05), and this response was consistently observed in the SAECs derived from 3 different donors (Fig. 6A). Since experimental models of fibrosis and EMT have demonstrated that Snail is expressed downstream of p38MAPK signaling (21, 34), we wanted to know whether blockade of p38MAPK activation in SAECs regulates SNAI1 expression. SAECs were pretreated with p38MAPK inhibitor and then treated with TGF-β1. We observed that the robust TGF-β1-mediated induction of SNAI1 was suppressed by blockade of p38MAPK activation (Fig. 6B). To examine the specific role of SNAI1 in mediating CIP down-regulation, we used an RNAi approach with nontargeting and SNAI1-specific siRNA sequences. The transfected SAECs were treated with TGF-β1. We observed significant down-regulation of TGF-β1-mediated induction of SNAI1 transcripts (Fig. 6C). Subsequently, we immunoblotted protein lysates against Snail, CD55, CD46, and cleaved/total PARP, using β-actin as the loading control. SNAI1-specific siRNA efficiently blocked TGF-β1-induced Snail expression. SAECs lacking Snail expression were protected from TGF-β1-mediated down-regulation of the CIPs CD46 and CD55 (Fig. 6D). We also observed that TGF-β1-mediated induction of cleaved PARP expression was blocked due to silencing of SNAI1 expression (Fig. 6D). Collectively, these findings suggest that TGF-β1-induced Snail expression is suppressed by blocking p38MAPK activation and implicate Snail in TGF-β1-mediated down-regulation of the CIPs CD46 and CD55.
Figure 6.

RNAi-mediated gene silencing of SNAI1 protects against loss of the CIPs CD46 and CD55 in SAECs. A) SAECs were treated with or without TGF-β1 (10 ng/ml) for the indicated times. RNA lysates were used to synthesize cDNA, which was then subjected to quantitative reverse transcriptase PCR (qRT-PCR) for SNAI1 and normalized with GAPDH. Values represent means ± sem (n=3). *P < 0.05 vs. baseline; unpaired t test. B) SAECs were pretreated with p38MAPK inhibitor (p38 In; SB203580; 6 μM) for1 h and then treated with TGF-β1 (10 ng/ml) for 6 h or were not treated. SNAI1 was analyzed by qRT-PCR (endogenous control, GAPDH). Values represent means ± sem (n=3). *P < 0.05, **P < 0.01 vs. TGF-β1; 1-way ANOVA with Bonferroni post hoc test. C) SAECs were transfected with nontargeting or SNAI1-specific siRNA sequences for 24 h, followed by treatment with TGF-β1 (10 ng/ml) for 6 h, or were not treated. SNAI1 was analyzed by qRT-PCR (endogenous control, GAPDH). Values represent means ± sem (n=3–4). *P < 0.05, **P < 0.01 vs. TGF-β1; 1-way ANOVA with Bonferroni post hoc test. D) SAECs were transfected as in C, followed by treatment with TGF-β1 (10 ng/ml) for 48 h, or were not treated. Protein lysates were immunoblotted against Snail, CD46, CD55, and total/cleaved PARP (β-actin, loading control). Data are representative of analyses of cells from 3 independent donors.
p38MAPK inhibition blocks C3a- and C5a-mediated down-regulation of the CIPs CD46 and CD55 in SAECs
We had defined the role of p38MAPK and Snail in the protection of CIP expression, and others have reported that a receptor/ligand complex of C5a may lead to Gi protein-dependent alternative activation of p38MAPK in human mast cells (35). We now sought to find out whether C3a and/or C5a can induce p38MAPK activation and downstream Snail expression in SAECs. Expanding on the dose-response studies in Fig. 4, we treated SAECs with C3a for various times. Initially, under the same conditions, C3a and C5a indeed down-regulated the CIPs in SAECs, as shown in Fig. 4. As expected, in response to C3a (Fig. 7A) and C5a (Fig. 7B), we observed a gradual down-regulation of CD46 and CD55 by 6 h and a robust early activation of p38MAPK at 30 min. Because both C3a and C5a induced p38MAPK activation and Snail expression, we next investigated the effects of pharmacologic inhibition of p38MAPK on C3a-/C5a-mediated loss of CIPs in SAECs (Fig. 7C). Pharmacologic blockade of p38MAPK prevented loss of CIPs in response to C3a and C5a. We next assessed the effect of C3a and C5a on Snail, the transcriptional repressor of E-CAD. Interestingly, temporal studies of the effects of C3a on SAECs showed that, whereas C3a had a biphasic induction effect on Snail with concomitant loss of E-CAD as early as 30 min and again at 24 h (Fig. 7D), C5a at 100 nM, had a gradual induction effect on Snail expression, with concomitant loss of E-CAD (Fig. 7E). We next used a p38MAPK inhibitor to determine whether ablation of p38MAPK activation would block the expression of C3a- and C5a-mediated Snail expression. Figure 7F shows that although p38MAPK inhibitor blocked C3a-mediated SNAI1 transcripts, there was no conclusive effect of the inhibitor on C5a-mediated SNAI1 expression. Together, these results suggest that C3a- and C5a-mediated down-regulation of CIPs may occur via common signaling pathways also triggered by TGF-β1.
Figure 7.
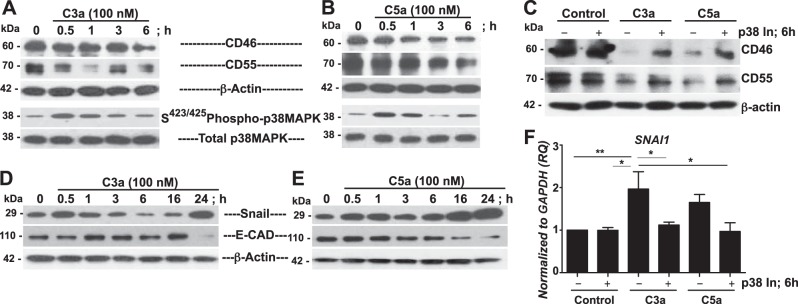
Blockade of p38MAPK activation protects against C3a- and C5a-mediated loss of the CIPs CD46 and CD55 in SAECs. A, B) SAECs were treated with C3a (A) or C5a (B) for the indicated times. Cell lysates were immunoblotted for CD46, CD55, Ser423/425, and total p38MAPK (β-actin, loading control). C) SAECs were pretreated with p38MAPK inhibitor (p38 In; SB203580; 6 μM) for 1 h and then treated with or without C3a or C5a (100 nM) for 6 h. Cell lysates were immunoblotted for CD46 and CD55 (β-actin, loading control). D, E) SAECs were treated with or without C3a (D) or C5a (E) for the indicated times. Cell lysates were immunoblotted for E-CAD and Snail (β-actin, loading control). F) SAECs were treated as in C. SNAI1 was analyzed by quantitative reverse transcriptase PCR (qRT-PCR; endogenous control, GAPDH). Data represent means ± sem (n=3). Data are representative of analyses of cells from 3 independent donors. *P < 0.05, **P < 0.01 vs. C3a; 1-way ANOVA with Newman-Keuls post hoc test.
Crosstalk between TGF-β1 and the anaphylatoxins C3a and C5a in SAECs
To investigate potential crosstalk between TGF-β1 and the anaphylatoxins, we examined the temporal expression of the TGFb and SMAD7 transcripts in response to C3a and C5a in SAECs derived from 3 different donor lungs. We observed a ∼2-fold induction of TGFb at 24 h in response to C3a (P < 0.007; Fig. 8A); however, C5a did not have a comparable effect on TGFb gene expression (Fig. 8B). Interestingly, Fig. 8B demonstrates that both C3a (P<0.007) and C5a (P<0.0001) significantly suppressed the gene expression of the TGF-β1 inhibitor SMAD7 (50% in 6 h; Fig. 8B). Furthermore, we examined the temporal regulation of specific receptors C3aR and C5aR in response to TGF-β1. Although TGF-β1 induced protein expression of C3aR at 6 h, which was sustained even at 24 h, that of C5aR was gradual, with up-regulation observed from 3 to 24 h (Fig. 8C). We next investigated the effect of pharmacologic blockade of C3aR and C5aR in response to C3a and C5a, respectively. We used SB290157 (C3aRA, a selective competitive inhibitor) to block C3aR and PMX205 (C5aRA, an allosteric, noncompetitive inhibitor) to block C5aR. We observed that C3aRA and C5aRA protected against loss of CIPs and induction of Snail expression, in response to C3a (Fig. 8D) and C5a (Fig. 8E), respectively. Collectively, these results suggest that TGF-β1, C3a, and C5a drive epithelial injury and eventual fibrosis via a feed-forward loop.
Figure 8.

Crosstalk among C3a, C5a, and TGF-β1. SAECs were treated with or without C3a or C5a (100 nM) for the indicated times. A, B) TGFb (A) and SMAD7 (B) were analyzed by quantitative reverse transcriptase PCR (qRT-PCR; endogenous control, GAPDH). Values represent means ± sem (n=3). *P < 0.05; unpaired t test. C) SAECs were treated with or without TGF-β1 (10 ng/ml) for the indicated times. Cell lysates were immunoblotted against specific receptors C3aR and C5aR (β-actin, loading control). D, E) SAECs were pretreated for 1 h with C3aRA (SB209157; 50, 100 μM; D) or C5aRA (PMX205; 1, 5 μM; E) and then treated with or without C3a or C5a (100 nM) for 6 h, respectively. Cell lysates were immunoblotted for CD46, CD55, and Snail (β-actin, loading control). Data are representative analyses of cells from 3 independent donors.
DISCUSSION
In this study, we asked whether there were potential links among TGF-β1, complement activation, and epithelial injury that would eventually lead to pulmonary fibrosis. Datasets presented in this study (Fig. 9) show that loss of CIPs resulted in epithelial injury, leading to apoptosis, as demonstrated by cleaved PARP. TGF-β1-mediated down-regulation of CIPs may occur via the p38MAPK/Snail signaling axis, which may also be triggered by C3a and C5a. Furthermore, C3a- and C5a-mediated epithelial injury may be enhanced due to TGF-β1-mediated induction of their respective receptors. TGF-β1, in turn, was expressed by C3a- or C5a-injured epithelial cells with significant down-regulation of mothers against decapentaplegic homolog 7 (SMAD7), the inhibitory Smad. Pharmacologic blockade of these receptors protect against TGF-β1-, C3a-, and C5a-mediated loss of CIPs. Thus, TGF-β1 can facilitate complement activation by down-regulating CIPs, which is complicated by anaphylatoxin-induced suppression of CIPs while facilitating TGF-β1-mediated signaling, eventually resulting in tissue injury.
Figure 9.

Model of the crosstalk between compliment activation and TGF-β1 in augmenting epithelial injury. TGF-β1 mediates epithelial injury by down-regulating the CIPs via the p38MAPK/Snail axis in primary normal human SAECs. Both C3a and C5a are capable of down-regulating CIPs, directly and by inducing the p38MAPK/Snail axis. Furthermore, whereas both C3a and C5a effectively suppress SMAD7, only C3a induces TGF-β1 expression. TGF-β1 contributes to the signaling potency of C3a and C5a by up-regulating the expression of their respective receptors. The crosstalk collectively amplifies epithelial injury and tissue damage.
To the best of our knowledge, this is the first report to clearly demonstrate the clinical relevance of the expression profile of the CIPs and the anaphylatoxins in IPF and link it with underlying molecular mechanisms leading to epithelial injury. Although CIPs are significantly down-regulated in IPF vs. non-IPF human lung biopsy tissue, as suggested by immunoblot analysis of the whole-lung homogenates, it is possible that the decrease is due to substantial epithelial injury in the diseased lung. Whereas CD46 blocks the formation of activated C3 convertases (36), CD55 “accelerates” the “decay” of preformed C3 convertase and blocks the formation of the membrane attack complex (37). We also report that IPF tissues expressed higher levels of C3a and C5a in the lungs and in the systemic circulation. Collectively, the data presented link complement activation to loss of CIPs and eventual tissue injury.
Emerging concepts of repeated epithelial injury have superseded the concept of chronic inflammation in the pathogenesis of IPF, with several studies establishing the critical role of TGF-β1 in initiating or exacerbating epithelial injury/IPF (38–40). Our observations in vitro suggest that although down-regulation of CIPs in response to TGF-β1 occurs at 72 h, this effect is observed even earlier in response to both C3a and C5a, even at 6 h (CD46; Fig. 7) or 24 h (CD46 and CD55; Fig. 4). Interestingly, this trend was confirmed in at least 3 different normal donor lungs. Although cleaved PARP was up-regulated in the cells treated with TGF-β1, C3a, or C5a, we conclusively demonstrated that down-regulation or loss of the individual CIPs via RNAi, led to increased cleaved PARP formation, which may eventually render the cell apoptotic. Notably, the levels of C3a and C5a used in this study (∼90.91 and ∼83.3 ng/ml, respectively) were equivalent to the levels found locally in the lungs of patients with IPF (Fig. 3C, D).
p38MAPK has been widely implicated in epithelial injury (22, 32, 41). Accordingly, we observed a role of p38MAPK in protecting against the loss of CIPs. We eliminated the possibility of the effect of 2 other MAPKs, ERK and JNK, by using specific inhibitors. We found that blockade of ERK and JNK under the same conditions did not protect against the loss of CIPs (data not shown). Cumulatively, these data suggest one more mechanism by which p38MAPK induces epithelial injury.
Typically, epithelial remodeling and repair go through a series of events that include epithelial cell–cell and cell–matrix adhesion contacts, reorganization of the actin cytoskeleton, induction of mesenchymal gene expression, and acquisition of motile capacity. These events are tightly controlled by several transcriptional factors, such as Twist, NF-κB, Rho, Rac, Snail, and GSK-3β (42). In this context, SNAI1 has been implicated in the repression of epithelial targets in IPF (20) and a selective increase in paracellular ion permeability in tight junctions (43). Our study confirms the finding a previously reported study in kidney tubulointerstitial epithelial cells that Snail expression is indeed downstream of p38MAPK (21). We therefore targeted Snail expression and confirmed the role of Snail in complement activation. Both C3a and C5a robustly activated p38MAPK, similar to the effects of TGF-β1. Interestingly, C3a induced Snail in a biphasic manner, with early induction at 30 min, which declined by 6 h, and late induction again at 24 h, consistent with concomitant E-CAD expression. Furthermore, C5a induced Snail expression early (at 30 min), but the subsequent induction was more gradual. Overall, Snail induction by both C3a and C5a was temporally earlier than that mediated by TGF-β1. Notably, although C3a robustly induced Snail expression in all donors, the heterogeneity in the various donors and a possible discrepancy in temporal induction of Snail by C5a resulted in inconclusive data in our study. Our previous study implicated p38MAPK in EMT (22), but this is the first report to demonstrate that blockade of p38MAPK protects against C3a- and C5a-mediated loss of CIPs.
Because we defined the role of TGF-β1-associated molecular targets such as p38MAPK and Snail in the loss of CIPs and showed that TGF-β1-mediated loss of CIPs was temporally delayed compared with loss of C3a and C5a, we also examined the role of C3a and C5a in TGF-β1 regulation. The TGF-β/Smad2/3 signaling pathway has been implicated in clinical IPF (44). TGF-β1 signaling occurs via type I and II receptor–mediated phosphorylation, whereby activated TGF-β1 receptor I phosphorylates Smad2 and Smad3 [receptor (R)-Smads] at C termini. R-Smads translocate to the nucleus and trigger fibrogenesis. Whereas the R-Smad linker region is phosphorylated by MAPK, activation is antagonized by inhibitory Smad7 (45) overexpression, which down-regulates TGF-β-induced activity and fibrosis (46). Liu et al. (47) have reported the induction of TGF-β1 in response to both C3a and C5a in normal human proximal tubular epithelial cells. However, in our study, we detected a consistent response of selective induction of TGF-β1 by C3a, but not by C5a, in cells from at least 3 different donors. Furthermore, to our knowledge, this is the first report to demonstrate the down-regulation of the inhibitory Smad7 via C3a and C5a treatment of SAECs. Accordingly, it is possible that blockade of receptors specific to C3a and C5a alleviates TGF-β expression and the downstream signaling cascade. Interestingly, we did not detect C3a-mediated induction of the TGF-β-specific intracellular signaling molecule Smad 2/3 in SAECs. Overall, these data provide additional insight into the molecular mechanism and crosstalk between TGF-β1 and complement activation in the pathogenesis of IPF.
C3a and C5a exert their functions by binding to their G-protein-coupled receptors, C3aR and C5aR, present on most myeloid and parenchymal cells (48). C3aR and C5aR have similar structures, but their functions may overlap, be disparate, or be mutually exclusive events, prompting their being called the “salt and pepper of immune response” by Steven Sacks (49). Specifically, C3aR signaling is implicated in proteinuric nephropathy (15) and fatal asthma (50). C5aR signaling has been implicated in renal allograft survival (51), tubulointerstitial fibrosis (52), and acute lung injury (53, 54). To the best of our knowledge, our current report is the first to demonstrate up-regulation of C3aR and C5aR expression. Pharmacologic approaches to blocking C3aR and C5aR protected against C3a- and C5a-mediated loss of CIPs and induction of Snail. With inhibition of C3aR, we also observed that, whereas loss of CD55 was prevented, CD46 expression was not as robust with blockade of C3aR. This finding may have been due to the effects of C3a-mediated TGFb expression at that particular time point. Besides, it is possible that because of nonspecific effects of the inhibitor, we detected loss of CIP with the higher dose of either inhibitor alone. We also observed induction of C5L2 (C5ar2) in SAECs, in response to TGF-β1, (data not shown). One of the mechanisms behind C3aR- and C5aR-mediated downstream signaling in response to circulating immune complexes in vitro may be via p38MAPK activation; however, their varied effects both in vitro and in vivo on epithelial injury and mesenchymal functions remain to be established.
In summary, our findings suggest that immune complexes cause complement activation, which in conjunction with TGF-β1 accelerate epithelial injury via shared signaling pathways, thus triggering a positive feedback mechanism. Our results further suggest that targeting the complement cascade and a previously unexplored combination strategy of blocking signaling by C3aR and C5aR in addition to that of TGF-β1 may be a novel therapeutic approach in IPF.
Acknowledgments
The authors thank K. P. Lipking for analyzing the immunostaining intensity in the clinical tissues.
The study was funded in part by U.S. National Institutes of Health grants NIH-NHLBI-LTRC CS11-99-0002 and NIH-NHLBI R01 HL109288 to R.V. and NIH-NHLBI R01 HL096845 to D.S.W. D.S.W. is a cofounder of ImmuneWorks, Inc., a biotechnology company involved in developing therapeutics for various forms of lung diseases.
The other authors declare no conflicts of interests.
Footnotes
- ATII
- alveolar type II
- C3a
- complement component 3a
- C3aR
- complement component 3a receptor
- C5a
- complement component 5a
- C5aR
- complement component 5a receptor
- CD46
- cluster of differentiation 46
- CD55
- cluster of differentiation 55
- CIP
- complement inhibitory protein
- E-CAD
- E-cadherin
- EMT
- epithelial–mesenchymal transition
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- IL-17A
- interleukin-17A
- IPF
- idiopathic pulmonary fibrosis
- p38MAPK
- mitogen-activated protein kinase
- PARP
- poly(ADP-ribose) polymerase
- PCR
- polymerase chain reaction
- RNAi
- RNA interference
- SABM
- small airway basal medium
- SAEC
- small airway epithelial cell
- siRNA
- small interference RNA
- SMAD7
- mothers against decapentaplegic homolog 7
- TGF-β1
- transforming growth factor β, isoform 1
REFERENCES
- 1. Katzenstein A. L., Myers J. L. (1998) Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 157, 1301–1315 [DOI] [PubMed] [Google Scholar]
- 2. Gross T. J., Hunninghake G. W. (2001) Idiopathic pulmonary fibrosis. N Engl. J. Med. 345, 517–525 [DOI] [PubMed] [Google Scholar]
- 3. Pittet J. F., Griffiths M. J., Geiser T., Kaminski N., Dalton S. L., Huang X., Brown L. A., Gotwals P. J., Koteliansky V. E., Matthay M. A., Sheppard D. (2001) TGF-beta is a critical mediator of acute lung injury. J. Clin. Invest. 107, 1537–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Polosukhin V. V., Degryse A. L., Newcomb D. C., Jones B. R., Ware L. B., Lee J. W., Loyd J. E., Blackwell T. S., Lawson W. E. (2012) Intratracheal bleomycin causes airway remodeling and airflow obstruction in mice. Exp. Lung Res. 38, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou B., Buckley S. T., Patel V., Liu Y., Luo J., Krishnaveni M. S., Ivan M., DeMaio L., Kim K. J., Ehrhardt C., Crandall E. D., Borok Z. (2012) Troglitazone attenuates TGF-beta1-induced EMT in alveolar epithelial cells via a PPARgamma-independent mechanism. PloS One 7, e38827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willis B. C., Liebler J. M., Luby-Phelps K., Nicholson A. G., Crandall E. D., du Bois R. M., Borok Z. (2005) Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 166, 1321–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim K. K., Kugler M. C., Wolters P. J., Robillard L., Galvez M. G., Brumwell A. N., Sheppard D., Chapman H. A. (2006) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 103, 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dreisin R. B., Schwarz M. I., Theofilopoulos A. N., Stanford R. E. (1978) Circulating immune complexes in the idiopathic interstitial pneumonias. N. Engl. J. Med. 298, 353–357 [DOI] [PubMed] [Google Scholar]
- 9. Jansen H. M., Schutte A. J., Elema J. D., Giessen M. V., Reig R. P., Leeuwen M. A., Sluiter H. J., The T. H. (1984) Local immune complexes and inflammatory response in patients with chronic interstitial pulmonary disorders associated with collagen vascular diseases. Clin. Exp. Immunol. 56, 311–320 [PMC free article] [PubMed] [Google Scholar]
- 10. Phan S. H., Thrall R. S. (1982) Inhibition of bleomycin-induced pulmonary fibrosis by cobra venom factor. Am. J. Pathol. 107, 25–28 [PMC free article] [PubMed] [Google Scholar]
- 11. Addis-Lieser E., Kohl J., Chiaramonte M. G. (2005) Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J. Immunol. 175, 1894–1902 [DOI] [PubMed] [Google Scholar]
- 12. Kohl J., Gessner J. E. (1999) On the role of complement and Fc gamma-receptors in the Arthus reaction. Mol. Immunol. 36, 893–903 [DOI] [PubMed] [Google Scholar]
- 13. Suzuki H., Lasbury M. E., Fan L., Vittal R., Mickler E. A., Benson H. L., Shilling R., Wu Q., Weber D. J., Wagner S. R., Lasaro M., Devore D., Wang Y., Sandusky G. E., Lipking K., Pandya P., Reynolds J., Love R., Wozniak T., Gu H., Brown K. M., Wilkes D. S. (2013) Role of complement activation in obliterative bronchiolitis post-lung transplantation. J. Immunol. 191, 4431–4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drouin S. M., Kildsgaard J., Haviland J., Zabner J., Jia H. P., McCray P. B., Jr., Tack B. F., Wetsel R. A. (2001) Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J. Immunol. 166, 2025–2032 [DOI] [PubMed] [Google Scholar]
- 15. Tang Z., Lu B., Hatch E., Sacks S. H., Sheerin N. S. (2009) C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J. Am. Soc. Nephrol. 20, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Braun M. C., Reins R. Y., Li T. B., Hollmann T. J., Dutta R., Rick W. A., Teng B. B., Ke B. (2004) Renal expression of the C3a receptor and functional responses of primary human proximal tubular epithelial cells. J. Immunol. 173, 4190–4196 [DOI] [PubMed] [Google Scholar]
- 17. Varsano S., Frolkis I., Ophir D. (1995) Expression and distribution of cell-membrane complement regulatory glycoproteins along the human respiratory tract. Am. J. Respir. Crit. Care Med. 152, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 18. Varsano S., Frolkis I., Rashkovsky L., Ophir D., Fishelson Z. (1996) Protection of human nasal respiratory epithelium from complement-mediated lysis by cell-membrane regulators of complement activation. Am. J. Respir. Cell Mol. Biol. 15, 731–737 [DOI] [PubMed] [Google Scholar]
- 19. Quigg R. J., Nicholson-Weller A., Cybulsky A. V., Badalamenti J., Salant D. J. (1989) Decay accelerating factor regulates complement activation on glomerular epithelial cells. J. Immunol. 142, 877–882 [PubMed] [Google Scholar]
- 20. Jayachandran A., Konigshoff M., Yu H., Rupniewska E., Hecker M., Klepetko W., Seeger W., Eickelberg O. (2009) SNAI transcription factors mediate epithelial-mesenchymal transition in lung fibrosis. Thorax 64, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 21. Vidyasagar A., Reese S., Acun Z., Hullett D., Djamali A. (2008) HSP27 is involved in the pathogenesis of kidney tubulointerstitial fibrosis. Am. J. Physiol. Renal Physiol. 295, F707–F716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vittal R., Fan L., Greenspan D. S., Mickler E. A., Gopalakrishnan B., Gu H., Benson H. L., Zhang C., Burlingham W., Cummings O. W., Wilkes D. S. (2012) IL-17 induces type V collagen overexpression and EMT via TGF-beta dependent pathways in obliterative bronchiolitis. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L401–L414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vittal R., Fan L., Greenspan D. S., Mickler E. A., Gopalakrishnan B., Gu H., Benson H. L., Zhang C., Burlingham W., Cummings O. W., Wilkes D. S. (2013) IL-17 induces type V collagen overexpression and EMT via TGF-beta-dependent pathways in obliterative bronchiolitis. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vittal R., Fisher A., Gu H., Mickler E. A., Panitch A., Lander C., Cummings O. W., Sandusky G. E., Wilkes D. S. (2013) Peptide-mediated inhibition of MK2 ameliorates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 49, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vittal R., Mickler E. A., Fisher A. J., Zhang C., Rothhaar K., Gu H., Brown K. M., Emtiazdjoo A., Lott J. M., Frye S. B., Smith G. N., Sandusky G. E., Cummings O. W., Wilkes D. S. (2013) Type V collagen induced tolerance suppresses collagen deposition, TGF-beta and associated transcripts in pulmonary fibrosis. PloS One 8, e76451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meliconi R., Senaldi G., Sturani C., Galavotti V., Facchini A., Gasbarrini G., Vergani D. (1990) Complement activation products in idiopathic pulmonary fibrosis: relevance of fragment Ba to disease severity. Clin. Immunol. Immunopathol. 57, 64–73 [DOI] [PubMed] [Google Scholar]
- 28. Akram K. M., Lomas N. J., Spiteri M. A., Forsyth N. R. (2013) Club cells inhibit alveolar epithelial wound repair via TRAIL-dependent apoptosis. Eur. Respir. J.41, 683–694 [DOI] [PubMed] [Google Scholar]
- 29. Fujita T., Maruyama M., Araya J., Sassa K., Kawagishi Y., Hayashi R., Matsui S., Kashii T., Yamashita N., Sugiyama E., Kobayashi M. (2002) Hydrogen peroxide induces upregulation of Fas in human airway epithelial cells via the activation of PARP-p53 pathway. Am. J. Respir. Cell Mol. Biol. 27, 542–552 [DOI] [PubMed] [Google Scholar]
- 30. Boulares A. H., Zoltoski A. J., Sherif Z. A., Jolly P., Massaro D., Smulson M. E. (2003) Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 28, 322–329 [DOI] [PubMed] [Google Scholar]
- 31. Singhera G. K., MacRedmond R., Dorscheid D. R. (2008) Interleukin-9 and -13 inhibit spontaneous and corticosteroid induced apoptosis of normal airway epithelial cells. Exp. Lung Res. 34, 579–598 [DOI] [PubMed] [Google Scholar]
- 32. Yadav U. C., Ramana K. V., Srivastava S. K. (2013) Aldose reductase regulates acrolein-induced cytotoxicity in human small airway epithelial cells. Free Radic. Biol. Med. 65C, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wettstein G., Bellaye P. S., Kolb M., Hammann A., Crestani B., Soler P., Marchal-Somme J., Hazoume A., Gauldie J., Gunther A., Micheau O., Gleave M., Camus P., Garrido C., Bonniaud P. (2013) Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. 27, 1549–1560 [DOI] [PubMed] [Google Scholar]
- 35. Nishiura H., Tokita K., Li Y., Harada K., Woodruff T. M., Taylor S. M., Nsiama T. K., Nishino N., Yamamoto T. (2010) The role of the ribosomal protein S19 C-terminus in Gi protein-dependent alternative activation of p38 MAP kinase via the C5a receptor in HMC-1 cells. Apoptosis 15, 966–981 [DOI] [PubMed] [Google Scholar]
- 36. Liszewski M. K., Farries T. C., Lublin D. M., Rooney I. A., Atkinson J. P. (1996) Control of the complement system. Adv. Immunol. 61, 201–283 [DOI] [PubMed] [Google Scholar]
- 37. Molina H., Wong W., Kinoshita T., Brenner C., Foley S., Holers V. M. (1992) Distinct receptor and regulatory properties of recombinant mouse complement receptor 1 (CR1) and Crry, the two genetic homologues of human CR1. J. Exp. Med. 175, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Henderson W. R., Jr., Chi E. Y., Ye X., Nguyen C., Tien Y. T., Zhou B., Borok Z., Knight D. A., Kahn M. (2010) Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 107, 14309–14314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kapanci Y., Desmouliere A., Pache J. C., Redard M., Gabbiani G. (1995) Cytoskeletal protein modulation in pulmonary alveolar myofibroblasts during idiopathic pulmonary fibrosis: possible role of transforming growth factor beta and tumor necrosis factor alpha. Am. J. Respir. Crit. Care Med. 152, 2163–2169 [DOI] [PubMed] [Google Scholar]
- 40. Li M., Krishnaveni M. S., Li C., Zhou B., Xing Y., Banfalvi A., Li A., Lombardi V., Akbari O., Borok Z., Minoo P. (2011) Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Invest. 121, 277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bakin A. V., Rinehart C., Tomlinson A. K., Arteaga C. L. (2002) p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206 [DOI] [PubMed] [Google Scholar]
- 42. Thiery J. P., Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell. Biol. 7, 131–142 [DOI] [PubMed] [Google Scholar]
- 43. Carrozzino F., Soulie P., Huber D., Mensi N., Orci L., Cano A., Feraille E., Montesano R. (2005) Inducible expression of Snail selectively increases paracellular ion permeability and differentially modulates tight junction proteins. Am. J. Physiol. Cell Physiol. 289, C1002–C1014 [DOI] [PubMed] [Google Scholar]
- 44. Fernandez I. E., Eickelberg O. (2012) The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc. Am. Thorac. Soc. 9, 111–116 [DOI] [PubMed] [Google Scholar]
- 45. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C. Y., Shyu A. B., Muller M., Gaestel M., Resch K., Holtmann H. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18, 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y. Y., Grinnell B. W., Richardson M. A., Topper J. N., Gimbrone M. A., Jr., Wrana J. L., Falb D. (1997) The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 47. Liu F., Gou R., Huang J., Fu P., Chen F., Fan W. X., Huang Y. Q., Zang L., Wu M., Qiu H. Y., Wei D. P. (2011) Effect of anaphylatoxin C3a, C5a on the tubular epithelial-myofibroblast transdifferentiation in vitro. Chin. Med. J. 124, 4039–4045 [PubMed] [Google Scholar]
- 48. Loverre A., Tataranni T., Castellano G., Divella C., Battaglia M., Ditonno P., Corcelli M., Mangino M., Gesualdo L., Schena F. P., Grandaliano G. (2011) IL-17 expression by tubular epithelial cells in renal transplant recipients with acute antibody-mediated rejection. Am. J. Transplant. 11, 1248–1259 [DOI] [PubMed] [Google Scholar]
- 49. Sacks S. H. (2010) Complement fragments C3a and C5a: the salt and pepper of the immune response. Eur. J. Immunol. 40, 668–670 [DOI] [PubMed] [Google Scholar]
- 50. Fregonese L., Swan F. J., van Schadewijk A., Dolhnikoff M., Santos M. A., Daha M. R., Stolk J., Tschernig T., Sterk P. J., Hiemstra P. S., Rabe K. F., Mauad T. (2005) Expression of the anaphylatoxin receptors C3aR and C5aR is increased in fatal asthma. J. Allergy Clin. Immunol. 115, 1148–1154 [DOI] [PubMed] [Google Scholar]
- 51. Li Q., Peng Q., Xing G., Li K., Wang N., Farrar C. A., Meader L., Sacks S. H., Zhou W. (2010) Deficiency of C5aR prolongs renal allograft survival. J. Am. Soc. Nephrol. 21, 1344–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boor P., Konieczny A., Villa L., Schult A. L., Bucher E., Rong S., Kunter U., van Roeyen C. R., Polakowski T., Hawlisch H., Hillebrandt S., Lammert F., Eitner F., Floege J., Ostendorf T. (2007) Complement C5 mediates experimental tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 18, 1508–1515 [DOI] [PubMed] [Google Scholar]
- 53. Bosmann M., Grailer J. J., Ruemmler R., Russkamp N. F., Zetoune F. S., Sarma J. V., Standiford T. J., Ward P. A. (2013) Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 27, 5010–5021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sun L., Guo R. F., Gao H., Sarma J. V., Zetoune F. S., Ward P. A. (2009) Attenuation of IgG immune complex-induced acute lung injury by silencing C5aR in lung epithelial cells. FASEB J. 23, 3808–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]