

Abstract
The acid-sensitive outwardly rectifying (ASOR) anion channel has been found in non-neuronal cell types and was shown to be involved in acidotoxic death of epithelial cells. We have recently shown that the ASOR channel is sensitive to temperature. Here, we extend those results to show that temperature-sensitive ASOR anion channels are expressed in cortical neurons and involved in acidotoxic neuronal cell death. In cultured mouse cortical neurons, reduction of extracellular pH activated anionic currents exhibiting phenotypic properties of the ASOR anion channel. The neuronal ASOR currents recorded at pH 5.25 were augmented by warm temperature, with a threshold temperature of 26 °C and the Q10 value of 5.6. After 1 h exposure to acidic solution at 37 °C, a large population of neurons suffered from necrotic cell death which was largely protected not only by ASOR channel blockers but also by reduction of temperature to 25 °C. Thus, it is suggested that high temperature sensitivity of the neuronal ASOR anion channel provides, at least in part, a basis for hypothermic neuroprotection under acidotoxic situations associated with a number of pathological brain states.
Keywords: temperature sensitivity, acid-sensitive anion channel, acidotoxicity, hypothermia
Introduction
Strong acidification of extracellular solution has been shown to activate a voltage-dependent anion channel, called the acid-sensitive outwardly rectifying (ASOR) anion channel,1 in a variety of non-neuronal cells.1-10 This channel is phenotypically characterized by activation by severe extracellular acidification alone as well as by strong outward rectification1-10 and was shown to be involved in the genesis of acidotoxic necrotic cell death in human epithelial cells.1 Our recent study11 showed that the epithelial ASOR anion channel is sensitive to temperature.
In the brain, strong acidosis with extracellular pH below 6 is a common feature of ischemia, seizure, trauma and hyperglycemia, and it contributes to neuronal injury.12-15 Hypothermia is known to be the most effective therapeutic means for protecting the brain against ischemic, excitotoxic or traumatic brain injury,16-18 but the precise mechanisms of hypothermic neuroprotection are still unclear.16,19,20 In light of these facts, it is interesting to investigate whether brain neurons express temperature-sensitive ASOR anion channels that are involved in acidotoxic neuronal cell death. Thus, in this addendum to our previous study,11 we demonstrate that mouse brain neurons express the ASOR anion channel, which is sensitive to temperature and involved in the genesis of acidotoxic necrosis, and are protected by hypothermia in a manner sensitive to blockers of the ASOR anion channel.
Results
ASOR anion channel is expressed in cortical neurons and is temperature-sensitive
In the whole-cell patch-clamp configuration using the extracellular and intracellular (pipette) solutions rich with N-methyl D-glucamine (NMDG+), the currents recorded from mouse cortical neurons were found to be augmented by reducing the extracellular pH from 7.4 to 4.5 at 25 °C in a reversible manner, as shown in Figure 1A. The acid-induced current exhibited strong outward rectification (Fig. 1B). When the extracellular Cl− concentration ([Cl]o) was reduced by replacement with an equimolar amount of aspartate−, the Erev values were shifted to more positive potentials with a slope of −56.5 mV/decade (Fig. 1C), which is close to the ideal value for a genuine Cl− electrode. Similar anion replacement studies showed that the halide anion selectivity of the acid-activated current component is I−: Br−: Cl−: methansulfonate−: aspartate− = 4.47 ± 0.20: 1.60 ± 0.04: 1: 0.11 ± 0.01: 0.10 ± 0.01 (n = 7−9), indicating low-field anion selectivity. The acid-activated currents recorded at pH 4.5 and 25 °C were rapidly suppressed by a stilbene-derivative Cl− channel blocker, 4,4'-diisothiocyanostilbene-2,2'-disulphonic acid (DIDS), with a half-maximal inhibition concentration (IC50) of 4.7 μM at +60 mV (Fig. 2A). The currents were also sensitive to a bisphenol-derivative drug, phloretin, with IC50 of 22.0 μM at +60 mV (Fig. 2B). These biophysical and pharmacological properties of neuronal acid-activated currents are phenotypically identical to those of ASOR anion channel expressing in a variety of non-neuronal cells.1-10
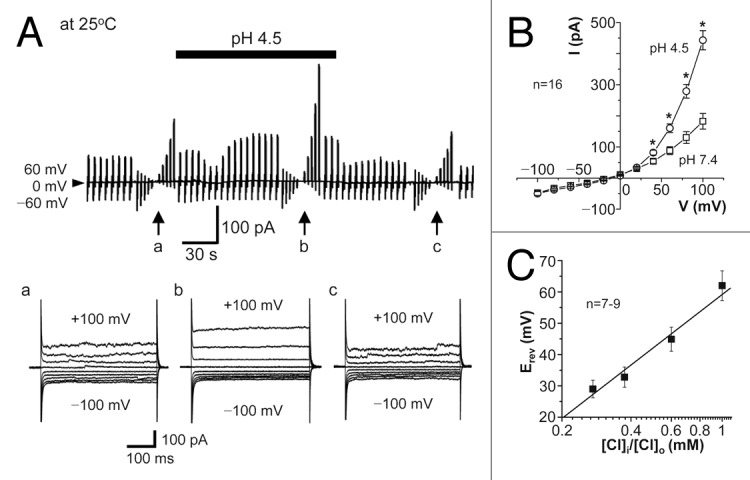
Figure 1. Whole-cell anionic currents activated by extracellular acidification in cortical neurons at 25 °C. (A) Top: Representative record of whole-cell currents before, during (at bar), and after exposure to acidic solution (pH 4.5). Alternating pulses (0.25 s duration, every 5 s) of ± 60 mV or step pulses (0.5 s duration, every 3 s) of ± 100 mV in 20-mV increments (at arrows) were applied. Bottom: Expanded traces of current responses to step pulses (applied at a, b, and c). (B) I-V relationships for the mean currents (with the SEM bars) of steady-state peak responses to step pulses before (pH 7.4) and during (pH 4.5) exposure to acidic solution. Asterisks indicate significant differences between data recorded at pH 7.4 and pH 4.5 (P < 0.05). (C) Relation between the mean reversal potential (Erev) for the acid-activated current components and the logarithm of [Cl]i/[Cl]o. The slope determined by a linear regression analysis is −56.5 mV/decade.

Figure 2. Sensitivity of ASOR anion currents recorded at 25 °C in cortical neurons to DIDS (A) and phloretin (B). Top: Representative records of ASOR currents before and during application of 100 μM DIDS and 300 μM phloretin. Alternating pulses were applied as in Figure 1A. Bottom: Concentration-dependent inhibition of ASOR currents (acid-activated components) recorded at +60 mV and 25 °C by DIDS and phloretin. The data are fitted by the Hill equation with the indicated IC50 values.
Since our previous study11 showed high sensitivity of the human epithelial ASOR channel currents to temperature, we next observed the effects of continuous changes in temperature of the extracellular solution from 22 to 38 °C on the mouse neuronal ASOR channel currents recorded at pH 5.25. The currents gradually decreased and increased with reducing and raising the temperature, respectively (Fig. 3A). When the logarithms of acid-activated component of the currents were plotted against the reciprocal of the absolute temperature, the resultant curve was asymptotically approximated to two lines at ≤ 32 °C (Fig. 3B). The transition temperature estimated from the intersecting point of these two lines is 25.6 °C, and the 10° temperature coefficient (Q10) values are 5.6 at warm temperature between 32 and 26 °C and 2.2 at cool temperature between 26 and 22 °C. Activation of the mouse neuronal ASOR currents recorded at pH 5.25 became more prominent by increasing temperature from 25 °C to 37 °C (Fig. 3C). The activation curves shifted to higher pH by such a temperature increase with the pH value for half-maximal activation (EC50) of 4.86 at 25 °C and 5.38 at 37 °C (Fig. 3D). Thus, it is concluded that mouse cortical neurons functionally express the ASOR anion channel which is sensitive to temperature.

Figure 3. Effects of temperature changes on the ASOR anion channel currents recorded in cortical neurons. (A) Representative trace (top) of ASOR current activation during application of alternating pulses from 0 to ± 60 mV at pH 5.25 in response to a ramp increase in temperature (bottom) from 22 to 38 °C. (B) Arrhenius plot for the mean ASOR currents (with the SEM bars) recorded at +60 mV and pH 5.25. The whole-cell current (I) and temperature were simultaneously recorded as done in A. (Q10 values and two lines, see in the text). (C) Representative current responses to step pulses (left) and mean peak currents recorded at +100 mV (right) at 37 and 25 °C (at pH 5.25). Asterisk indicates a significant difference (P < 0.01) between the mean peak values (with the SEM bars) recorded at 37 and 25 °C. (D) Shift of pH dependence of relative mean ASOR currents (recorded at +60 mV) by a temperature increase from 25 to 37 °C. The data are fitted by the Hill equation with the EC50 values given on the figures.
Temperature sensitivity of the ASOR anion channel in mouse cortical neurons is involved in hypothermic protection against acidotoxic necrosis in vitro
Double staining studies in cultured cortical neurons using propidium iodide (PI) and Hoechst 33342 showed that injury induced by 1 h exposure to control serum-free neutral-pH electrolyte solution looked only slight at both 25 and 37 °C, but this was markedly enhanced at pH 5.25 (Fig. 4A). The % of PI-positive neurons at pH 5.25 was ~20% at 25 °C, but this value further increased to ~50% (P < 0.01) by increasing the temperature to 37 °C (Fig. 4B). Acidosis-induced neuronal injury was largely prevented by DIDS (10 μM) or phloretin (50 μM) at 37 °C (P < 0.01; Figure 4B, lower panel), whereas that was not significantly affected by DIDS or phloretin at 25 °C (Fig. 4B, upper panel). These data indicate that acidosis-induced neuronal injury is sensitive to temperature and to the ASOR channel blockers.
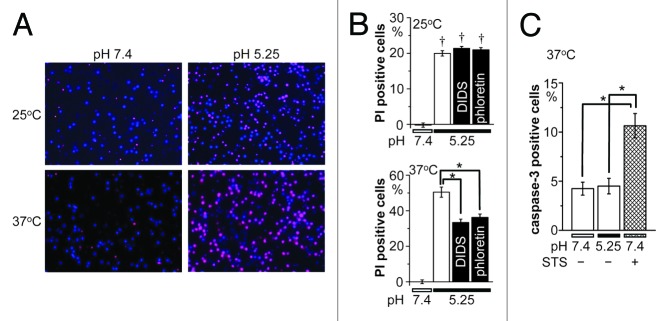
Figure 4. Acidotoxic necrotic death of cortical neurons and its sensitivity to hypothermia and anion channel blockers. (A) Representative fluorescence micrographs of cortical neurons stained with Hoechst 33342 (blue) for nuclei of all neurons and with PI for nuclei of injured neurons 1 h after exposure to control (pH 7.4) or acidic (pH 5.25) solution at 25 or 37 °C. (B) Summarized data showing the fraction of PI-positive neurons at 25 °C (top) and 37 °C (bottom) in neutral pH or acidic solution in the absence or presence of 10 μM DIDS or 50 μM phloretin. (C) Caspase-3 activity after exposure to control (pH 7.4) or acidic (pH 5.25) solution for 1 h at 37 °C in the absence or presence of 0.5 μM STS. Each column represents the mean (with the SEM bar) of 28−96 (B) or 5−10 (C) samples. Daggers indicate significant differences (P < 0.01) between the data obtained at 25 and 37 °C (B). Single asterisks indicate significant differences (P < 0.01) between the data obtained at 37 °C without and with anion channel blockers (B) or between those in the presence and absence of STS (C).
Activation of caspase-3 was not observed in cortical neurons exposed to acidic solution (pH 5.25) for 1 h at 37 °C but was largely observed after 1 h exposure to a mitochondrion-mediated apoptosis inducer, staurosporine (STS, 0.5 μM), as summarized in Figure 4C. Thus, it appears that acidosis-induced neuronal injury represents necrotic, not apoptotic, cell death. As necrotic cell death is known to be associated with sustained cell swelling, called necrotic volume increase (NVI),21 we then measured the cross-sectional area (CSA) of the soma of cortical neurons after switching the pH value of bathing solution from 7.4 to 5.25. As shown in Figure 5A, acidosis induced cell swelling at both 25 and 37 °C with exhibiting peak swelling after around 2.5 min followed by sustained moderate swelling. The peak swelling at 37 °C was significantly larger than that at 25 °C (P < 0.01; Figure 5B). Application of DIDS (10 μM) or phloretin (50 μM) almost completely inhibited acidosis-induced cell swelling at 25 °C and also largely suppressed that at 37 °C (Fig. 5). These results may indicate that acidosis-induced neuronal swelling is mediated by activation of ASOR anion channels sensitive to temperature, DIDS and phloretin.

Figure 5. Acidotoxic cell swelling (NVI) and its sensitivity to hypothermia and anion channel blockers in cortical neurons. (A) Time courses of cell size (CSA) changes before (pH 7.4) and after application (started at arrows) of acidic (pH 5.25) solution at 25 °C (top) or 37 °C (bottom) in the absence (open symbols) or presence (filled symbols) of 10 μM DIDS or 50 μM phloretin. Insets: Representative microscopic images of a neuron before (0 min) and 2.5 min after the beginning of exposure to acidic solution at 25 °C (top photos) and 37 °C (bottom photos). (B) Summarized data showing the peak cell swelling (CSA recorded at 2.5 min) relative to the control cell size (CSA recorded at 0 min) at 25 °C (top) or 37 °C (bottom) in the absence (open column) or presence of 10 μM DIDS or 50 μM phloretin. Asterisks indicate significant difference (P < 0.01) between the data obtained at pH 5.25 without anion channel blockers and those obtained at pH 7.4 without anion channel blockers or at pH 5.25 with anion channel blockers. Daggers indicate significant difference (P < 0.01) between the data obtained at 25 and 37 °C.
Discussion
The acid-sensitive outwardly rectifying (ASOR) anion channel was here, for the first time, found to be functionally expressed in brain neurons. The ASOR anion channel in mouse cortical neurons exhibited sensitivity to acid (pH ≤ 6), steep outward rectification, low-field anion selectivity (with permeability sequence of Cl− < Br− < I−) and sensitivity to DIDS and phloretin. These properties are very similar to those observed in human epithelial cells.1 In addition, the neuronal ASOR channel showed high sensitivity to temperature. The current was augmented by warm temperature with threshold temperature (transition temperature) of 25.6 °C and a Q10 value of 5.6, which were evaluated by the Arrhenius plot. The transition temperature of mouse neuronal ASOR anion channel is lower than that of the human epithelial ASOR anion channel (32 °C)11 and the ANO1 anion channel (44 °C).22 The Q10 value is also smaller than those of the human epithelial ASOR channel (8.8)11 and ANO1 channel (19.4).22 However, it is much higher than those (1.2−1.7) in most ion channels,23 indicating that the gating process of ASOR anion channel is different from a diffusion-limited process but may be mediated by some conformational rearrangement of channel proteins (including some inter-subunit interaction) between the closed and open state. A difference in temperature sensitivity between the human epithelial and mouse neuronal ASOR channels may be due to differences in the species and/or tissues. The precise reason of the difference awaits molecular identification of the ASOR channel protein.
As was the case in human epithelial cells,1 mouse cortical neurons suffered from necrotic cell injury characterized by stainability with PI and lack of caspase-3 activation after 1 h incubation in acidic solution (pH 5.25). Since such an acidotoxic neuronal injury was largely rescued by ASOR anion channel blockers (DIDS and phloretin) and to lowered temperature below the transition temperature, it appears that the neuronal ASOR anion channel is involved in the genesis of acidotoxic injury in neuronal cells in vitro. In brains, extracellular pH may fall below 6 under a variety of pathologic conditions including ischemia, seizure, excitotoxicity, trauma, hyperglycemia and tumor.12-15,24,25 Thus, it is highly possible that the ASOR anion channel activity contributes, at least in part, to the pathogenesis of acidotoxic necrosis in brains in vivo. Voltage-insensitive, acid-sensitive Na+-permeable cation channel, ASIC, was also shown to be involved in necrotic neuronal cell death under acidotoxic conditions.26,27 Therefore, there is a possibility that both acid-activated cation entry via ASIC and anion entry via ASOR anion channels into neurons participate, in a manner restraint to electroneutrality, in the NVI process which is known to be induced by water inflow driven mainly by NaCl influx.19,28-30
Neuroprotective efficacy of hypothermia is well established not only by animal studies but also by preclinical experience and early clinical trials.16-20 The protective effects of hypothermia cannot be explained only by slowing of cerebral metabolism but must involve other more important mechanisms.19,20 In brain neurons, high temperature sensitivity of the neuronal ASOR anion channel and hence of acidotoxic neuronal cell death may provide one of multiple bases for hypothermia protection from pathological situations coupled to severe acidosis.
Methods and Materials
The experimental protocol was approved in advance by the Ethics Review Committee for Animal Experimentation of the National Institute for Physiological Sciences. Neuronal cell cultures derived from mouse embryonic cerebral cortices were prepared, as previously described.31 In brief, pregnant C57BL/6NCr mice (Japan SLC, Inc.) were anesthetized with halothane (Takeda Pharmaceutical Co.) and killed by cervical dislocation. Then the uteri were immediately removed and placed in cold PBS. The 16-d-old embryos were obtained, and their cerebral cortices were removed from their brain, trimmed in Leibobitz’s L-15 medium (Gibco BRL), and then digested using EBSS (Gibco) containing 0.8 mM EDTA, 20 mM glucose and 10 U/ml papain (Worthington Biochemical, Lakewood, NJ) for 20 min at 37 °C. The tissue were once washed with Neurobasal-A medium (Gibco) supplemented with 0.5 mM L-glutamine, 25 μM glutamate, 1/50-diluted B-27 Supplement (50X; Gibco), 100 U/ml penicillin plus 0.1 mg/ml streptomycin (Gibco) in the presence of 1 mg/ml DNase I (Roche Diagnostics Co.) and then were washed 5 times with Neurobasal-A medium in the absence of DNase I. The cells were isolated by mechanical trituration through a Pasteur pipette. The cell suspension thus obtained was plated (1 × 106 cells/cm3) on 0.2% polyethylenemine-coated coverslips or slide plates. After 3 d, the cells were provided for experiments. Whole-cell patch-clamp experiments at controlled temperature were performed, as previously reported.11 Pipette (intracellular) solution consisted of (in mM): 30 NMDG-Cl, 80 NMDG-aspartate, 50 HEPES, 2 MgSO4, 1 EGTA, and 25 mannitol (300 mosmol/kg⋅H2O, pH 7.4). Extracellular bathing solution contained (in mM): 110 NMDG-Cl, 12 HEPES, 5 MgSO4, and 88 mannitol (330 mosmol/kg⋅H2O, pH 7.4). To prepare acidic bathing solutions, HEPES was replaced with MES. The osmolality was measured by using a freezing-point depression osmometer (OM802; Vogel, Giessen, Germany). To study anion selectivity permeability sequence, Cl− in the bath solution was replaced by I−, Br−, methansulfonate− and aspartate−. Necrotic and apoptotic cell death was monitored by stainability with PI and Hoechst 33342, as previously described,1 by using In Cell Analyzer 1000 (GE Healthcare). Apoptotic cell death was monitored with FLICA and Hoechst 33342, as previously described,32 by using a fluorescence microscope (IX70; Olympus). Cell size was monitored by measuring the CSA of the soma of cortical neurons, as previously reported.31 Statistical analyses were made by ANOVA and the paired or unpaired Student t test. Data are presented as means ± SEM of n observations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research from MEXT. The authors thank K Shigemoto and N Yasui for technical assistance, RZ Sabirov for discussion, and T Okayasu for secretarial assistance.
Glossary
Abbreviations:
- ASOR
acid-sensitive outwardly rectifying
- [Cl]o
extracellular Cl− concentration
- CSA
cross-sectional area
- DIDS
4,4'-diisothiocyanostilbene-2,2'-disulphonic acid
- EC50
value for half-maximal activation
- Erev
reversal potential
- IC50
half-maximal inhibition concentration
- NMDG
N-methyl D-glucamine
- NVI
necrotic volume increase
- PI
propidium iodide
- Q10
10° temperature coefficient
- STS
staurosporine
References
- 1.Wang H-Y, Shimizu T, Numata T, Okada Y. Role of acid-sensitive outwardly rectifying anion channels in acidosis-induced cell death in human epithelial cells. Pflugers Arch. 2007;454:223–33. doi: 10.1007/s00424-006-0193-z. [DOI] [PubMed] [Google Scholar]
- 2.Auzanneau C, Thoreau V, Kitzis A, Becq F. A Novel voltage-dependent chloride current activated by extracellular acidic pH in cultured rat Sertoli cells. J Biol Chem. 2003;278:19230–6. doi: 10.1074/jbc.M301096200. [DOI] [PubMed] [Google Scholar]
- 3.Fu ZJ, Li XZ, Wang QR, Shi L, Zhang LQ, Pan XL. Extracellular acidic pH-activated, outward rectifying chloride currents can be regulated by reactive oxygen species in human THP-1 monocytes. Biochem Biophys Res Commun. 2013;432:701–6. doi: 10.1016/j.bbrc.2013.01.090. [DOI] [PubMed] [Google Scholar]
- 4.Kucherenko YV, Mörsdorf D, Lang F. Acid-sensitive outwardly rectifying anion channels in human erythrocytes. J Membr Biol. 2009;230:1–10. doi: 10.1007/s00232-009-9179-z. [DOI] [PubMed] [Google Scholar]
- 5.Lambert S, Oberwinkler J. Characterization of a proton-activated, outwardly rectifying anion channel. J Physiol. 2005;567:191–213. doi: 10.1113/jphysiol.2005.089888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda JJ, Filali MS, Collins MM, Volk KA, Lamb FS. The ClC-3 Cl-/H+ antiporter becomes uncoupled at low extracellular pH. J Biol Chem. 2010;285:2569–79. doi: 10.1074/jbc.M109.018002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS. Activation of swelling-activated chloride current by tumor necrosis factor-α requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem. 2010;285:22864–73. doi: 10.1074/jbc.M109.099838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nobles M, Higgins CF, Sardini A. Extracellular acidification elicits a chloride current that shares characteristics with ICl(swell) Am J Physiol Cell Physiol. 2004;287:C1426–35. doi: 10.1152/ajpcell.00549.2002. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Ma W, Zhu L, Ye D, Li Y, Liu S, Li H, Zuo W, Li B, Ye W, et al. ClC-3 is a candidate of the channel proteins mediating acid-activated chloride currents in nasopharyngeal carcinoma cells. Am J Physiol Cell Physiol. 2012;303:C14–23. doi: 10.1152/ajpcell.00145.2011. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto S, Ehara T. Acidic extracellular pH-activated outwardly rectifying chloride current in mammalian cardiac myocytes. Am J Physiol Heart Circ Physiol. 2006;290:H1905–14. doi: 10.1152/ajpheart.00965.2005. [DOI] [PubMed] [Google Scholar]
- 11.Sato-Numata K, Numata T, Okada T, Okada Y. Acid-sensitive outwardly rectifying (ASOR) anion channels in human epithelial cells are highly sensitive to temperature and independent of ClC-3. Pflugers Arch. 2013;465:1535–43. doi: 10.1007/s00424-013-1296-y. [DOI] [PubMed] [Google Scholar]
- 12.Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-A. [DOI] [PubMed] [Google Scholar]
- 13.Kraig RP, Pulsinelli WA, Plum F. Heterogeneous distribution of hydrogen and bicarbonate ions during complete brain ischemia. Prog Brain Res. 1985;63:155–66. doi: 10.1016/S0079-6123(08)61981-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rehncrona S. Brain acidosis. Ann Emerg Med. 1985;14:770–6. doi: 10.1016/S0196-0644(85)80055-X. [DOI] [PubMed] [Google Scholar]
- 15.Siesjö BK. Acidosis and ischemic brain damage. Neurochem Pathol. 1988;9:31–88. doi: 10.1007/BF03160355. [DOI] [PubMed] [Google Scholar]
- 16.Gunn AJ, Thoresen M. Hypothermic neuroprotection. NeuroRx. 2006;3:154–69. doi: 10.1016/j.nurx.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu L, Yenari MA. Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci. 2007;12:816–25. doi: 10.2741/2104. [DOI] [PubMed] [Google Scholar]
- 18.Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci. 2012;13:267–78. doi: 10.1038/nrn3174. [DOI] [PubMed] [Google Scholar]
- 19.Lyden PD, Krieger D, Yenari M, Dietrich WD. Therapeutic hypothermia for acute stroke. Int J Stroke. 2006;1:9–19. doi: 10.1111/j.1747-4949.2005.00011.x. [DOI] [PubMed] [Google Scholar]
- 20.Polderman KH. Application of therapeutic hypothermia in the ICU: opportunities and pitfalls of a promising treatment modality. Part 1: Indications and evidence. Intensive Care Med. 2004;30:556–75. doi: 10.1007/s00134-003-2152-x. [DOI] [PubMed] [Google Scholar]
- 21.Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, Back SK, Na HS, Harfe BD, Wang F, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015–21. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- 23.Hille B. Ion channels of excitable membranes - Third edition. Sunderland: Sinauer Associates Inc, 2001. [Google Scholar]
- 24.Evelhoch JL. pH and therapy of human cancers. Novartis Found Symp. 2001;240:68–80, discussion 80-4, 152-3. doi: 10.1002/0470868716.ch5. [DOI] [PubMed] [Google Scholar]
- 25.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–65. [PubMed] [Google Scholar]
- 26.Xiong Z-G, Zhu X-M, Chu X-P, Minami M, Hey J, Wei W-L, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 27.Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci U S A. 2004;101:6752–7. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barros LF, Hermosilla T, Castro J. Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:401–9. doi: 10.1016/S1095-6433(01)00438-X. [DOI] [PubMed] [Google Scholar]
- 29.Okada Y, Maeno E, Mori S. Essential role of anion channel in induction of apoptotic and necrotic cell death. In: Yuan JX-J, ed. Ion Channels in the Pulmonary Vasculature. Boca Raton, FL: Taylor & Francis, 2005:527-44. [Google Scholar]
- 30.Okada Y, Maeno E, Shimizu T, Manabe K, Mori S, Nabekura T. Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch. 2004;448:287–95. doi: 10.1007/s00424-004-1276-3. [DOI] [PubMed] [Google Scholar]
- 31.Inoue H, Mori S, Morishima S, Okada Y. Volume-sensitive chloride channels in mouse cortical neurons: characterization and role in volume regulation. Eur J Neurosci. 2005;21:1648–58. doi: 10.1111/j.1460-9568.2005.04006.x. [DOI] [PubMed] [Google Scholar]
- 32.Dezaki K, Maeno E, Sato K, Akita T, Okada Y. Early-phase occurrence of K+ and Cl- efflux in addition to Ca 2+ mobilization is a prerequisite to apoptosis in HeLa cells. Apoptosis. 2012;17:821–31. doi: 10.1007/s10495-012-0716-3. [DOI] [PubMed] [Google Scholar]