

Abstract
B-myb belongs to the myb family of transcription factors that include A-myb and c-myb. While A-myb and c-myb are tissue-specific, B-myb is broadly expressed in rapidly dividing cells of developing adult mammals. Results of our study showed that increased B-myb expression of was associated with the progression of breast cancer and that B-myb protein levels were significantly elevated in matched metastases. High B-myb levels also predict shorter overall survival of breast cancer patients. Moreover, B-myb stimulated transcription of target genes that promoted entry into the S and M-phases of the cell cycle, cell proliferation, migration and invasion in breast cancer. Taken together, our results strongly demonstrated that B-myb had a critical role in both cell cycle progression and tumorigenesis, and might serve as a novel potential target in the diagnosis and/or treatment of human breast cancer.
Keywords: B-myb, cell cycle, proliferation, migration, invasion, breast cancer
Introduction
Breast cancer is one of the most common cancer affecting women worldwide. Despite the recent development of various therapeutic strategies, the progression for this cancer still remains poor. The precise genetic changes those are responsible for nasopharyngeal carcinoma progressions are largely unknown. The development and progression of breast cancer may involve accumulation of multiple genetic alterations over a long period of time [1-4]. Thus, more efforts are needed to understand its molecular pathway in order to develop an effective therapy to achieve cure.
B-myb is homologous to c-myb in the DNA binding domain, and its pattern of expression is not restricted to hematopoieticcells. In fact, B-myb transcripts have been detected in the majority of cell lines and tissues tested. Like c-myb, B-myb acts as a transactivating factor [5-7]. B-myb gene expression is found in all cell types and protein level is proportional to the degree of cell proliferation. This explains the strong expression of B-myb in embryonal stem cells, in developing mammalian tissues and in adult haematopietic precursor cells. The broad expression of B-myb in proliferating cells at least in part explains the striking phenotype of B-myb knockout mice, which show early embryonal death due to impaired inner cell mass formation.
Its expression is barely detectable in G0 and is induced at the G1/S transition of the cell cycle. Its protein levels parallel only in part mRNA expression and a pool of stable B-myb protein is detectable throughout the cell cycle. It is phosphorylated during S phase by the cyclin-dependent kinase cdk2 and evidence from several laboratories suggests that this modification activates B-myb [8]. Its phosphorylation may interfere with co-repressor binding therefore enhancing B-myb transcriptional activity. While cyclin/cdk2-directed phosphorylation activates B-MYB, it also induces accelerated protein turnover. B-myb has been shown to promote DNA replicationand transcriptional activation of genes, such as cyclin B1, essential for G2/M phase progression. Cyclin D1 interacts with the B-Myb transcriptional domain, quenching B-myb transactivation by interfering with CBP/p300 [9-13]. When cells exit quiescence in response to growth factors they generate a burst of Cyclin D1, required for further progression along the cell cycle. A plausible hypothesis is that cyclin D1 could co-ordinate B-myb with the cell cycle, maintaining it in a repressed state until cyclin D1 destruction in late G1. Phosphorylation by mitotic cyclins and cdk2 could then switch-on transcription of relevant B-myb-target genes in S or later cell cycle phases [2,14-16]. Our studies will firstly try to illustrate the mechanism that B-myb facilitates breast cancer and we could take into account B-myb serve as a potential therapeutic target in human breast cancer.
Materials and methods
Cell culture and siRNA transfection
MDA-MB-231 cell lines were maintained in RPMI1640 (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA). Cells were obtained from the cell bank of The Committee on Type Culture Collection of The Chinese Academy of Sciences (CCTCC, Shanghai, China). All siRNAs were chemically synthesized by Shanghai GenePharma (Shanghai, China). The sense sequence of the siRNA duplex for the negative control was 5’-UUCUCCGAACGUGU-CACGUTT-3’, and for BMYB was 5’-CAGACAAUGCUGUGAAGAATT-3’. The siRNAs were transfected into the indicated cells using Lipofectamine RNAiMAX reagent (invitrogen) according to the manufacturer’s instructions. Cells were collected and subjected to subsequent analysis 48-72 h after transfection.
Cell proliferation assay
Cell proliferation was measured using the CCK-8 (Dojindo Laboratories Japan). Briefly, 3000 cells per well were plated onto 96-well plates. At the indicated time points, 10 μL of the CCK-8 Solution was added and incubated for 2 h at 37°C. The absorption at 450 nm was measured using a microplate reader. All assays were performed in triplicates. The results are presented as the fold increase of absorption value relative to the corresponding zero time point samples.
Stable cell line generation
293T cells were transfected with the pQCXIP vector encoding each gene as well as the pVPack-GP and pVPack-Ampho vectors (Stratagene, Tokyo, Japan). The culture supernatant was collected 48 h later and applied to MDA-MB-231 cells with 2 μg/ml of polybrene (Sigma). Cells were cultured for 24 h, and then 1 μg/ml of puromycin (Sigma) was added to select for infected cells. To produce MDA-MB-231 cells that constitutively expressed shRNAs, oligonucleotides encoding shRNA specific for human ALX1 (5’-CCAATATTTCATGGGCCAT-3’) and luciferase (5’-CTTACGCTGAGTACTTCGA-3’) were cloned into the pSIREN-RetroQ retroviral vector (Clontech). Recombinant retrovirus was produced, and infected MDA-MB-231 cells were selected with 1 μg/ml puromycin for 3 days.
Soft agar colony formation assay
Colony formation ability was detremined by anchorage-independent soft agar assay on MDA-MB-231 cells. Briefly, 1.5 ml RPMI 1640 medium containing 0.6% agarose were added to each well of a six-well cell culture plates and allowed to solidify (base agar). 4×103 of MDA-MB-231 cells were then mixed with RPMI 1640 medium containing 0.35% agarose and added to the top of base agar. The cells were then cultured for 14 days at 37°C under 5% carbon dioxide. The plates were stained with 0.005% crystal violet for 1 hour, and the colonies were counted under microscope.
Transwell migration and invasion assay
The migration assay was conducted in a 24-well transwell cell culture apparatus fitted with multiporous polycarbonate membrane insert (8 um pore size) (Millipore). Briefly, cells were collected and resuspended in serum-free media at a density of 1×105 cells/ml. The top chamber of transwell was loaded with 100 ul of cell suspension, and the lower chamber was filled with 0.5 ml of media supplemented with 10% FBS as a chemoattrac-tant. After incubation at 37°C in 5% CO2 for 24 h, the filters were removed, rinsed two times with PBS, fixed with methanol, and stained with 0.5% crystal violet reagent. Cells on the upper side of the filter were wiped off with cotton swabs. Migrated cells on the lower side of the filter were determined by counting specified cross-sectional fields on the filters with a phase-contrast micro-scope. The transwell invasion assays were done under the same conditions as the transwell migration assays, but in Matrigel-coated (BD) transwells.
RNA isolation and qRT-PCR
Cells were collected 48-72 h after transfection. Total RNA was prepared from the indicated cells using the Total RNA Kit I (Omega Bio-Tek) according to the manufacturer’s instructions. The cDNA was generated from 1 μg of total RNA using PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa) following the manufacturer’s instructions. Real time PCR was carried out by using the SYBR® Premix Ex Taq™ (Perfect Real Time, TaKaRa) following the manufacturer’s instructions. The relative expression level of the target gene compared with that of the housekeeping gene, either GAPDH or β-actin, was calculated by the 2-∆∆Ct method (Bu et al., 2010; Livak and Schmittgen, 2001). The primer sequences used were listed in Supplement Table 1.
Table 1.
The primer sequences used for qRT-PCR
| Primer name | Primer sequence |
|---|---|
| GAPDH | F: 5’gatccctgtcaagaccccaa3’ |
| R: 5’ggaagtggagggttaacgga3’ | |
| B-myb | F: 5’ctatcctgagcccgaggaag3’ |
| R: 5’agtttcggaggggaagaagg3’ |
Immunoblotting analysis
The immunoblotting method has been also described in details previously (Bu et al., 2010). The primary antibodies used in the present study are Rabbit polyclonal anti-B-myb (ab89504, abcam) and rabbit polyclonal anti-GAPDH (Xianzhi Bio, Hangzhou, China) antibody. The secondary antibody used in study is Anti-rabbit IgG, HRP-linked Antibody (#7074S, Cell Signaling) and Anti-mouse IgG, HRP-linked Antibody (#7076, Cell Signaling).
Statistical analyses
All statistical analyses were carried out using the SPSS 16.0 statistical software package (SPSS Inc., Chicago, IL, USA). Two-sided independent Student’s t tests were performed to analyze the significance of the relationship between B-myb expression level and clinic-pathologic characteristics. P < 0.05 was considered statistically significant.
Results
B-myb expression is elevated in human breast cancer
To examine special expression to breast cancer progression, we analyzed the relationship of B-myb in tumors and lymph nodes tissues from 108 patients. Each sample was assigned an immunoreactivity score ranging from 0 to 6. Representative samples are shown in Figure 1A along with date analysis (Figure 1). Primary tumors and corresponding lymph node metastases exhibited diffuse cytoplasmic staining for B-myb. Over-expression B-myb levels also predict shorter overall survival of breast cancer patients. Paired comparisons of immunoreactivity scores between primary and metastatic tumors were significant (P < 0.001).
Figure 1.
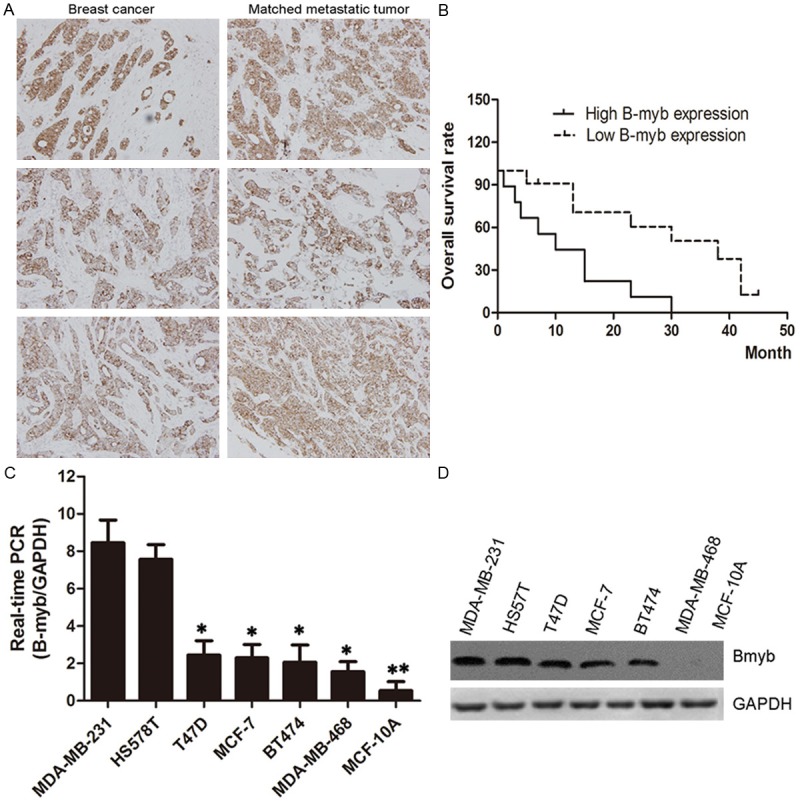
Expression of B-myb in breast cancer patient specimens and cell lines. A. Expression of IBP in primary breast cancer and matched lymph node tumors (×400); B. Kaplan-Meier plots of B-myb expression in 20 cases of breast cancer patients. Overall survival rate was performed by log-rank test. P < 0.05 indicate significant differences between two groups; C and D. Expression of B-myb in breast cancer cell lines. Equal amounts of proteins from seven non- or low metastatic breast cancer cell lines (MDA-MB-468, T47D, MCF-10A and MCF-7) and two highly metastatic breast cancer cell lines (MDA-MB-231 and HS578T) were evaluated by immunoblot analysis with antibodies against B-myb and glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
We also examined the expression of B-myb in normal human mammary epithelial cells, seven non or low metastatic breast cancer cell lines (MDA-MB-468, MDA-MB-231, T47D, HS578T, BT474, MCF-10A and MCF-7) and two highly metastatic cell lines (MDA-MB-231 and HS578T). Higher levels of B-myb RNA and protein were observed in breast cancer cells, especially over-expressed in metastatic cancer cells (Figure 3A and 3B). These findings indicated that B-myb is highly expressed in metastatic breast cancer cells. This correlation also shows that B-myb might have a crucial role in breast cancer metastasis.
Figure 3.
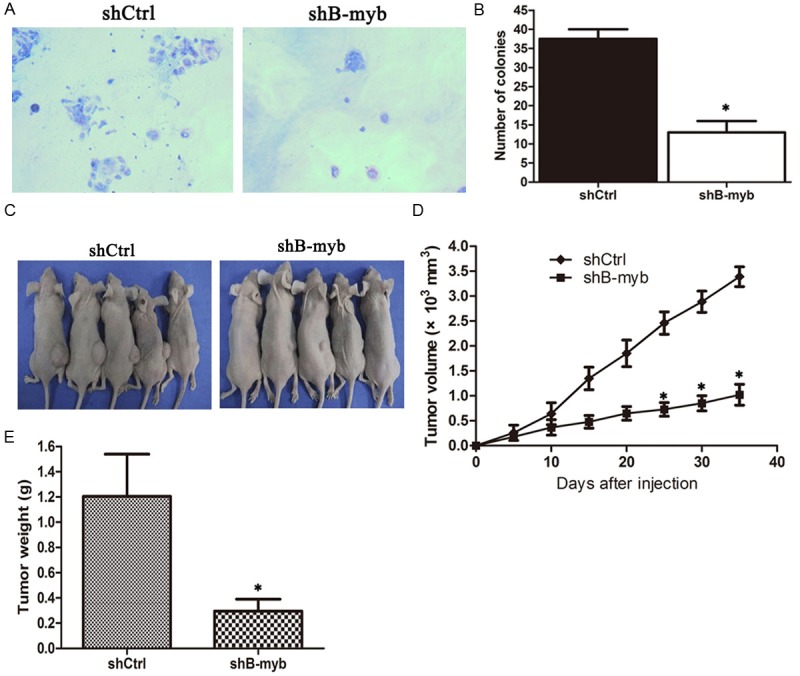
B-myb depletion inhibits breast tumorgenecity in vitro and in vivo. (A) Colony formation assay for B-myb depletion cells and control cells; (B) Numbers of Colonies for B-myb knockdown cells and control cells; (C-E) B-myb knockdown cells and control cells were injected subcutaneously into the dorsal flanks of nude mice, respectively. The tumor weight was measured at the end of the experiment and (D) The tumor growth curve. The tumor size was measured about twice a week for tumor growth curve construction. *P < 0.01.
B-myb depletion affects the cell cycle progression
To validate the positive functional involvement of B-myb in breast cancer, the B-myb expression was depleted via siRNA-mediated silencing in three different breast cancer cell lines, MDA-MB-231, MCF-7 and HS578T, and the cell cycle profile and cellular proliferation were subsequently analyzed. Quantitative RT-PCR and Western blot analysis demonstrated that the B-myb expression was significantly inhibited at both mRNA and protein levels in three cell lines (Figure 2A and 2B). Cell cycle analysis revealed that silencing B-myb expression resulted into a remarkable S phase arrest and a mild G2/M arrest in MDA-MB-231, HS578T and MCF-7 (Figure 2C).
Figure 2.
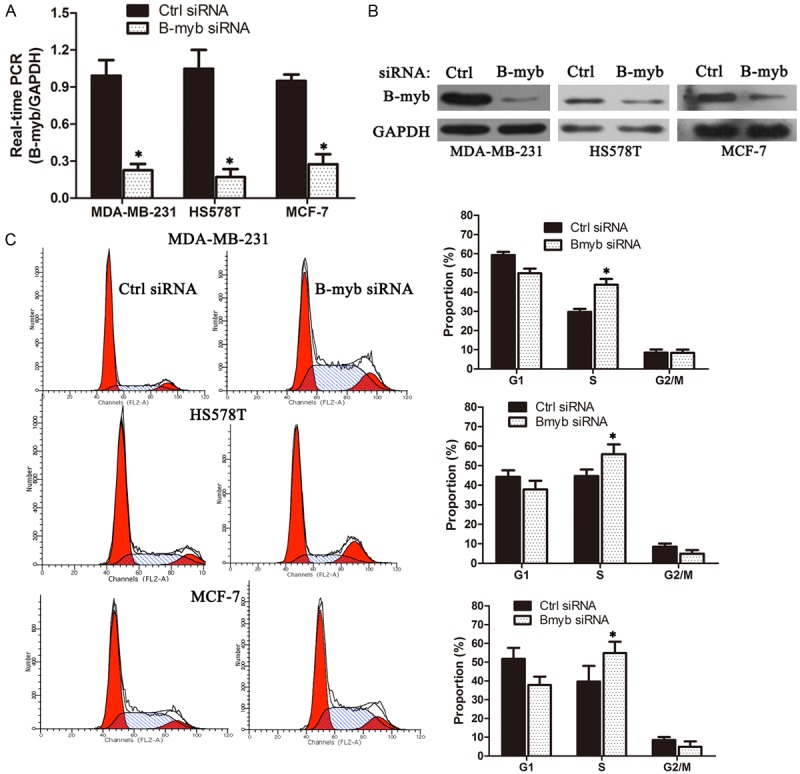
B-myb depletion affects cell cycle progression in breast cancer cells. A, B. siRNA-mediated B-myb depletion. MDA-MB-231, HS578T and MCF-7 cells were transiently transfected with the control siRNA and B-myb siRNA, respectively. Four-eight hours after transfection, total RNA and whole cell lysates were prepared and subjected to qRT-PCR and immunoblotting, respectively; C. Cell cycle analysis after B-mybdepletion. *P < 0.01.
B-myb depletion inhibits breast tumorigenesis in vitro and in vivo
To further test whether the inhibition of B-myb expression affected cancer cell growth in vivo, we generated MDA-MB-231 cells that constitutively expressed short hairpin RNA (shRNA) targeting B-myb shCtrl or shB-myb. Quantitative RT-PCR and Western blot analysis confirmed that the B-myb expression was significantly knockdown at both mRNA and protein levels in the stable B-myb knockdown cells (Supplementary data). As shown in Figure 3A and 3B, colony formation assay demonstrated that the stable B-myb knockdown cells showed a significantly reduced colony formation in both number and size, as compared with the control cells.
ShCtrl and shB-myb cells were subcutaneously injected to the femoral area of nude mice and tumor formation was examined. Both cell lines formed 6 subcutaneous tumors of 7 injected sites. The tumor formation of shB-myb cells was suppressed compared with the tumor formation of shCtrl cells (Figure 2C). Mice were sacrificed 36 days after tumor cell injection and the tumor weight was determined. The average tumor weight of shB-myb cells was significantly reduced compared with that of shCtrl cells (Figure 2D).
B-myb depletion reduces migration and invasion ability in breast cancer cells
We further examined whether inhibition of B-myb expression affected cell migration and invasion ability in breast cancer cells. The cell migration and invasion ability was evaluated by wound healing assay and matrigel invasion assay. As shown in Figure 4A, wound healing assay revealed that knockdown of B-myb significantly decreased the rate of lateral migration into a wound introduced in a confluent monolayer of cells compared with controls. Consistently, transwell migration assay showed that knockdown of B-myb also significantly inhibited the cellular transmigration ability compared with controls (Figure 4B). In the matrigel invasion assay, we also found that knockdown of B-myb significantly decreased the number of cells that penetrated through the Matrigel-coated membrane (Figure 4C). Therefore, these results strongly indicated that PRR11 might also regulate the cell migration and invasion ability of breast cancer cells.
Figure 4.
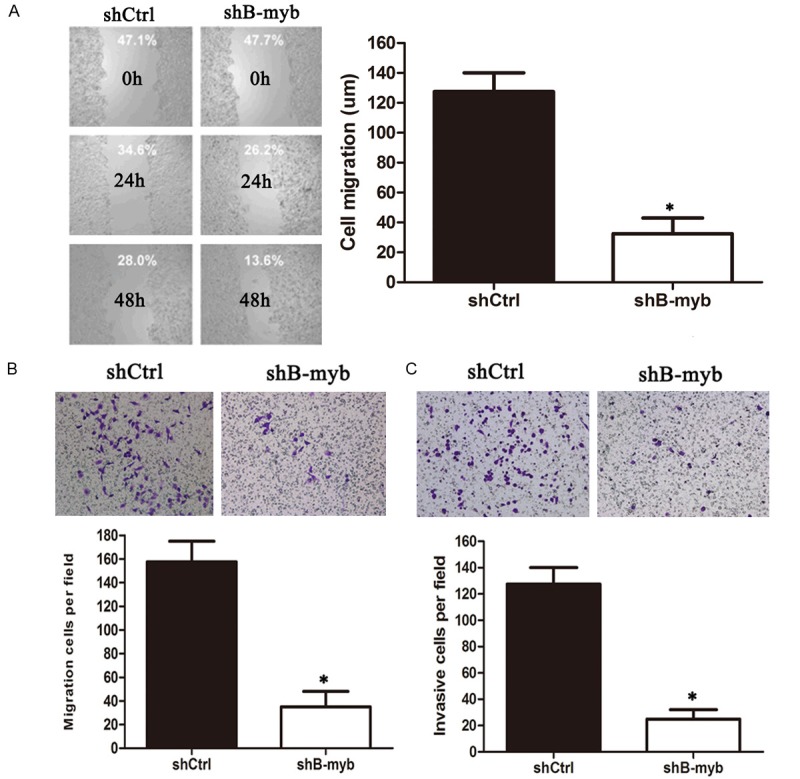
B-myb depletion inhibits migration and invasion in breast cancer cells. MDA-MB-231 cells were transiently transfected with the control shRNA and B-myb shRNA, respectively. Following siRNA transfection, cells were subjected to Would healing assay (A), transwell migration (B) and matrigel invasion assay (C), respectively. *P < 0.01.
Discussion
We defined B-myb as an oncogene frequently overexpressed in breast cancer cells that promotes cell migration and invasion. We confirmed the increased expression of B-myb in breast cancer, in particular in advanced cancer. We demonstrated that, as shown by the results of FACS profiles, silencing of B-myb expression resulted in significant S phase arrest in breast cancer cells and mild G2/M arrest in MDA-MB-231 and MCF-7 cells, indicating that B-myb regulates cell cycle progression especially S phase progression. Consistently, silencing of B-myb expression.
We performed a series of functional experiments with BMyb overexpression or depletion.the results indicated that B-myb promoted the cell cycle, cell proliferation, migration and invasion of MDA-MB-231 cells.then to further clarified the mechanism that B-myb promote breast cancer progression. qPCR was used to detect the mRNA expression levels of the cell cycle-related genes and the cell motility genes when BMyb overexpression or depletion. B-myb stimulated transcription of target genes that promoted entry into the S and M-phases of the cell cycle, cell proliferation, migration and invasion in breast cancer.
A series of experiments in vitro showed that the BMyb promoted the progress of breast cancer, now we will further clarify the mechanism of B-myb promoting breast cancer progression. We found out some genes associated with cell cycle and cell movement through the analysis of microarray data downloaded. qPCR was used to study whether the genes were regulated by B-Myb. B-myb has been reported is a growth-regulated gene. Its expression is barely detectable in G0 and is induced at the G1/S transition of the cell cycle and Its protein levels parallel only in part mRNA expression and a pool of stable B-myb protein is detectable throughout the cell cycle. Besides B-myb is ubiquitously expressed and functions at the G1/S transition of the cell cycle.
According to the provenance of c-myb as a cellular proto-oncogene and the ability of B-myb to regulate the expression of cell cycle genes, it is not surprising that B-myb is involved in cell proliferation and carcinogenesis [17,18]. B-myb expression is required for entry into S-phase and can overcome growth inhibitory signals. In addition, B-Myb overexpression occurs in several cancers and has been linked to aggressive tumor growth and poor outcomes in neuroblastomas and other tumors. Conversely, B-myb repression can inhibit the proliferation of normal and tumor cells. We confirmed B-myb promoted cell proliferation and the progress of breast cancer by CCK-8 and colony formation assay when B-myb overexpression or depletion.Transwell showed B-myb promoted cancer cell invasion and migration. There were no relevant literature that reported the mechanisms that B-myb promoted breast cancer invasion and migration. We will firstly study and try to preliminary clarify the mechanisms that B-myb regulation of invasion and migration in breast cancer.
In summary, we studied the significance of the relationship between B-myb and clinic-pathologic characteristics and assessed the value of B-myb worked as an independent factor affecting prognosis. Our study strongly indicated that B-myb had a critical role in cell cycle progression, tumorigenesis, migration and invasion of breast cancer, and It might serve as a potential target in the diagnosis and/or treatment of human breast cancer.
Acknowledgements
This study was supported by the grants from Taizhou science and technology program (102KY13).
Disclosure of conflict of interest
None.
References
- 1.Joaquin M, Watson RJ. Cell cycle regulation by the B-Myb transcription factor. Cell Mol Life Sci. 2003;60:2389–2401. doi: 10.1007/s00018-003-3037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sala A. B-MYB, a transcription factor implicated in regulating cell cycle, apoptosis and cancer. Eur J Cancer. 2005;41:2479–2484. doi: 10.1016/j.ejca.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Sala A, Watson R. B-Myb protein in cellular proliferation, transcription control, and cancer: latest developments. J Cell Physiol. 1999;179:245–250. doi: 10.1002/(SICI)1097-4652(199906)179:3<245::AID-JCP1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Lam EW, Robinson C, Watson RJ. Characterization and cell cycle-regulated expression of mouse B-myb. Oncogene. 1992;7:1885–1890. [PubMed] [Google Scholar]
- 5.Lam EW, Bennett JD, Watson RJ. Cell-cycle regulation of human B-myb transcription. Gene. 1995;160:277–281. doi: 10.1016/0378-1119(95)00184-8. [DOI] [PubMed] [Google Scholar]
- 6.Sala A, Kundu M, Casella I, Engelhard A, Calabretta B, Grasso L, Paggi MG, Giordano A, Watson RJ, Khalili K, Peschle C. Activation of human B-MYB by cyclins. Proc Natl Acad Sci U S A. 1997;94:532–536. doi: 10.1073/pnas.94.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziebold U, Bartsch O, Marais R, Ferrari S, Klempnauer KH. Phosphorylation and activation of B-Myb by cyclin A-Cdk2. Curr Biol. 1997;7:253–260. doi: 10.1016/s0960-9822(06)00121-7. [DOI] [PubMed] [Google Scholar]
- 8.Charrasse S, Carena I, Brondani V, Klempnauer KH, Ferrari S. Degradation of B-Myb by ubiquitin-mediated proteolysis: involvement of the Cdc34-SCF (p45Skp2) pathway. Oncogene. 2000;19:2986–2995. doi: 10.1038/sj.onc.1203618. [DOI] [PubMed] [Google Scholar]
- 9.Cesi V, Tanno B, Vitali R, Mancini C, Giuffrida ML, Calabretta B, Raschella G. Cyclin D1-dependent regulation of B-myb activity in early stages of neuroblastoma differentiation. Cell Death Differ. 2002;9:1232–1239. doi: 10.1038/sj.cdd.4401103. [DOI] [PubMed] [Google Scholar]
- 10.Horstmann S, Ferrari S, Klempnauer KH. Regulation of B-Myb activity by cyclin D1. Oncogene. 2000;19:298–306. doi: 10.1038/sj.onc.1203302. [DOI] [PubMed] [Google Scholar]
- 11.Joaquin M, Watson RJ. The cell cycle-regulated B-Myb transcription factor overcomes cyclin-dependent kinase inhibitory activity of p57(KIP2) by interacting with its cyclin-binding domain. J Biol Chem. 2003;278:44255–44264. doi: 10.1074/jbc.M308953200. [DOI] [PubMed] [Google Scholar]
- 12.Bar-Shira A, Pinthus JH, Rozovsky U, Goldstein M, Sellers WR, Yaron Y, Eshhar Z, Orr-Urtreger A. Multiple genes in human 20q13 chromosomal region are involved in an advanced prostate cancer xenograft. Cancer Res. 2002;62:6803–6807. [PubMed] [Google Scholar]
- 13.Thorner AR, Hoadley KA, Parker JS, Winkel S, Millikan RC, Perou CM. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene. 2009;28:742–751. doi: 10.1038/onc.2008.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bu Y, Suenaga Y, Okoshi R, Sang M, Kubo N, Song F, Nakagawara A, Ozaki T. NFBD1/MDC1 participates in the regulation of G2/M transition in mammalian cells. Biochem Biophys Res Commun. 2010;397:157–162. doi: 10.1016/j.bbrc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- 15.Nomura N, Takahashi M, Matsui M, Ishii S, Date T, Sasamoto S, Ishizaki R. Isolation of human cDNA clones of myb-related genes, A-myb and B-myb. Nucleic Acids Res. 1988;16:11075–11089. doi: 10.1093/nar/16.23.11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gualdrini F, Corvetta D, Cantilena S, Chayka O, Tanno B, Raschella G, Sala A. Addiction of MYCN amplified tumours to B-MYB underscores a reciprocal regulatory loop. Oncotarget. 2010;1:278–288. doi: 10.18632/oncotarget.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arsura M, Introna M, Passerini F, Mantovani A, Golay J. B-myb antisense oligonucleotides inhibit proliferation of human hematopoietic cell lines. Blood. 1992;79:2708–2716. [PubMed] [Google Scholar]
- 18.Zhu W, Giangrande PH, Nevins JR. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 2004;23:4615–4626. doi: 10.1038/sj.emboj.7600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Song F, Chen X, Li Y, Fan J, Wu X. Bmi-1 regulates epithelial-to-mesenchymal transition to promote migration and invasion of breast cancer cells. Int J Clin Exp Pathol. 2014;7:3057–3064. [PMC free article] [PubMed] [Google Scholar]