

Abstract
Nonalcoholic fatty liver disease (NAFLD) is today considered the most common form of chronic liver disease, affecting a high proportion of the population worldwide. NAFLD encompasses a large spectrum of liver damage, ranging from simple steatosis to steatohepatitis, advanced fibrosis and cirrhosis. Obesity, hyperglycemia, type 2 diabetes and hypertriglyceridemia are the most important risk factors. The pathogenesis of NAFLD and its progression to fibrosis and chronic liver disease is still unknown. Accumulating evidence indicates that mitochondrial dysfunction plays a key role in the physiopathology of NAFLD, although the mechanisms underlying this dysfunction are still unclear. Oxidative stress is considered an important factor in producing lethal hepatocyte injury associated with NAFLD. Mitochondrial respiratory chain is the main subcellular source of reactive oxygen species (ROS), which may damage mitochondrial proteins, lipids and mitochondrial DNA. Cardiolipin, a phospholipid located at the level of the inner mitochondrial membrane, plays an important role in several reactions and processes involved in mitochondrial bioenergetics as well as in mitochondrial dependent steps of apoptosis. This phospholipid is particularly susceptible to ROS attack. Cardiolipin peroxidation has been associated with mitochondrial dysfunction in multiple tissues in several physiopathological conditions, including NAFLD. In this review, we focus on the potential roles played by oxidative stress and cardiolipin alterations in mitochondrial dysfunction associated with NAFLD.
Keywords: Oxidative stress, Cardiolipin, Mitochondrial bioenergetics, Antioxidants, Nonalcoholic fatty liver disease
Core tip: Nonalcoholic fatty liver disease (NAFLD) is considered the most common form of chronic liver disease. Mitochondrial dysfunction and oxidative stress play a key role in the physiopathology of NAFLD. Mitochondrial respiratory chain is the main source of reactive oxygen species, which may damage mitochondrial proteins, lipids and mitochondrial DNA. Cardiolipin, a phospholipid of the inner mitochondrial membrane, plays an important role in mitochondrial bioenergetics and in apoptosis. Cardiolipin abnormalities have been associated with mitochondrial dysfunction in several physiopathological conditions, including NAFLD. In this review, we focus on the potential roles played by oxidative stress and cardiolipin alterations in mitochondrial dysfunction in NAFLD.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common form of chronic liver disease, affecting a high proportion of the population worldwide[1,2]. NAFLD refers to the wide spectrum of liver damage including several pathological conditions such as nonalcoholic steatohepatitis (NASH), advanced fibrosis and cirrhosis[3-5]. A number of predisposing factors have been related to NAFLD, such as obesity, diabetes, dyslipidemia, drugs and parenteral nutrition[6,7]. However, the pathogenesis of NAFLD and its progression to fibrosis and chronic liver disease is still unknown. The leading hypothesis for nonalcoholic-induced liver disease is the two-hits model[8]. The first hit is an initial metabolic alteration, like insulin resistance, hyperglycemia and hyperlipidemia and the accumulation of triglyceride in hepatocytes, leading to steatosis. The second hit triggers the progression to more severe liver pathologies like steatohepatitis, fibrosis and cirrhosis of liver. Examples of the second hit could be genetic and environmental factors like diet, smoke and pollutants leading to mitochondrial dysfunction. All these factors are recognized to induce oxidative stress, mainly via mitochondrial alterations. Mitochondrial dysfunction impairs fat homeostasis in liver and also leads to an overproduction of reactive oxygen species (ROS). These reactive oxygen species are considered to play an important role in inducing lethal hepatocyte injury associated with NAFLD[9-13]. Oxidative stress may cause damage at cellular level, such as membrane lipid peroxidation, cell degeneration and necrosis, cell death by apoptosis[14-16], proinflammatory cytokine expression, liver stellate cell activation and fibrogenesis[11,17-19].
It is well known that the mitochondrial respiratory chain is an important source of ROS at subcellular level and hence a potential contributing factor to NAFLD. Results obtained in our laboratory demonstrated enhanced mitochondrial ROS formation associated with an impairment of the mitochondrial respiratory chain in rats fed a choline-deficient diet (CDD)[20-22]. Similar results have been reported in patients with nonalcoholic steatosis[23]. In addition, it has been shown that patients with steatohepatitis present with ultrastructural mitochondrial alterations[24,25] and impairment of hepatic ATP synthesis[26].
Cardiolipin is a dimeric phospholipid found almost exclusively in the inner mitochondrial membrane (IMM) where it plays a pivotal role in several reactions and processes involved in mitochondrial bioenergetics, primarily the process of the oxidative phosphorylation[27-31]. In addition, cardiolipin is believed to participate in several steps of the intrinsic (mitochondrial) pathway of the apoptotic process[32-35], in mitochondrial morphology and dynamics, including fusion and fission processes, as well as in protein translocation, insertion and assembly into mitochondria[36,37]. Cardiolipin alterations are considered an important contributing factor in mitochondrial dysfunction in multiple tissues in several physiopathological situations[37-39]. Cardiolipin oxidation has been shown to be involved in mitochondrial dysfunction in insulin resistance[40], obesity[41] and NAFLD[22]. In this review, we focus on the potential roles played by oxidative stress and cardiolipin alterations in mitochondrial dysfunction associated with nonalcoholic fatty liver disease.
MITOCHONDRIAL ABNORMALITIES IN NAFLD
The mechanisms responsible for NAFLD are still not fully elucidated. Reduced capacity to oxidize fatty acids, increased delivery and transport of free fatty acids (FFAs) into the liver and enhanced hepatic fatty acid synthesis are considered important factors in the pathogenesis of fatty liver. Structural and functional mitochondrial alterations have been shown to contribute to the pathogenesis of NAFLD. Structural alterations include morphological changes, ultrastructural lesions and depletion of mitochondrial DNA (mtDNA), while functional alterations include the activity of respiratory chain complexes and the mitochondrial β-oxidation[19]. Abnormal morphological changes in liver mitochondria have been observed in patients and animal models of NASH[24,42]. Results obtained by electronic microscopy revealed that mitochondria in a mouse model with fatty oxidation defect and hepatic steatosis are large and swollen, limited in number, and that the mitochondrial matrix has paracrystalline inclusions and hypodensity[13]. These ultrastructural mitochondrial perturbations in patients with NAFLD may be indicative of altered mitochondrial functions, primarily the process of oxidative phosphorylation[23,43]. NAFLD, often found in patients with insulin resistance, obesity and type 2 diabetes, is associated with decreased oxygen consumption and ATP generation, reduced total mtDNA and mtDNA transcription factor A, and reduced content of respiratory proteins in the fat, muscle and liver[44]. MtDNA depletion in hepatocytes impairs mitochondrial function and causes hepatic steatosis and other liver injury[45]. Patients with NASH have decreased expression of mtDNA encoded polypeptides[46] and impaired activity of respiratory chain complexes[23].
Another important factor underlying the mitochondrial dysfunction found in NAFLD patients and animal models is tumor necrosis factor (TNF-α). The involvement of this cytokine in the pathogenesis of NASH is well documented[4]. High blood TNF-α levels have been found in patients with nonalcoholic fatty liver disease[47]. The levels of TNF-α in liver tissue of ob/ob mice were much higher than in normal mice[48]. The likely sources of hepatic TNF-α are hepatocytes and Kupffer cells[49]. TNF-α induces mitochondrial perturbations such as swelling with a lighter matrix and abnormal morphological alterations in the membrane. In addition, TNF-α-induced mitochondrial swelling causes a bursting of the membrane leading to an interference between respiratory chain complexes I and III[50]. Treatment of ob/ob mice with anti-TNF-α has a beneficial effect on mitochondrial respiratory chain complexes (I, III, IV and V) activity, α-oxidation activity and liver histology[48]. TNF-α may also alter electron transport chain (ETC) by inducing hypoxia-inducible factor -1α and mtDNA damage[18,48,51].
MITOCHONDRIA AS SOURCE OF ROS PRODUCTION
Oxidative stress is a condition due to an altered balance between the production of ROS and/or reactive nitrogen species (RNS) and the antioxidant defences capacity. The resulting imbalance appears to be involved in various physiopathological situations in which an oxidative insult causes tissue damage and cell death. The effects of ROS and RNS are not always injurious but, under physiological conditions, they represent essential signaling roles and physiological regulatory mechanisms in several vital cellular processes[52,53].
The redox state of the ETC components is considered the main factor involved in mitochondrial ROS generation. Electron transfer through the mitochondrial respiratory chain generates an electrochemical gradient which is utilized to synthesize ATP. In certain metabolic situations, such as high-fat or high-glucose states, inhibited ETC complexes activity or exposure to xenobiotics, the mitochondrial electrochemical gradient is high. Under these conditions the life of superoxide-generating electron transport intermediates, such as the ubisemiquinone radical, is prolonged, facilitating the transfer of electrons one at a time to oxygen, thus increasing the superoxide anion (O2•-) generation.
Mitochondria are the primary source of cellular reactive oxygen species. It has been reported that approximately 0.2%-2% of the oxygen consumed by the cell is converted by mitochondria to ROS, mainly through the production of superoxide anion (O2•-)[54]. Mitochondria consume around 90% of a cell’s oxygen to produce ATP through the process of oxidative phosphorylation. This process, however, comes with additional cost, the production of potentially harmful ROS. The mitochondrial electron transport chain is considered the main source of ROS. The primary oxygen radical species generated by mitochondria is superoxide anion which is then converted to hydrogen peroxide (H2O2) by spontaneous dismutation or by superoxide dismutase (SOD), present both within the mitochondria and in the cytosol. Hydrogen peroxide, in turn, is converted into water by glutathione peroxidase or catalase, otherwise, in the presence of divalent cations such as iron, H2O2 can undergo Fenton’s reaction to produce hydroxyl radical •OH.
Mechanisms of ROS production by mitochondrial respiratory chain complexes and the sites of their production have been reviewed extensively[55]. The two major sites of O2•- production are complex I (NADH-coenzyme Q reductase) and complex III (ubiquinone-cytochrome c oxidoreductase). Mitochondria can produce superoxide anion, predominantly from complex I, likely at the site of FMN[55] and from complex III, at the site of the unstable ubisemiquinone molecules[56,57] or the cytochrome b[58]. ROS are also generated, to a lesser extent, outside of the mitochondrion by nonenzymatic and/or enzymatic reactions. Reactions involved in extra-mitochondrial ROS production include NADPH oxidase, xanthine oxidase, D-amino oxidase, the P-450 cytochromes and proline and lysine hydroxylase, uncoupled NO synthase. ROS may also be generated by FFA oxidation in peroxisomes and microsomes and from Kupffer cell activation. In the obese and in patients with NASH, the pro-oxidant cytochrome P450 2E1 (CYP2E1) isoform activity is increased[59], likely induced by FFA or ketones. This microsomal enzyme, in addition to its involvement in the degradation of xenobiotics, promotes FFA β-oxidation, during which ROS are generated[60]. Kupffer cells may also generate ROS via the NADPH-oxidase system[61].
Mitochondrial ROS generation might be regulated by nitric oxide (NO)[62]. This compound can be converted to other RNS such as nitroxyl anion (NO-) or the toxic peroxynitrite (ONOO-)[62]. It has been reported that mitochondria can produce NO from mitochondrial nitric oxide synthase (iNOS)[62,63]; however, this has been questioned[64]. NO may affect mitochondrial respiratory chain function by reacting with cytochrome c oxidase, thus interrupting electron transfer to oxygen[65]. In addition, peroxynitrite, a product resulting from NO reaction with O2•-, inhibits the activity of various proteins, including electron transport chain complexes[66]. Mechanism through which peroxynitrite exert this effect are varied and include its oxidative potential[67] and its ability to damage DNA[68]. Oxidizing reactivity of ONOO- is comparable to that of •OH[69]. Endogenous NO can regulate mitochondrial production of ROS by two mechanisms: at low levels NO can increase O2•- and H2O2 generation by modulating the rate of oxygen consumption at the level of cytochrome c oxidase[70], whereas at high levels, NO inhibits H2O2 production by reacting with O2•-, resulting in ONOO- formation[71]. NO and other RNS are increased in response to chronic alcohol consumption through induction of iNOS[72]. Studies also report increased iNOS expression in the liver of ob/ob mice[48]. iNOS induction is linked to alterations in the NO-dependent control of mitochondrial respiration (at the level of cytochrome c oxidase), which potentially contributes to the development of alcoholic steatohepatitis. The importance of understanding the role of NO in the modulation of the mitochondrial electron transport chain activity in hepatic physiology and pathophysiology is supported by various studies, indicating that localized NO production can occur within the organelle through a specific mitochondrial NOS isoform or via metabolism of nitrite to NO[73,74].
Within the mitochondrial matrix, manganese-SOD converts O2•- to H2O2. Hydrogen peroxide can be further metabolized by glutathione peroxidase (Gpx I) and peroxiredoxin (Prx III) or can diffuse from the mitochondria into the cytosol. O2•- is unable to cross the mitochondrial membrane except in its protonated form, which is a small fraction of this radical at physiological pH[75]. The O2•- present in the intermembrane space might be scavenged by the oxidized form of cytochrome c or can diffuse into the cytosol through the voltage dependent anion channel (VDAC)[76].
Important mitochondrial antioxidant defence systems are represented by glutathione (GSH) and multiple GSH-linked antioxidant enzymes. Among these are Gpx 1, mainly located in the cytosol, and Gpx 4, also known as phospholipid hydroperoxide glutathione peroxidase, predominantly linked to the mitochondrial membrane. These enzymes catalyze the reduction of H2O2 and lipid hydroperoxides.
Depletion of mitochondrial GSH has been implicated in the development of alcoholic liver disease in that GSH participates in pathways responsible for ROS detoxification[77]. It has been also proposed that depletion of mitochondrial GSH sensitizes hepatocytes from alcohol-fed animals to TNFα-induced cell death[78]. Conflicting effects of NAFLD on mitochondrial GSH levels have also been reported. For example, decreased[48] and increased[79] levels of mitochondria GSH have been reported in a murine model of NASH such as the ob/ob mice. It is possible that the age of animals and their diet could explain, at least in part, the differences observed in mitochondrial GSH[80,81]. Depletion of mitochondrial GSH levels could also be due to its reduced uptake by mitochondria as a result of enhanced levels of cholesterol within the inner mitochondrial membrane[82] and the decrease in synthesis of S-adenosylmethionine, the major methyl donor in liver and precursor to GSH[83]. Recently, lower hepatic expression of the Mu-class glutathione S- transferase was reported in obese patients with steatosis compared to obese individuals without NAFLD[84]. Thus, low levels of GSH and decreased expression and/or activity of some glutathione-related enzymes may have additional deleterious effects on fatty liver. These findings demonstrate that although GSH is a potential player in the pathogenesis of fatty liver disease, additional studies are necessary to delineate the precise role of mitochondrial GSH alterations in the pathogenesis of alcohol and non-alcohol mediated mitochondrial dysfunction and liver injury.
MITOCHONDRIAL OXIDATIVE DAMAGE IN NAFLD
Mitochondrial ROS generation is considered an important contributing factor to various liver diseases via the accumulation of mtDNA mutations that could lead to lethal cell injury[85] through the impairment of several bioenergetic reactions involved in the oxidative phosphorylation. In addition, due to its close proximity to the ETC, the lack of protective histones and incomplete repair mechanisms, mtDNA is highly prone to oxidative damage, leading to DNA breaks and the occurrence of somatic of mtDNA mutations[55]. Mitochondrial DNA encodes 13 proteins involved in ETC functioning, as well as the two mitochondrial ribosomal RNAs and all the mitochondrial transfer RNAs required for the synthesis of mtDNA-encoded polypeptides within mitochondria.
During NASH, different types of mtDNA damage have been detected, including deletions, point mutations and increased 8-hydroxydeoxyguanosine level[24,86]. Reduced levels of mtDNA have also been reported in patients with NASH[87] that seem to be responsible for the impairment of mitochondrial function and development and progression of liver steatosis and other liver injuries. Reduced expression of mtDNA-encoded polypeptides has been reported in patients with NASH, further supporting a role for mtDNA depletion in mitochondrial dysfunction[88]. Interestingly, although loss of mtDNA has been demonstrated in the livers of patients with NASH, mtDNA level was shown to be markedly enhanced in patients with fatty liver but without inflammation or fibrosis[89]. Increased levels of mutations in the mitochondrially encoded subunit I and subunit II genes of complex I and complex IV, respectively, have been detected in the livers of NASH patients compared to patients with simple fatty livers[90]. The accumulation of mtDNA mutations may lead to mitochondrial respiratory chain dysfunction, which in turn results in an increase in mitochondrial ROS production and subsequent accumulation of more mtDNA mutations. This triggers a vicious cycle of oxidative damage in which mitochondrial dysfunction produces larger amounts of reactive oxygen species which in turn can induce further oxidative damage to mitochondrial structure and function, as well as to other cellular components. Damage to mtDNA can be propagated as mitochondria and cell divide[91]. Therefore, oxidative damage to mtDNA could have severe implications for mitochondrial dysfunction in fatty liver, allowing the physiological consequences of the damage to be amplified.
In addition to mtDNA and proteins, phospholipid constituents of the mitochondrial membranes are particularly prone to ROS-induced oxidative attack, especially the long chain polyunsaturated fatty acids. Phospholipids are the most abundant lipid components of the mitochondrial membranes where they play multiple roles. Phospholipids modulate the membrane permeability barrier, the structural and functional properties of membrane-associated enzymatic activities and provide a matrix for the assembly and function of a variety of catalytic processes. Polyunsaturated fatty acids (PUFA) are essential components of mitochondrial phospholipids. The presence of high concentration of PUFA in their structure render mitochondrial phospholipids a prime target for reactions with oxidizing agents and enables them to participate in long free radical chain reactions, producing a variety of by-products of lipid peroxidation process, such as hydroperoxides and endoperoxides. These compounds may undergo fragmentation to produce several reactive intermediates, among them, malondialdehyde and the most reactive 4-hydroxy-trans-2-nonenal. These two by-products of lipid peroxidation have been shown to interact with and inactivate ETC components, including cytochrome c oxidase, by forming adducts with this enzyme[92], contributing to triggering mitochondrial dysfunction in NASH[93,94].
Phospholipid peroxidation is considered an important contributing factor in mitochondrial dysfunction in several physiopathological conditions as well as during the aging process[95]. In fact, phospholipid peroxidation alters the structural organization of the mitochondrial bilayer, perturbing membrane fluidity and permeability. These alterations, in turn, affect mitochondrial ETC activity, the oxidative phosphorylation process, inner membrane barrier properties, maintenance of mitochondrial membrane potential and mitochondrial Ca2+ buffer capacity.
Cardiolipin is a phospholipid component of the inner mitochondrial membrane, rich in unsaturated fatty acids, particularly linoleic acid in heart and liver tissues or arachidonic and docosahexaenoic acid in brain tissue mitochondria[96]. Due to its high content of unsaturated fatty acids, cardiolipin is highly sensitive to ROS-induced oxidative damage. In fact, the presence of a methylene bridge between two double bonds of CL fatty acids makes these compounds highly prone to oxidative damage. In addition, CL molecules seem to be a primary target for ROS attack due to their location in the inner mitochondrial membrane in the proximity of the sites of ROS generation, notably the ETC complexes. Normal CL can be measured directly by a normal-phase HPLC technique with UV detection at 206 nm, while oxidized cardiolipin form can be detected with the same HPLC technique with detection at 235 nm (indicative of conjugated dienes)[96]. Results obtained in our laboratory have demonstrated an oxidation/depletion of cardiolipin in hepatic mitochondria isolated from rat fed with CDD, an experimental animal model of non-alcoholic fatty liver[22].
ROLE OF CARDIOLIPIN IN MITOCHONDRIAL BIOENERGETICS
Cardiolipin plays pleiotropic functions in mitochondria. This phospholipid is particularly abundant in biological membranes involved in the generation of an electrochemical gradient that is used to produce ATP, such the bacterial plasma membrane and the inner mitochondrial membrane[28]. The association between CL and these energy-transducing membranes suggests a critical role for CL in mitochondrial bioenergetics. Cardiolipin has been shown to interact with and to be required for optimal functioning of several inner mitochondrial membrane proteins and enzymes[27-31,97], including among others, the ETC complexes involved in the oxidative phosphorylation process and ADP/ATP translocator. Crystallographic studies have demonstrated the presence of few tightly bound CL molecules in each of the crystal structures of complex III, complex IV and ADP/ATP carrier[98]. These results suggest that CL is an integral component of these proteins, the presence of which is critical to folding, structure and function.
Mitochondrial respiratory chain complexes are organized into higher oligomeric structures, generically called supercomplexes or respirosomes[99]. Cardiolipin, due to its dimeric structure, seems to affect the assembly, stabilization and functional activity of such respiratory supercomplexes. Indeed, in mitochondria lacking CL, a respiratory supercomplex formed by complex III and IV is destabilized[100]. Similarly, dimeric organization of ADP/ATP carrier and other ADP/ATP carrier-containing supercomplexes are destabilized in CL-deficient mitochondria[101]. Altogether, these findings indicate the potential role played by CL in the supramolecular organization of proteins involved in mitochondrial bioenergetics. Thus, abnormalities in CL structure, content and fatty acids composition may have a deleterious effect in mitochondrial function in several physiopathological conditions[37,38,96,102], including fatty liver diseases[22].
CARDIOLIPIN OXIDATION AND RESPIRATORY CHAIN DYSFUNCTION IN NAFLD
As discussed earlier in this review, CL molecules seem to be required for functional activity of several mitochondrial inner membrane proteins, including respiratory chain complexes involved in the oxidative phosphorylation process. Cardiolipin oxidation may significantly affect the respiratory chain complexes activity and mitochondrial function. Results from our laboratory showed that exposure of beef heart submitochondrial particles to mitochondrial-mediated ROS generation or ROS generating systems resulted in an oxidation and depletion of mitochondrial CL and in a significant decrease of the complexes I, III and IV activity[103-105]. Moreover, exogenously added CL-liposomes were able to significantly prevent the ROS-mediated decrease of the activity of these respiratory chain complexes[103-105]. Thus, the oxidation and depletion of mitochondrial CL seem to be responsible for the ROS-induced alterations of the respiratory chain complexes I, III and IV activities.
Choline deficient diet (CDD) is a well-known experimental model to induce fatty liver in rats[106]. The depletion of choline interferes with the synthesis and/or secretion of very low-density lipoproteins and thus inhibits the transport of triglycerides outside of the hepatocytes, resulting in fatty liver in rats. Steatosis induced by the CDD is characterized by pathological and biochemical similarities with fatty liver in humans. Reactive oxygen species have been shown to have an important role in hepatic tissue injury in fatty liver[18,19]. Alterations in mitochondrial respiratory chain activity seem to be involved in the oxygen radicals production and thus they are considered a potential contributing factor in the pathogenesis of NAFLD[12,20,23]. Due to the fact that ROS are a highly reactive and short lived species, it is likely that mitochondrial-mediated ROS production leads to primary reactions and damage to cellular components located near the site of their production. Therefore, mitochondria could be considered both as a major source of ROS production and as a major target of their oxidative attack. The effect of ROS should be greatest at the level of mitochondrial membrane constituents, especially the protein complexes involved in the electron transport chain and phospholipid constituents rich in unsaturated fatty acids, particularly cardiolipin. Results obtained in our and other laboratories have shown that the activity of complex I is significantly diminished in liver mitochondria isolated from CDD rats compared to control animals[20,22]. Complex I has been shown to also be affected in the liver of patients with NASH[23]. The molecular mechanism underlying this alteration in complex I activity was also explored. In vitro experiments demonstrated that some CL molecules are linked to complex I and are required for functional activity of this enzyme complex[97,107]. As mentioned above, the content of CL decreased in liver mitochondria from CDD animals, while the level of oxidized cardiolipin increased[22]. Furthermore, the lowered activity of complex I in liver mitochondria from CDD animals could be restored to the level of control liver by the addition of exogenous CL. Other major phospholipid components of the mitochondrial membranes, such as phosphatidylcholine and phosphatidylethanolamine and peroxidized CL, were unable to replace CL in this effect. Moreover, oxidized cardiolipin markedly inhibited the activity of complex I in beef heart submitochondrial particles (personal observations). These results suggest that the observed dysfunction of complex I in hepatic mitochondria from CDD animals could be attributed, at least in part, to ROS-induced cardiolipin alterations.
The activity of complex I is considered a rate-limiting step for the mitochondrial ETC and thus is an important factor for the regulation of the oxidative phosphorylation process. In addition, complex I is an important locus of mitochondrial ROS production and therefore a likely source of reactive oxygen species in fatty liver diseases. The impairment of complex I, observed in liver mitochondria from CDD animals and attributable to ROS induced cardiolipin damage, may generate more superoxide radicals, triggering a vicious cycle of ROS-induced mitochondrial membrane damage that leads to mitochondrial malfunction and subsequently to hepatocyte injury (Figure 1). Long-chain free fatty acids which accumulate in nonalcoholic fatty liver are considered the initial source of this ROS damaging cycle[18]. These compounds have been shown to promote ROS production in mitochondria, although the mechanism(s) underlying this effect is still unclear[108,109].
Figure 1.
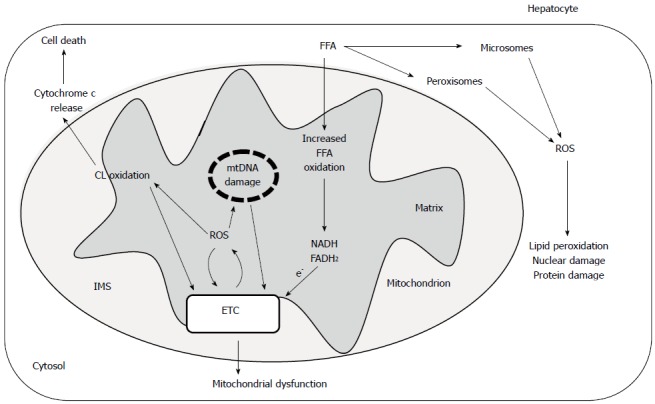
Possible role of reactive oxygen species and cardiolipin oxidation in hepatic mitochondrial dysfunction in nonalcoholic steatohepatitis.
It is now accepted that individual components of the ETC may exist as large macromolecular assemblies, or so called supercomplexes[99]. Several biochemical and biophysical lines of evidence support the existence of such supercomplexes. Such supercomplex organization of ETC would likely increase the efficiency of electron/proton translocation and hence that of ATP synthesis[101]. Another important consequence of the supercomplex organization of ETC is that of minimizing the production of ROS[110]. Experimental evidence indicates that cardiolipin molecules participate in the structural organization and stabilization of these supercomplexes[100]. As mentioned above, the content of CL declines, while that of its oxidized form increases in liver mitochondria from CDD animals. On this basis, it could be predicted that these CL alterations might be involved in the destabilization of the respiratory chain supercomplexes in fatty liver. Thus, the CL-dependent alterations of the ETC organization in supercomplexes could be another factor contributing to ROS generation and to mitochondrial bioenergetic decay in NAFLD. Cardiolipin-dependent destabilization of mitochondrial respiratory chain supercomplexes has been already reported in some pathophysiological situations, such as Barth syndrome[111] and diabetes[112].
APOPTOSIS IN NAFLD
Cell death in the liver and peripheral tissues has emerged as an important mechanism involved in the development and progression of NAFLD. An increase in hepatocyte cell death by apoptosis is typically present in patients with NAFLD and in experimental models of steatohepatitis[15,16]. Apoptosis may be executed by two fundamental pathways[113]: the extrinsic pathway is mediated by death receptors on the cellular surface and the intrinsic pathway is organelle-based. The extrinsic pathway is initiated by death receptors, including Fas, TNF-receptors and TNF-related apoptosis-inducing ligand receptors. These receptors trigger intracellular cascades that activate death-inducing proteolytic enzymes, especially caspases. Apoptosis can be initiated by mitochondria in the intrinsic pathway. In liver cells, mitochondrial dysfunction plays a critical role by amplifying the apoptotic signal and integrating both pathways into a final common pathway. Mitochondrial dysfunction results in the release of several proapoptotic proteins into the cytosol, including cytochrome c. This hemoprotein then forms an activation complex with apoptotic protease activating factor 1 and caspase 9, known as the apoptosome. This complex activates the downstream effectors caspases 3, 6 and 7, which execute the final apoptotic changes[114]. The mitochondrial events are regulated by the Bcl-2 family proteins, which comprises the antiapoptotic proteins Bcl-2 and Bcl-xL, the proapoptotic proteins, such as Bax and Bak, and the BH3-only proteins, such as Bad, Bim and Bid, which provide crosstalk between intrinsic and extrinsic pathways[115]. Indeed, activated caspase 8 cleaves the cytosolic protein Bid to a truncated active fragment t-Bid, which translocates to mitochondria and induces cytochrome c release[116]. Cardiolipin has been shown to be involved in several steps of the intrinsic pathway of the apoptotic process[32,33], including mitochondrial permeability transition pore (MPTP) and cytochrome c release from mitochondria[117].
CARDIOLIPIN OXIDATION, MPTP AND CYTOCHROME C RELEASE FROM MITOCHONDRIA
MPTP is defined as the sudden increase of mitochondrial inner membrane permeability to low molecular weight solutes (1.5 kDa) in response to many stimuli, including high levels of Ca2+ and oxidant stress. MPTP activation by a combination of elevated intramitochondrial Ca2+ and oxidative stress promotes the collapse of transmembrane ion gradients, resulting in membrane depolarization, uncoupling of oxidative phosphorylation and ATP depletion[118,119]. Under conditions of low ATP levels, the cells are unable to maintain structural and functional integrity, including ion homeostasis. This results in outer membrane permeabilization and cytochrome c release into the cytoplasm to initiate pro-apoptotic signals. The duration of MPTP opening directly affects maintenance of ATP stores and cellular integrity. If MPTP opening is transient, the cell can recover; while in conditions of prolonged MPTP opening and ATP depletion, irreversible damage and cell death occur, predominantly through necrosis. A number of molecules were accepted as key structural components of the MPTP, including cyclophilin-D, a peptidyl-prolyl cis-trans isomerase, in the matrix, ADP/ATP and phosphate carriers in the inner membrane and VDAC (also known as porin) in the outer membrane[120]. Very recently, it was reported that reconstituted dimers of the F0F1 ATP synthase, incorporated into lipid bilayers, form Ca2+-activated channels with properties identical to those of the mitochondrial megachannel, the electrophysiological equivalent of the MPTP, indicating dimers of the F0F1ATP synthase to be a new putative component of the MPTP[121].
Ca2+ ions are considered the main inducers of the MPTP opening. Our studies have shown that exogenous added oxidized CL sensitized mitochondria to Ca2+-induced MPTP opening[117]. Similarly, oxidation of endogenous CL by tert-Butyl hydroperoxide resulted in MPTP opening[122]. Ca2+ and oxidized CL seem to play a coordinated role in MPTP opening by interacting with components of the MPTP, probably ADP/ATP carrier. It was recently reported that hepatic mitochondria from CDD rats were more vulnerable to Ca2+-induced MPTP activation in comparison to the respective control animals[123]. Pretreatment with cyclosporine A completely prevented Ca2+-dependent mitochondrial swelling. It is conceivable that oxidized CL, the level of which increases in mitochondria from CDD animals[22], could be a contributing factor to the enhanced susceptibility of these mitochondria to Ca2+-dependent MPTP opening. Interestingly, the induction of MPTP opening by oxidized CL and Ca2+ is associated with the release of cytochrome c from mitochondria[117].
Release of cytochrome c, as well as other proteins, from the mitochondria to the cytosol appears to play a central role in the induction of the apoptotic cascade that ultimately leads to the programmed cell death[117]. Cardiolipin has been implicated in the process of apoptosis in animal cells through its interaction with cytochrome c[32,33,117]. This hemoprotein is normally bound to the outer leaflet of the inner mitochondrial membrane, primarily to CL molecules[124]. The strength of the binding of cytochrome c to mitochondria is dependent on the fatty acyl chain composition of CL. Oxidation of CL promotes the detachment of cytochrome c from mitochondrial membrane[32,35]. Experimental evidence indicates that cytochrome c release from mitochondria during apoptosis occurs via a two-step process. This mechanism first involves the detachment of cytochrome c from the outer leaflet of the inner mitochondrial membrane, followed by permeabilization of the outer mitochondrial membrane and the release of cytochrome c into the cytosol space[33]. Oxidized cardiolipin, in the presence of Ca2+, induces the mitochondrial permeability transition pore opening[117]. In addition, CL oxidation seems to promote the permeabilization of the outer mitochondrial membrane, probably interacting with Bcl2 family proteins, such as Bax and Bid[125]. Thus, oxidized CL may have a critical role in several steps of the apoptotic process such as: the translocation of t-Bid to mitochondria with subsequent oligomerization of Bax and Bak leading to membrane permeabilization; cytochrome c dissociation from the inner mitochondrial membrane; and activation of MPTP, followed by the release of this hemoprotein from the mitochondrial intermembrane space to the cytosol (Figure 1). The fact that CL oxidation/depletion has been detected in liver mitochondria from CDD animals[22] suggests a potential role of these CL alterations in the apoptotic process associated with fatty liver. Indeed, apoptosis is an important mechanism of cell death in patients with NASH[126]. Thus, modulation of CL vulnerability to peroxidation may be a potential target for sensitizing cells to apoptosis in fatty liver. Another consequence of cytochrome c release from mitochondria is that it interferes with electron transport through ETC, resulting in ROS formation and triggering a new vicious cycle, which will worsen the disturbance.
MITOCHONDRIAL-TARGETED ANTIOXIDANTS IN NAFLD
Several therapeutic agents have been tested for their ability to alleviate fatty liver diseases; however, no proven drug therapies have emerged as being highly effective. As mitochondrial oxidative stress is considered an important factor in the etiology of fatty liver, the majority of drug discovery efforts to date have centered on compounds that prevent mitochondrial ROS overproduction. Both traditional and new antioxidant compounds have been shown to reduce the development and/or reverse the pathological effects of fatty liver disease, presumably through their capability to decrease oxidative stress in tissues. Antioxidants which have been shown to be particularly effective in treating fatty liver disease include N-acetyl-L-cysteine, zinc, ursodeoxycholic acid, α-tocopherol (vitamin E), lipoic acid, carnitine, S-adenosyl-L-methionine and polyphenolic compounds, such as silybin and resveratrol, just to name a few. Further information on antioxidant therapy for treatment of fatty liver diseases can be found in the following excellent review articles[127-132].
Despite a large number of experimental studies encouraging the antioxidant therapy in the treatment of NAFLD, there are few clinical trials that support the efficacy of these compounds. Some antioxidants have been shown to have a positive effect and others as ineffective, probably due to the fact that these compounds do not reach the site of free radical generation, especially when mitochondria are the primary source of ROS. It should be considered that in certain physiological conditions the effect of ROS are not injurious but may have important signaling roles in physiological regulatory mechanisms. For example, mitochondrial-mediated ROS production has been shown to play an important signaling role in the glucose-stimulated insulin secretory pathway in B-cells and is also involved in insulin signaling and sensitivity[133]. Recently, new antioxidant compounds have been synthesized by attaching them to a lipophilic triphenylphosphonium moiety in order to target these agents to the mitochondrion[134]. These new modified antioxidant agents have been shown to accumulate several hundredfold in the mitochondrion in response to the mitochondrial membrane potential. Indeed, mitochondrial-targeted ubiquinone, modified by conjugation to lipophilic cations, was shown to reduce cardiac ischemia/reperfusion injury[135].
As discussed above, CL alterations, especially CL oxidation, may be involved in mitochondrial dysfunction, as well as in mitochondrial-mediated steps of the apoptotic process in NAFLD, such as MPTP opening and cytochrome c release from mitochondria. Therefore, compounds that target CL on the IMM preventing its oxidation would be particularly useful in improving mitochondrial function and reducing ROS generation. New mitochondrial-targeted antioxidant agents are cationic derivatives of plastoquinones which are selectively accumulated within mitochondria[136]. The mechanism by which these antioxidant compounds exert their antioxidant activity includes, in particular, prevention of mitochondrial cardiolipin oxidation by ROS attack. Although these mitochondria-targeted antioxidant agents can decrease mitochondrial ROS production, recent studies indicate they impair several reactions involved in mitochondrial bioenergetics[137]. Therefore, more studies are needed to demonstrate the safety and efficacy of these antioxidant agents in diseases for which mitochondrial dysfunction and oxidative stress are proposed to be important factors in their etiology, including NAFLD.
Melatonin, a hormonal product of the pineal gland, has been shown to protect against physiopathological states characterized by ROS overproduction[138]. The mechanism(s) by which this compound exerts these protective effects are not well established. Experimental evidence indicates that some of the protective effects of melatonin may be produced through its action at mitochondrial level by preventing ROS-induced cardiolipin oxidation[139,140]. In this respect, our studies have demonstrated that melatonin is able to prevent cardiolipin peroxidation in mitochondria isolated from heart and brain tissues. This effect may underlie the protection afforded by melatonin against mitochondrial dysfunction in heart ischemia/reperfusion[141] and brain aging[142].
Another new family of agents that target CL in the inner mitochondrial membrane are the SS (Szeto-Schiller) peptide antioxidants[143]. These compounds have been shown to optimize cristae architecture, improve mitochondrial bioenergetics and reduce ROS production. In addition, these antioxidant peptides have been evaluated in numerous preclinical disease models characterized by bioenergetic failure. Additional studies are needed to better investigate the potential efficacy of these new CL-targeted antioxidants in diseases for which oxidant stress, CL abnormalities and mitochondrial dysfunction play a role in pathology, including fatty liver diseases.
CONCLUSION
Strong evidence indicates that the pathogenesis of nonalcoholic fatty liver disease is linked to mitochondrial structural and functional alterations. ROS overproduction and altered oxidative phosphorylation are important factors involved in mitochondrial dysfunction. Oxidative damage to mitochondria alters mitochondrial respiratory chain complexes and mitochondrial DNA to partially block the flow of electrons. The subsequent increase in mitochondrial ROS formation triggers a vicious cycle of damage amplification. CL, the signature phospholipid of the mitochondria, is particularly prone to ROS-induced oxidative damage. This phospholipid plays a pivotal role in modulating the activity of a variety of mitochondrial bioenergetic reactions and processes, especially oxidative phosphorylation and coupled respiration. Cardiolipin is also an important player in different stages of the mitochondrial apoptotic process. A number of studies have demonstrated the involvement of CL alterations in mitochondrial dysfunction in multiple tissues in a variety of physiopathological states. In this review, we highlighted the potential roles played by CL alterations in mitochondrial bioenergetics decay and in the mitochondrial apoptotic process associated with nonalcoholic fatty liver disease. Thus, CL oxidation seems to affect the activity of several mitochondrial bioenergetic reactions in fatty liver, including electron transport chain and mitochondrial permeability transition, as well as the release of cytochrome c from mitochondria. The development of new and effective antioxidant strategies aimed at reducing the production of oxidants and hence CL oxidation in mitochondria offers great promise for the prevention and treatment of liver disease.
Footnotes
P- Reviewer: Ahmed M, Mustonen AM, Weiss RS S- Editor: Qi Y L- Editor: Roemmele A E- Editor: Wang CH
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Wieckowska A, Feldstein AE. Nonalcoholic fatty liver disease in the pediatric population: a review. Curr Opin Pediatr. 2005;17:636–641. doi: 10.1097/01.mop.0000172816.79637.c5. [DOI] [PubMed] [Google Scholar]
- 3.Ludwig J, McGill DB, Lindor KD. Review: nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 1997;12:398–403. [Google Scholar]
- 4.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 5.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 6.Falck-Ytter Y, Younossi ZM, Marchesini G, McCullough AJ. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis. 2001;21:17–26. doi: 10.1055/s-2001-12926. [DOI] [PubMed] [Google Scholar]
- 7.Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 8.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 9.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497–1507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- 12.García-Ruiz C, Baulies A, Mari M, García-Rovés PM, Fernandez-Checa JC. Mitochondrial dysfunction in non-alcoholic fatty liver disease and insulin resistance: cause or consequence? Free Radic Res. 2013;47:854–868. doi: 10.3109/10715762.2013.830717. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malassagne B, Ferret PJ, Hammoud R, Tulliez M, Bedda S, Trébéden H, Jaffray P, Calmus Y, Weill B, Batteux F. The superoxide dismutase mimetic MnTBAP prevents Fas-induced acute liver failure in the mouse. Gastroenterology. 2001;121:1451–1459. doi: 10.1053/gast.2001.29590. [DOI] [PubMed] [Google Scholar]
- 15.Alkhouri N, Carter-Kent C, Feldstein AE. Apoptosis in nonalcoholic fatty liver disease: diagnostic and therapeutic implications. Expert Rev Gastroenterol Hepatol. 2011;5:201–212. doi: 10.1586/egh.11.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Syn WK, Choi SS, Diehl AM. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:565–580. doi: 10.1016/j.cld.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- 18.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Hensley K, Kotake Y, Sang H, Pye QN, Wallis GL, Kolker LM, Tabatabaie T, Stewart CA, Konishi Y, Nakae D, et al. Dietary choline restriction causes complex I dysfunction and increased H(2)O(2) generation in liver mitochondria. Carcinogenesis. 2000;21:983–989. doi: 10.1093/carcin/21.5.983. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira CP, da Costa Gayotto LC, Tatai C, Della Bina BI, Janiszewski M, Lima ES, Abdalla DS, Lopasso FP, Laurindo FR, Laudanna AA. Oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, in rats fed with a choline-deficient diet. J Cell Mol Med. 2002;6:399–406. doi: 10.1111/j.1582-4934.2002.tb00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta. 2007;1767:1260–1267. doi: 10.1016/j.bbabio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 24.Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430–434. doi: 10.1016/s0168-8278(99)80033-6. [DOI] [PubMed] [Google Scholar]
- 25.Le TH, Caldwell SH, Redick JA, Sheppard BL, Davis CA, Arseneau KO, Iezzoni JC, Hespenheide EE, Al-Osaimi A, Peterson TC. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology. 2004;39:1423–1429. doi: 10.1002/hep.20202. [DOI] [PubMed] [Google Scholar]
- 26.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 27.Hoch FL. Cardiolipins and biomembrane function. Biochim Biophys Acta. 1992;1113:71–133. doi: 10.1016/0304-4157(92)90035-9. [DOI] [PubMed] [Google Scholar]
- 28.Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- 29.Houtkooper RH, Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol Life Sci. 2008;65:2493–2506. doi: 10.1007/s00018-008-8030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta. 2014;1837:408–417. doi: 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Klingenberg M. Cardiolipin and mitochondrial carriers. Biochim Biophys Acta. 2009;1788:2048–2058. doi: 10.1016/j.bbamem.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, et al. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic Biol Med. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalvez F, Gottlieb E. Cardiolipin: setting the beat of apoptosis. Apoptosis. 2007;12:877–885. doi: 10.1007/s10495-007-0718-8. [DOI] [PubMed] [Google Scholar]
- 35.Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001;509:435–438. doi: 10.1016/s0014-5793(01)03206-9. [DOI] [PubMed] [Google Scholar]
- 36.Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Cardiolipin and mitochondrial function in health and disease. Antioxid Redox Signal. 2014;20:1925–1953. doi: 10.1089/ars.2013.5280. [DOI] [PubMed] [Google Scholar]
- 38.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 39.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med. 2010;48:1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, Liu X, Jefferson LS, Xiong J, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12:154–165. doi: 10.1016/j.cmet.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aoun M, Feillet-Coudray C, Fouret G, Chabi B, Crouzier D, Ferreri C, Chatgilialoglu C, Wrutniak-Cabello C, Cristol JP, Carbonneau MA, et al. Rat liver mitochondrial membrane characteristics and mitochondrial functions are more profoundly altered by dietary lipid quantity than by dietary lipid quality: effect of different nutritional lipid patterns. Br J Nutr. 2012;107:647–659. doi: 10.1017/S000711451100331X. [DOI] [PubMed] [Google Scholar]
- 42.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 43.Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999;116:1184–1193. doi: 10.1016/s0016-5085(99)70022-3. [DOI] [PubMed] [Google Scholar]
- 44.Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demeilliers C, Maisonneuve C, Grodet A, Mansouri A, Nguyen R, Tinel M, Lettéron P, Degott C, Feldmann G, Pessayre D, et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology. 2002;123:1278–1290. doi: 10.1053/gast.2002.35952. [DOI] [PubMed] [Google Scholar]
- 46.Santamaria E, Avila MA, Latasa MU, Rubio A, Martin-Duce A, Lu SC, Mato JM, Corrales FJ. Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proc Natl Acad Sci USA. 2003;100:3065–3070. doi: 10.1073/pnas.0536625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 48.García-Ruiz I, Rodríguez-Juan C, Díaz-Sanjuan T, del Hoyo P, Colina F, Muñoz-Yagüe T, Solís-Herruzo JA. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology. 2006;44:581–591. doi: 10.1002/hep.21313. [DOI] [PubMed] [Google Scholar]
- 49.Crespo J, Cayón A, Fernández-Gil P, Hernández-Guerra M, Mayorga M, Domínguez-Díez A, Fernández-Escalante JC, Pons-Romero F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163. doi: 10.1053/jhep.2001.29628. [DOI] [PubMed] [Google Scholar]
- 50.Sánchez-Alcázar JA, Schneider E, Martínez MA, Carmona P, Hernández-Muñoz I, Siles E, De La Torre P, Ruiz-Cabello J, García I, Solis-Herruzo JA. Tumor necrosis factor-alpha increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. J Biol Chem. 2000;275:13353–13361. doi: 10.1074/jbc.275.18.13353. [DOI] [PubMed] [Google Scholar]
- 51.Nagakawa Y, Williams GM, Zheng Q, Tsuchida A, Aoki T, Montgomery RA, Klein AS, Sun Z. Oxidative mitochondrial DNA damage and deletion in hepatocytes of rejecting liver allografts in rats: role of TNF-alpha. Hepatology. 2005;42:208–215. doi: 10.1002/hep.20755. [DOI] [PubMed] [Google Scholar]
- 52.Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, Van Breusegem F. ROS signaling: the new wave? Trends Plant Sci. 2011;16:300–309. doi: 10.1016/j.tplants.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 53.D’Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 54.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 57.Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- 58.Nohl H, Stolze K. Ubisemiquinones of the mitochondrial respiratory chain do not interact with molecular oxygen. Free Radic Res Commun. 1992;16:409–419. doi: 10.3109/10715769209049191. [DOI] [PubMed] [Google Scholar]
- 59.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–133. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 60.Schattenberg JM, Wang Y, Singh R, Rigoli RM, Czaja MJ. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. J Biol Chem. 2005;280:9887–9894. doi: 10.1074/jbc.M410310200. [DOI] [PubMed] [Google Scholar]
- 61.Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem. 1998;273:11038–11043. doi: 10.1074/jbc.273.18.11038. [DOI] [PubMed] [Google Scholar]
- 63.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 64.Venkatakrishnan P, Nakayasu ES, Almeida IC, Miller RT. Absence of nitric-oxide synthase in sequentially purified rat liver mitochondria. J Biol Chem. 2009;284:19843–19855. doi: 10.1074/jbc.M109.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 66.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 67.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 68.Szabó C. DNA strand breakage and activation of poly-ADP ribosyltransferase: a cytotoxic pathway triggered by peroxynitrite. Free Radic Biol Med. 1996;21:855–869. doi: 10.1016/0891-5849(96)00170-0. [DOI] [PubMed] [Google Scholar]
- 69.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 70.Sarkela TM, Berthiaume J, Elfering S, Gybina AA, Giulivi C. The modulation of oxygen radical production by nitric oxide in mitochondria. J Biol Chem. 2001;276:6945–6949. doi: 10.1074/jbc.M007625200. [DOI] [PubMed] [Google Scholar]
- 71.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 72.Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, et al. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol. 2006;291:G857–G867. doi: 10.1152/ajpgi.00044.2006. [DOI] [PubMed] [Google Scholar]
- 73.Haynes V, Elfering S, Traaseth N, Giulivi C. Mitochondrial nitric-oxide synthase: enzyme expression, characterization, and regulation. J Bioenerg Biomembr. 2004;36:341–346. doi: 10.1023/B:JOBB.0000041765.27145.08. [DOI] [PubMed] [Google Scholar]
- 74.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- 75.Gus’kova RA, Ivanov II, Kol’tover VK, Akhobadze VV, Rubin AB. Permeability of bilayer lipid membranes for superoxide (O2-.) radicals. Biochim Biophys Acta. 1984;778:579–585. doi: 10.1016/0005-2736(84)90409-7. [DOI] [PubMed] [Google Scholar]
- 76.Madesh M, Hajnóczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.García-Ruiz C, Morales A, Ballesta A, Rodés J, Kaplowitz N, Fernández-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pastorino JG, Hoek JB. Ethanol potentiates tumor necrosis factor-alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 79.Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA, Diehl AM. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys. 2000;378:259–268. doi: 10.1006/abbi.2000.1829. [DOI] [PubMed] [Google Scholar]
- 80.Armeni T, Pieri C, Marra M, Saccucci F, Principato G. Studies on the life prolonging effect of food restriction: glutathione levels and glyoxalase enzymes in rat liver. Mech Ageing Dev. 1998;101:101–110. doi: 10.1016/s0047-6374(97)00167-x. [DOI] [PubMed] [Google Scholar]
- 81.Grattagliano I, Portincasa P, Cocco T, Moschetta A, Di Paola M, Palmieri VO, Palasciano G. Effect of dietary restriction and N-acetylcysteine supplementation on intestinal mucosa and liver mitochondrial redox status and function in aged rats. Exp Gerontol. 2004;39:1323–1332. doi: 10.1016/j.exger.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Llacuna L, Fernández A, Montfort CV, Matías N, Martínez L, Caballero F, Rimola A, Elena M, Morales A, Fernández-Checa JC, et al. Targeting cholesterol at different levels in the mevalonate pathway protects fatty liver against ischemia-reperfusion injury. J Hepatol. 2011;54:1002–1010. doi: 10.1016/j.jhep.2010.08.031. [DOI] [PubMed] [Google Scholar]
- 83.Martínez-Chantar ML, García-Trevijano ER, Latasa MU, Pérez-Mato I, Sánchez del Pino MM, Corrales FJ, Avila MA, Mato JM. Importance of a deficiency in S-adenosyl-L-methionine synthesis in the pathogenesis of liver injury. Am J Clin Nutr. 2002;76:1177S–1182S. doi: 10.1093/ajcn/76/5.1177S. [DOI] [PubMed] [Google Scholar]
- 84.Younossi ZM, Baranova A, Ziegler K, Del Giacco L, Schlauch K, Born TL, Elariny H, Gorreta F, VanMeter A, Younoszai A, et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology. 2005;42:665–674. doi: 10.1002/hep.20838. [DOI] [PubMed] [Google Scholar]
- 85.Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, Schaffer D, Schaffer SW. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol. 2008;294:C413–C422. doi: 10.1152/ajpcell.00362.2007. [DOI] [PubMed] [Google Scholar]
- 86.Nomoto K, Tsuneyama K, Takahashi H, Murai Y, Takano Y. Cytoplasmic fine granular expression of 8-hydroxydeoxyguanosine reflects early mitochondrial oxidative DNA damage in nonalcoholic fatty liver disease. Appl Immunohistochem Mol Morphol. 2008;16:71–75. doi: 10.1097/PAI.0b013e31803156d5. [DOI] [PubMed] [Google Scholar]
- 87.Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology. 2010;52:1992–2000. doi: 10.1002/hep.23927. [DOI] [PubMed] [Google Scholar]
- 88.Haque A, Nishikawa M, Qian W, Mashimo M, Hirose M, Nishiguchi S, Inoue M. Lack of mitochondrial DNA enhances growth of hepatocellular carcinoma in vitro and in vivo. Hepatol Res. 2006;36:209–216. doi: 10.1016/j.hepres.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 89.Chiappini F, Barrier A, Saffroy R, Domart MC, Dagues N, Azoulay D, Sebagh M, Franc B, Chevalier S, Debuire B, et al. Exploration of global gene expression in human liver steatosis by high-density oligonucleotide microarray. Lab Invest. 2006;86:154–165. doi: 10.1038/labinvest.3700374. [DOI] [PubMed] [Google Scholar]
- 90.Kawahara H, Fukura M, Tsuchishima M, Takase S. Mutation of mitochondrial DNA in livers from patients with alcoholic hepatitis and nonalcoholic steatohepatitis. Alcohol Clin Exp Res. 2007;31:S54–S60. doi: 10.1111/j.1530-0277.2006.00287.x. [DOI] [PubMed] [Google Scholar]
- 91.Van Remmen H, Hamilton ML, Richardson A. Oxidative damage to DNA and aging. Exerc Sport Sci Rev. 2003;31:149–153. doi: 10.1097/00003677-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 92.Musatov A, Carroll CA, Liu YC, Henderson GI, Weintraub ST, Robinson NC. Identification of bovine heart cytochrome c oxidase subunits modified by the lipid peroxidation product 4-hydroxy-2-nonenal. Biochemistry. 2002;41:8212–8220. doi: 10.1021/bi025896u. [DOI] [PubMed] [Google Scholar]
- 93.Pessayre D, Fromenty B, Mansouri A. Mitochondrial injury in steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1095–1105. doi: 10.1097/00042737-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 94.Chen J, Petersen DR, Schenker S, Henderson GI. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res. 2000;24:544–552. [PubMed] [Google Scholar]
- 95.Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, et al. Pathological aspects of lipid peroxidation. Free Radic Res. 2010;44:1125–1171. doi: 10.3109/10715762.2010.498478. [DOI] [PubMed] [Google Scholar]
- 96.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 97.Musatov A, Robinson NC. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic Res. 2012;46:1313–1326. doi: 10.3109/10715762.2012.717273. [DOI] [PubMed] [Google Scholar]
- 98.Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 100.Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005;280:29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci. 2012;37:32–41. doi: 10.1016/j.tibs.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int. 2011;58:447–457. doi: 10.1016/j.neuint.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 103.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett. 2000;466:323–326. doi: 10.1016/s0014-5793(00)01082-6. [DOI] [PubMed] [Google Scholar]
- 104.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion. 2001;1:151–159. doi: 10.1016/s1567-7249(01)00011-3. [DOI] [PubMed] [Google Scholar]
- 105.Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–141. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- 106.Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012;28:159–165. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and NADH: ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- 108.Schönfeld P, Wojtczak L. Fatty acids decrease mitochondrial generation of reactive oxygen species at the reverse electron transport but increase it at the forward transport. Biochim Biophys Acta. 2007;1767:1032–1040. doi: 10.1016/j.bbabio.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 109.Srivastava S, Chan C. Hydrogen peroxide and hydroxyl radicals mediate palmitate-induced cytotoxicity to hepatoma cells: relation to mitochondrial permeability transition. Free Radic Res. 2007;41:38–49. doi: 10.1080/10715760600943900. [DOI] [PubMed] [Google Scholar]
- 110.Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 2013;19:1469–1480. doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 112.Ferreira R, Guerra G, Padrão AI, Melo T, Vitorino R, Duarte JA, Remião F, Domingues P, Amado F, Domingues MR. Lipidomic characterization of streptozotocin-induced heart mitochondrial dysfunction. Mitochondrion. 2013;13:762–771. doi: 10.1016/j.mito.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 113.Yuan J, Horvitz HR. A first insight into the molecular mechanisms of apoptosis. Cell. 2004;116:S53–S56, 1 p following S59. doi: 10.1016/s0092-8674(04)00028-5. [DOI] [PubMed] [Google Scholar]
- 114.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 115.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 116.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 117.Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett. 2006;580:6311–6316. doi: 10.1016/j.febslet.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 118.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 119.Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 120.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 121.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Petrosillo G, Moro N, Ruggiero FM, Paradies G. Melatonin inhibits cardiolipin peroxidation in mitochondria and prevents the mitochondrial permeability transition and cytochrome c release. Free Radic Biol Med. 2009;47:969–974. doi: 10.1016/j.freeradbiomed.2009.06.032. [DOI] [PubMed] [Google Scholar]
- 123.Teodoro JS, Rolo AP, Duarte FV, Simões AM, Palmeira CM. Differential alterations in mitochondrial function induced by a choline-deficient diet: understanding fatty liver disease progression. Mitochondrion. 2008;8:367–376. doi: 10.1016/j.mito.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 124.Rytömaa M, Mustonen P, Kinnunen PK. Reversible, nonionic, and pH-dependent association of cytochrome c with cardiolipin-phosphatidylcholine liposomes. J Biol Chem. 1992;267:22243–22248. [PubMed] [Google Scholar]
- 125.Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato AA, Fujii Y. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 126.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 127.Tome S, Lucey MR. Review article: current management of alcoholic liver disease. Aliment Pharmacol Ther. 2004;19:707–714. doi: 10.1111/j.1365-2036.2004.01881.x. [DOI] [PubMed] [Google Scholar]
- 128.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 129.Comar KM, Sterling RK. Review article: Drug therapy for non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2006;23:207–215. doi: 10.1111/j.1365-2036.2006.02751.x. [DOI] [PubMed] [Google Scholar]
- 130.Federico A, Trappoliere M, Loguercio C. Treatment of patients with non-alcoholic fatty liver disease: current views and perspectives. Dig Liver Dis. 2006;38:789–801. doi: 10.1016/j.dld.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 131.Portincasa P, Grattagliano I, Palmieri VO, Palasciano G. Current pharmacological treatment of nonalcoholic fatty liver. Curr Med Chem. 2006;13:2889–2900. doi: 10.2174/092986706778521878. [DOI] [PubMed] [Google Scholar]
- 132.Serviddio G, Bellanti F, Sastre J, Vendemiale G, Altomare E. Targeting mitochondria: a new promising approach for the treatment of liver diseases. Curr Med Chem. 2010;17:2325–2337. doi: 10.2174/092986710791698530. [DOI] [PubMed] [Google Scholar]
- 133.Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, Collins S, Andersen ME. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244:77–83. doi: 10.1016/j.taap.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 135.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 136.Skulachev VP, Antonenko YN, Cherepanov DA, Chernyak BV, Izyumov DS, Khailova LS, Klishin SS, Korshunova GA, Lyamzaev KG, Pletjushkina OY, et al. Prevention of cardiolipin oxidation and fatty acid cycling as two antioxidant mechanisms of cationic derivatives of plastoquinone (SkQs) Biochim Biophys Acta. 2010;1797:878–889. doi: 10.1016/j.bbabio.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 137.Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Acuña Castroviejo D, Escames G, Carazo A, León J, Khaldy H, Reiter RJ. Melatonin, mitochondrial homeostasis and mitochondrial-related diseases. Curr Top Med Chem. 2002;2:133–151. doi: 10.2174/1568026023394344. [DOI] [PubMed] [Google Scholar]
- 139.Paradies G, Petrosillo G, Paradies V, Reiter RJ, Ruggiero FM. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J Pineal Res. 2010;48:297–310. doi: 10.1111/j.1600-079X.2010.00759.x. [DOI] [PubMed] [Google Scholar]
- 140.Peng TI, Hsiao CW, Reiter RJ, Tanaka M, Lai YK, Jou MJ. mtDNA T8993G mutation-induced mitochondrial complex V inhibition augments cardiolipin-dependent alterations in mitochondrial dynamics during oxidative, Ca(2+), and lipid insults in NARP cybrids: a potential therapeutic target for melatonin. J Pineal Res. 2012;52:93–106. doi: 10.1111/j.1600-079X.2011.00923.x. [DOI] [PubMed] [Google Scholar]
- 141.Petrosillo G, Colantuono G, Moro N, Ruggiero FM, Tiravanti E, Di Venosa N, Fiore T, Paradies G. Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol. 2009;297:H1487–H1493. doi: 10.1152/ajpheart.00163.2009. [DOI] [PubMed] [Google Scholar]
- 142.Petrosillo G, De Benedictis V, Ruggiero FM, Paradies G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J Bioenerg Biomembr. 2013;45:431–440. doi: 10.1007/s10863-013-9505-0. [DOI] [PubMed] [Google Scholar]
- 143.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]