

Abstract
The nucleocapsid of measles virus is the template for viral RNA synthesis and is generated through packaging of the genomic RNA by the nucleocapsid protein (N). The viral polymerase associates with the nucleocapsid through a small, trihelical binding domain at the carboxyl terminus of the phosphoprotein (P). Translocation of the polymerase along the nucleocapsid during RNA synthesis is thought to involve the repeated attachment and release of the binding domain. We have investigated the interaction between the binding domain from measles P (amino acids 457–507) and the sequence it recognizes within measles N (amino acids 477–505). By using both solution NMR spectroscopy and x-ray crystallography, we show that N487–503 binds as a helix to the surface created by the second (α2) and third (α3) helices of P457–507, in an orientation parallel to the helix α3, creating a four-helix bundle. The binding interface is tightly packed and dominated by hydrophobic amino acids. Binding and folding of N487–503 are coupled. However, when not bound to P, N487–503 does not resemble a statistical random coil but instead exists in a loosely structured state that mimics the bound conformation. We propose that before diffusional encounter, the ensemble of accessible conformations for N487–503 is biased toward structures capable of binding P, facilitating rapid association of the two proteins. This study provides a structural analysis of polymerase–template interactions in a paramyxovirus and presents an example of a protein–protein interaction that must be only transiently maintained as part of its normal function.
Measles virus, a member of the paramyxovirus family, causes an acute infectious disease in humans. The virus is enveloped and possesses a negative-sense, single-stranded RNA genome ≈15,900 nt in length. Within the virion, the nucleocapsid protein (N) packages the genomic RNA into a helical protein–RNA complex termed the nucleocapsid. In the cytoplasm of an infected cell, the viral RNA polymerase uses the nucleocapsid as a template for both the transcription of messenger RNAs, encoding the individual viral proteins, as well as the replication and encapsidation of full-length copies of the viral genome. Unencapsidated RNA cannot act as a template for the polymerase. The replication of paramyxoviruses has been comprehensively reviewed (1–3).
The paramyxoviral polymerase has two components, the large protein and the phosphoprotein (P) (Fig. 1). The catalytic activities of the polymerase reside within the large protein, whereas P is responsible for, among other activities, binding the polymerase to the nucleocapsid (4, 5). P is a modular protein containing a number of functional domains separated by intrinsically disordered sequences (6). A coiled-coil domain oligomerizes P (7) while the extreme carboxyl terminus of P is involved in nucleocapsid binding. The structure of this region of measles P (amino acids 459–507) has been recently determined by x-ray crystallography and is a compact bundle of three α-helices (8). This region by itself constitutes the nucleocapsid-binding domain of P, being both necessary and sufficient for binding to nucleocapsid-like particles (8, 9).
Fig. 1.

Schematic diagram of polymerase attachment in measles virus. A helical nucleocapsid-binding domain (NBD) at the carboxyl terminus of P (amino acids 457–507) mediates binding of the polymerase to the nucleocapsid by attaching to a short and contiguous sequence (amino acids 477–505) within the tail of RNA-associated N. A coiled-coil within P holds the downstream NBDs in close proximity, with the sequence interleaving the coiled-coil and the downstream NBD likely to be largely unstructured (32). P is depicted as a tetramer; however, the oligomerization state of measles P has not been determined.
The RNA-associated N, to which P attaches, has a bipartite organization (Fig. 1). An amino terminal assembly domain (amino acids 1–375) is responsible for RNA packaging and organization of the helical nucleocapsid. A carboxyl-terminal tail, located on the nucleocapsid exterior, is not required for nucleocapsid assembly and appears to be intrinsically disordered (6, 10). In measles virus, the binding site for P has been mapped to amino acids 477–505 within the tail of N (9). By using isothermal titration calorimetry, it has been shown that measles P457–507 binds measles N477–505 with 1:1 stoichiometry and that the binding affinity is weak (Kd = 13 μM at 20°C and 35 μM at 37°C) (9). Data from a series of spectroscopic studies suggest that folding and binding of the tail of N are coupled (8, 10, 11), and a theoretical model of the interaction has been proposed in which an N-tail peptide binds as a helix into a hydrophobic groove on the surface of P457–507 (8). A schematic model of polymerase attachment in measles virus is shown in Fig. 1. Whereas it has been hypothesized that P will walk or cartwheel over the surface of the nucleocapsid during RNA synthesis, little is known from experimentation about the coupling of catalysis and movement of the viral polymerase.
Regardless of the details of translocation, the processive motion of the polymerase along the nucleocapsid requires the repeated attachment and release of the binding domain from measles P to the sequence it recognizes within the tail of N (Fig. 1). In this paper, we analyze the binding of measles P457–507 to measles N477–505 by using solution NMR spectroscopy and also describe an x-ray crystal structure of a chimeric molecule containing both binding domains. The domains are found in a complex that we show to be representative of the bound state. This study provides direct structural insights into polymerase–template interactions in a paramyxovirus and, more generally, an example of a fast-associating, weak-affinity protein–protein interaction that must be only transiently maintained as part of its normal function.
Methods
Sample Preparation. The expression and purification of P457–507 and N477–505, both unlabeled and isotopically substituted, was carried out as described (9). The protein P457–507 carries the mutation P458G to facilitate cleavage from its fusion partner during purification. N477–505 carries two nonnative amino acids (Gly and Ser, GS) at its amino terminus as well as a nonnative tyrosine at its carboxyl terminus, the latter facilitating quantitation of the protein by UV absorption. To make the chimeric P457–507(GS)4N486–505 (see Results), the coding sequence was created by using a two-stage PCR protocol and ligated into a variant of pET41a(+) (Novagen) as described for P457–507 (9). This procedure results in a vector expressing the required protein, fused to the carboxyl terminus of GST, with an interleaving tobacco etch virus protease cleavage site. The initial purification of the protein, and proteolytic cleavage to release P457–507(GS)4N486–505, was performed as described (9). The protein was further purified by cation-exchange chromatography by binding it to SP Sepharose HP resin, buffered with 25 mM Mops/KOH at pH 7/25 mM NaCl, and then displacing it by using a linear salt gradient. All protein concentrations were estimated from absorbance measurements at 280 nm (12).
NMR Spectroscopy, Data Processing, and Resonance Assignments. For spectroscopic measurements, samples were dialyzed against 10 mM NaH2PO4/Na2HPO4 at pH 5.7/100 mM NaCl/0.01% sodium azide. Ten percent (vol/vol) 2H2O was added to the samples before NMR data acquisition. All spectra were recorded at 20°C by using a Varian Inova 600-MHz spectrometer equipped with a triple-resonance pulsed-field gradient probe. NMR data were analyzed by using the programs nmrpipe (13) and sparky (T. G. Goddard and D. G. Kneller, University of California, San Francisco).
Backbone resonance assignments for P457–507 and N477–505 were made by using the standard triple-resonance experiments 3D HNCACB, CBCA(CO)NH, H(CCO)NH, and C(CO)NH. Assignments for N477–505 bound to P457–507 were made by repeating the same 3D experiments, using labeled N477–505 titrated with saturating amounts of unlabeled P457–507. 3D 15N-edited [1H,1H] NOESY spectra (mixing time 150 ms) and steady-state heteronuclear 1H–15N nuclear Overhauser enhancements (NOEs) (14) were recorded for both free and bound N477–505.
P457–507/N477–505 Titrations. The titration of P457–507 with N477–505, used for quantitative analysis, was performed in the following manner (15). Two samples were initially prepared, both 600 μl. Sample A contained 228 μM 15N-labeled P457–507. Sample B contained 15N-labeled P457–507 at the same concentration and unlabeled N477–505 at a concentration of 683 μM. The buffer composition was in each case identical (9 mM NaH2PO4/Na2HPO4 at pH 5.7/90 mM NaCl/0.009% sodium azide in 90% H2O/10% D2O). 2D 15N–1H heteronuclear sequential quantum correlation spectra (16, 17) were recorded on the A and B samples. These spectra represent the end points of the titration, with molar ratios of 1.0:0.0 and 1.0:3.0 P457–507/N477–505, respectively. Samples with intermediate values of the molar ratio were prepared by simultaneously removing an equal volume of liquid from each sample tube and then transferring the aliquots into the other tube (i.e., the aliquot withdrawn from tube A was transferred into tube B and vice versa). Spectra were recorded on these samples, and the volume exchange procedure was repeated until a total of 14 spectra had been obtained. With the titration performed in this fashion, neither the buffer composition nor the total concentration of the monitored species (P457–507) varied for any of the spectra recorded. A titration of N477–505 with P457–507 was performed in similar fashion.
Quantitative Analysis of NMR Titration Data. For individual resonances the peak position and linewidths at each point in the titration were estimated by fitting Lorentzian functions to the data, using the program sparky. We excluded from numerical analysis peaks whose chemical shift trajectories overlapped during the titration. Titration data were analyzed in terms of a simple bimolecular association scheme, as detailed in Results.
To determine the dissociation constant for the binding process, we analyzed resonances that experienced chemical shift perturbation during the titration, but little or no change in linewidth, and, hence were in very fast exchange (18). For one-to-one binding of a protein (P) to a ligand (L), when the fast exchange condition is satisfied, the observed chemical shift difference Δδobs is described by (19)
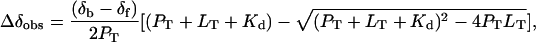 |
[1] |
where PT and LT are the total concentrations of protein and ligand, respectively, (δb - δf) is the total chemical shift difference between the bound and free state, and Kd is the equilibrium dissociation constant. PT and LT are both known, hence Kd and (δb - δf) can be determined by nonlinear least squares fitting. The program xcrvfit (R. Boyko and B. D. Sykes, University of Alberta, Edmonton, Alberta, Canada) was used for fitting.
The dissociation rate constant was estimated from resonances that underwent both chemical shift perturbation and line broadening during the course of the titration. For a nucleus in moderately fast exchange between the free and bound states, the observed linewidth Δνobs, at half height of the resonance (reported in Hz), is given by (19)
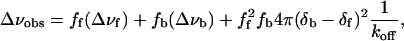 |
[2] |
where Δνf and Δνb are the linewidths in the free and bound states, (δb - δf) is the total chemical shift difference between the bound and free state, ff and fb are the fractions of the protein free and bound, and koff is the dissociation rate constant. Because the Kd was already determined, ff and fb could be calculated for each point in the titration. The raw titration data provided a good estimate for (δb - δf). The best-fit values for Δνf, Δνb, and koff were subsequently determined by a simple grid search, adjusting these parameters to minimize the sum of the squared differences between calculated and observed linewidths.
X-Ray Crystallography. Crystals of P457–507(GS)4N486–505 were grown by the vapor diffusion method. The protein (4.2 mM in 12.5 mM Mops/KOH at pH 7.0/100 mM NaCl) was equilibrated at room temperature against solutions containing 0.2 M 3-[(1,1-dimethyl-2-hydoxyethyl)amino]-2-hydroxypropanesulfonic acid (AMPSO)/KOH buffer at pH 9.1 and 0.5–1.0 M ammonium sulfate. Crystallization was insensitive to changes in pH (4.9–9.1) or buffer composition. Diffraction data were collected by the oscillation method on an R-axisIV (Rigaku, Tokyo) system, using a single crystal mounted in a thin-walled capillary and maintained at room temperature. Data integration and scaling were performed with the programs denzo and scalepack (Zbyszek Otwinowski, University of Texas, Austin, and Wladek Minor, University of Virginia, Charlottesville) (20). The unit cell dimensions were a = b = 42.2 Å, c = 81.8 Å, α = β = γ = 90° and the crystal space group was initially identified as either P41212 or P43212. There is a single molecule in the asymmetric unit of the crystal. Molecular replacement calculations with the program epmr (Agouron Pharmaceuticals, La Jolla, CA) (21), using the structure of the measles P nucleocapsid-binding domain (8), gave a clearly discriminated solution in space group P41212. The program xfit from the xtalview software package (The Scripps Research Institute, La Jolla, CA) (22) was used for interactive model building, and the structure was refined by using the program tnt (23). For calculation of Rfree, 5% of the data were randomly selected and excluded from all refinement procedures. Statistics associated with the x-ray diffraction data and model refinement are given in Table 1.
Table 1. X-ray diffraction data and model refinement statistics.
| Measurement | Value |
|---|---|
| X-ray diffraction data | |
| Outer resolution limit, Å | 2.0 |
| No. of observations | 5,306 |
| Mean redundancy | 7.1 |
| Completeness, % | 97.5 |
| Rmeasure (41) | 0.074 |
| Model refinement | |
| Rwork/Rfree, % | 23.0/27.5 |
| No. of atoms | 577 |
| No. of water molecules | 28 |
| Mean isotropic B factor, Å2: N moiety/P moiety | 50/34 |
| rms deviation from ideal geometry: Bond lengths, Å/bond angles, ° | 0.004/0.7 |
| Residues in most allowed regions of Ramachandran plot (42), % | 100 |
Results
Chemical Shift Changes on Binding of P457–507 to N477–505. After the sequence specific resonance assignment of P457–507 and N477–505 (see Methods), we determined the regions of the two molecules that were directly involved in binding by following chemical shift changes in the backbone amide resonances as the two proteins were titrated. A series of 2D 1H–15N heteronuclear sequential quantum correlation spectra were collected for both P457–507 and N477–505 (15N-labeled) in the presence of various concentrations of their unlabeled binding partners.
The chemical shift trajectories observed in the 2D spectra were linear, consistent with a two-state binding process. In both titrations, resonances were in fast to moderately fast exchange, with some of the resonances undergoing line broadening at the intermediate points of the titration. We define the total chemical shift change (in Hz) for the backbone amide resonances as
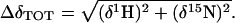 |
[3] |
For P457–507, we mapped the total chemical shift changes that occur on binding N477–505 onto the x-ray crystal structure (8). Substantial chemical shift changes occur in the last half of helix α2, helix α3, and their connecting loop (Fig. 2A). In contrast, residues within helix α1 are relatively unperturbed. These observations define the likely location for attachment of the measles N-tail peptide as the surface cleft created by helices α2 and α3 of P457–507, as predicted on the basis of surface hydrophobicity and sequence conser vation among the paramyxoviruses (8).
Fig. 2.

Mapping of the backbone amide chemical shift differences observed on binding of P457–507 and N477–505. (A) Ribbon diagram of the structure of P459–507, colored according to the total chemical shift differences observed on binding of N477–505. The figure is prepared with the program molmol (36). (B) Total chemical shift differences for N477–505, observed on binding of P457–507, plotted as a function of residue number.
For N477–505 we plotted the total chemical shift changes that occur on binding P457–507 as a function of residue number (Fig. 2B). Residues preceding Gln-483 are not influenced by binding and are unlikely to have any role in complex formation. Residues in the remainder of the tail peptide show various degrees of shift perturbation, with the largest chemical shifts observed for Ser-491 and Ala-492.
Strength and Kinetics of Binding. The titration data were further analyzed in terms of the simple binding scheme
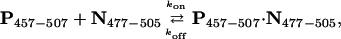 |
[4] |
where koff is the first-order rate constant for the unimolecular dissociation reaction, kon is the second-order rate constant for the bimolecular association reaction, and their ratio koff/kon is the equilibrium dissociation constant Kd. In the quantitative analysis described below we examined as many resonances as possible, detecting no inconsistencies that would suggest that this simple bimolecular association scheme is inappropriate.
To determine the equilibrium dissociation constant, we analyzed resonances that underwent modest chemical shift changes on binding. These resonances were in very fast exchange, exhibiting little or no change in linewidth over the course of the titration. Fig. 3A summarizes the model fitting process (Methods, Eq. 1) for one such resonance. The mean dissociation constant is 13.2 μM (sample SD 2.4 μM, n = 5). This result is in agreement with an independent estimate made by using isothermal titration calorimetry (9), where Kd was also determined to be 13 μM at 20°C.
Fig. 3.

Quantitative analysis of the interaction between P457–507 and N477–505. (A) Determination of the dissociation constant (Kd). The figure shows the changes in the chemical shift of the backbone amide 15N resonance for residue R461, observed during titration of P457–507 with N477–505. The solid line represents the fitted model (Eq. 1). For this resonance, the fitted parameters were (δb - δf) = 25.7 Hz, and Kd = 12.2 μM. (Inset) Relevant section of the 2D 15N–1H heteronuclear sequential quantum correlation spectra, with points 1, 4, 5, and 14 of the titration displayed. (B) Determination of the dissociation rate constant (koff). The figure shows changes in the linewidth of the backbone amide 1H resonance for residue K503, observed during titration of P457–507 with N477–505. The solid line represents the fitted model (Eq. 2). For this resonance, the chemical shift difference (δb - δf) between the free and bound state is 52 Hz, and the fitted parameters were Δνf = 12.5 Hz, Δνb = 14.5 Hz, and koff = 670 s-1. Inset shows the relevant section of the 2D 15N–1H heteronuclear sequential quantum correlation spectra, with points 1, 4, 5, and 14 of the titration displayed.
We also characterized the kinetics of the binding process by analyzing resonances in moderately fast exchange, which underwent small but measurable changes in linewidth over the course of the titration (Fig. 3B). For these resonances the increase in the observed linewidth caused by exchange between the free and bound states is controlled by the dissociation rate constant koff (Methods, Eq. 2). As expected (18), the maximal line broadening was seen at ≈1/3 saturation (Fig. 3B). The mean off rate was determined to be 640 s-1 (sample SD 70 s-1, n = 9). From this off rate, and the estimate for the dissociation constant, we calculate that the on rate for the binding process (kon = koff/Kd) is 0.5 × 108 M-1·s-1. Typical association rates for protein–protein interactions are in the order of 105 to 106 M-1·s-1 (24), hence association of the measles P-binding domain with the measles N-tail peptide proceeds relatively quickly and is approaching the diffusion-controlled limit.
Secondary Structure of the Measles N-Tail Peptide in the Free and Bound State Characterized by Solution NMR Spectroscopy. To assess the organization of N477–505 in the free and bound state, we examined several indicators of structure accessible by solution NMR spectroscopy. To make observations on N477–505 in the bound state we titrated labeled peptides (15N or 15N/13C) with saturating amounts of unlabeled P457–507.
First we examined the Cα chemical shifts (Fig. 4A), which are a reliable indicator of secondary structure (25), experiencing a downfield shift when located in α-helices and an upfield shift when located in β-strands. In accord with our previous observations, residues 477–484 were essentially unperturbed by the binding process, and their Cα chemical shifts showed little deviation from random coil values in either the free or bound state. Hence, these residues, which precede the sole proline residue (P485) in the tail peptide, are not involved in binding and do not appear to have any residual structure. In contrast, residues 486–503 were strongly perturbed by binding and showed large downfield shifts in their Cα resonances, consistent with organization of this region into a helix in the bound state. Intriguingly, these same resonances are also downshifted (relative to random coil values) in the free state, suggesting that this segment of N is not a statistical random coil when unbound.
Fig. 4.

Organization of N477–505 in the free and bound state. (A) Deviation of Cα chemical shifts from random coil values for N477–505 in the free (Left) and bound (Right) states. Random coil chemical shifts (25°C) were taken from Wishart et al. (37). (B) Steady-state heteronuclear 1H–15N NOEs for N477–505 in the free (Left) and bound (Right) states.
We also examined the magnitude of the steady-state heteronuclear 1H–15N NOE, as a measure of overall backbone flexibility (Fig. 4B). The steady-state 1H–15N NOE is highly sensitive to reorientation of the 1H–15N bond vector with negative, or small positive, NOE values indicating fast motion relative to the overall tumbling rate of the molecule. The observations are in accord with the chemical shift analysis. Residues 477–484 appear likely to lack any persistent structure in both the free and bound states, whereas residues 486–503 are both perturbed and markedly rigidified on binding. In the free state, there exists a core region of the peptide (residues 487–500) that shows restricted motion relative to the surrounding sequences.
Finally, we inspected 3D 15N-edited NOESY spectra (mixing time = 150 ms), looking for the Hα(i) → HN(i + 3) NOEs that are characteristic of helical geometry. For residues 487–500 of N in the bound state, we were able to unambiguously identify many of these medium-range NOEs. In contrast, in the free state, the same NOEs were not observed, indicating that although not a statistical random coil (Fig. 4), the unbound N is very loosely structured. This result is consistent with circular dichroism spectra of unbound N477–505, which do not display the features expected of a helical peptide (data not shown), as well as the circular dichroism titration studies carried out by others (8, 10, 11).
Crystallographic Structure Determination of P457–507 in Complex with N486–505. Our NMR spectroscopy data demonstrated that N487–503 bound to P457–507 as a helix, which was likely to be aligned with helix α3 of P (Fig. 2 A). However, it was not clear whether the helix from N would bind parallel or antiparallel to helix Pα3. To fully characterize the bound state, we carried out a crystallographic analysis of a chimeric protein in which amino acids 486–505 from N (N486–505) were fused to the carboxyl terminus of the nucleocapsid-binding domain of P (P457–507). The two binding elements were connected with a flexible linker, (GS)4, designed to be long enough to accommodate either parallel or antiparallel packing of the helix from N. Given the linker length, parallel packing could be achieved only through an intramolecular association, whereas antiparallel packing might arise from either intermolecular or intramolecular association.
The chimeric protein P457–507(GS)4N486–505 crystallized readily, and we determined the structure at 2-Å resolution by using the method of molecular replacement with the structure of P459–507 (8) as the search model. A schematic diagram of the complex observed in the crystal is shown in Fig. 5A. As expected the P–N complex resembles a four-helix bundle, with the helix contributed by N (helix Nα1) packed on the surface created by helices α2 and α3 of the binding domain from P. The packing angle between helix Nα1 and Pα2 is -154° and between helix Nα1 and Pα3 it is 10°. It is clear from consideration of both crystal packing and linker length that the complex observed in the crystal results from an intermolecular association with the N moiety from one protein binding to the P moiety of a separate molecule. There is almost no interpretable electron density for residues within the linker, so unambiguous assignment of the connectivity within the crystal is not possible, although there is sufficient space to accommodate this sequence.
Fig. 5.

Structure of the chimeric molecule P457–507(GS)4N486–505. (A) Ribbon diagram of the complex observed in the crystal. P is shown in blue and N is shown in green. The complex results from an intermolecular association, with the N moiety from one protein binding to the P moiety of a separate molecule. Residues within the flexible linker are disordered. (B) Burial of molecular surface in the complex. Upper shows two views of the molecular surface of the P–N complex (related by a 180° rotation), colored blue (P) or green (N) according to the contributing molecule. In Lower, either P or N has been removed and surface regions totally buried in the complex interior are colored white. Amino acids L481, L484, I488, F497, M504, and I504 from P, as well as S491, A494, L495, L498, and M501 from N each contribute more than 30 Å2 to the buried surface. (C) The sole direct hydrogen-bonding interaction between the N and P moieties in the complex. The side-chain atoms of Ser-491 (N) and Asp-493 (P), as well as the main-chain atoms of residues 488–490 (P) are shown in ball and stick representation. Apparent hydrogen bonds are indicated with yellow dashed lines. Ribbon diagrams were prepared by using the program ribbons 2.0 (38). Molecular surfaces were calculated and displayed by using the programs msms and msv (39) and buried surface areas were computed by using the program sims (40).
The complex observed by x-ray crystallography is consistent with the binding data obtained by using NMR spectroscopy. The residues of P indicated by NMR spectroscopy as being most perturbed by binding (Fig. 2A) are precisely those involved in forming the N–P interface in the complex (Fig. 5A). P is shown in approximately the same orientation in both figures to facilitate comparison. Similarly, those residues of N identified as being structured and helical in the bound state (Fig. 4) have this conformation in the crystal structure. The observations made by NMR spectroscopy, as well as our earlier isothermal titration calorimetry studies (9), strongly suggest that there is not more than one mode of binding (i.e., an ability of the helix Nα1 to bind in both parallel and antiparallel fashion).
The binding interface is tightly packed and dominated by hydrophobic amino acids (Fig. 5B), in agreement with earlier proposals that burial of hydrophobic side chains was likely to be driving the binding process (8, 9). The shape complementarity of the interface (shape correlation statistic, Sc = 0.71) (26) is comparable with that seen in other protein–protein interactions as, for example, in the subunit interfaces of oligomeric proteins. There is very little reconfiguration of P upon binding (rms deviation between unbound and bound forms = 0.46 Å for main-chain atoms and 1.6 Å for all atoms for residues 460–505). Association of the two molecules results in the burial of 400 Å2 of the molecular surface of P (13% of the total surface area of the molecule), and 350 Å2 of the surface of N (although binding and folding of N are coupled, so this number likely underestimates the surface area buried on binding). Within the complex, only two direct hydrogen bonds were identified between P and N, both of which involve the side-chain hydroxyl of Ser-491, from N (Fig. 5C).
Discussion
When the structure of the nucleocopsid binding domain of measles virus P was reported (8), a theoretical model of the interaction between this domain and the tail of measles virus N was also proposed. In this model, amino acids 489–506 of N were docked, in a helical conformation, onto the α2/α3 face of the binding domain from P. Whereas the model correctly identifies the secondary structure elements involved in binding, the helix contributed by N is bound in the reverse orientation to that observed in the crystal structure (Fig. 5A).
The P457–507–N486–505 complex is a four-helix bundle. The simplest and most commonly observed topology for such domains has an up-down-up-down arrangement of the helices, so that they run in alternating directions (27, 28). However, four-helical bundles with alternative topologies are also known. In the cytokine family, two long loops form crossovers, generating an up-up-down-down arrangement of helices. For the P457–507–N486–505 complex the helices of the bundle are contributed by different molecules, hence crossover loops are not required to achieve the relatively unusual arrangement of the helices. The observed packing angle between helix Nα1 and Pα3 (10°) is not commonly observed in protein structures (29), which may reflect that the complex is not optimized for stability.
The carboxyl-terminal tail of measles N (amino acids 400–525) lacks a defined tertiary structure (6). On the basis of a series of circular dichroism spectroscopic studies, it was earlier hypothesized that association of the tail of N with P induced folding of N (8, 10, 11). Our results confirm this idea but with two important caveats. First, the induced folding is quite localized, involving ≈18 aa from the 125-aa tail of N. Second, binding does not involve a complete disorder–order transition within N. Our observations on N477–505 made using NMR spectroscopy (Fig. 4) show that N486–503 is not a statistical random coil when unbound. Within this region, the ensemble of accessible conformations for N is likely to be biased toward structures capable of binding P, as reflected in the downfield shifting of the Cα resonances (Fig. 4A) and the restricted conformational freedom of residues 487–500 (Fig. 4B), which is observed even in the absence of P. This finding is in accord with earlier speculation that residual structure within the tail of N might be important for efficient binding of P (10).
Consistent with this interpretation, we find the kinetics of association of the two proteins are fast (calculated kon = 0.5 × 108 M-1·s-1). The fast binding kinetics and the weak binding affinity are likely dictated by the need for rapid movement of the polymerase along the nucleocapsid during RNA synthesis. Because there appears to be no topological restraint involved in tethering the polymerase to its template, as there is in many highly processive systems (30), the interactions between P and N must be delicately balanced to allow for both specific binding and movement. In this regard, it is possible that coupling the binding and folding of N allows for more precise control of the binding process than would be possible if both proteins were fully structured (31).
Given the structure of the P–N complex, it is also possible to attempt a more detailed interpretation of the thermodynamics of association, established by using isothermal titration calorimetry (9). We highlight two points. First, binding is associated with a change in heat capacity, ΔCP, of -510 cal/K·mol, based on measurement of ΔH at two temperatures. The large negative ΔCP is consistent with the burial of an appreciable hydrophobic surface area on complex formation. Second, the entropic contribution to the Gibbs free energy of association, ΔS, is quite unfavorable (-14 cal/K·mol at 20°C, and -42 cal/K·mol at 37°C). We suggest that this term is likely to be dominated by the loss in conformational freedom of the N-tail peptide on binding, particularly given the absence of significant rearrangement of P and paucity of direct hydrogen bonds in the complex (that would otherwise lead to entropic contributions through release of bound water).
In the case of measles virus (a Morbillivirus), the nucleocapsid-binding domain from P is very stable (9) and essentially acts as a folding template for the sequence it recognizes within the relatively unstructured tail of N. In Sendai virus (a Respirovirus) binding of P to the nucleocapsid appears likely to proceed in an entirely analogous fashion, with a structured and helical binding domain (32) recognizing a sequence within the tail of N (33–35). However, in the case of mumps virus (a Rubulavirus), we have earlier shown that the nucleocapsid-binding domain from P lacks persistent tertiary structure, and it recognizes a sequence not in the tail of N but within the structured amino-terminal assembly domain of the molecule (9). Thus, there is reason to think that our results may not extend across all of the Paramyxovirinae, and that the Rubulaviruses in particular may have evolved a subtly different binding mechanism. We anticipate that the helical binding element within N has migrated at some point in the evolution of these viruses, and more speculatively, that although binding and folding of the relevant domains from N and P will remain coupled in Rubulaviruses, the roles of the partners may be reversed.
Acknowledgments
We thank Dr. Walt Baase for suggesting structural studies on a chimeric molecule and Dr. Wendy Breyer for criticism of the manuscript. This work was supported in part by National Institutes of Health Grants GM57766 (to F.W.D.) and GM20066 (to B.W.M.).
Abbreviations: N, nucleocapsid protein; NOE, nuclear Overhauser enhancement; P, phosphoprotein.
References
- 1.Conzelmann, K. K. (1998) Annu. Rev. Genet. 32, 123-162. [DOI] [PubMed] [Google Scholar]
- 2.Sedlmeier, R. & Neubert, W. J. (1998) Adv. Virus Res. 50, 101-139. [DOI] [PubMed] [Google Scholar]
- 3.Curran, J. & Kolakofsky, D. (1999) Adv. Virus Res. 54, 403-422. [DOI] [PubMed] [Google Scholar]
- 4.Portner, A., Murti, K. G., Morgan, E. M. & Kingsbury, D. W. (1988) Virology 163, 236-239. [DOI] [PubMed] [Google Scholar]
- 5.Horikami, S. M. & Moyer, S. A. (1995) Virology 211, 577-582. [DOI] [PubMed] [Google Scholar]
- 6.Karlin, D., Ferron, F., Canard, B. & Longhi, S. (2003) J. Gen. Virol. 84, 3239-3252. [DOI] [PubMed] [Google Scholar]
- 7.Tarbouriech, N., Curran, J., Ruigrok, R. W. & Burmeister, W. P. (2000) Nat. Struct. Biol. 7, 777-781. [DOI] [PubMed] [Google Scholar]
- 8.Johansson, K., Bourhis, J. M., Campanacci, V., Cambillau, C., Canard, B. & Longhi, S. (2003) J. Biol. Chem. 278, 44567-44573. [DOI] [PubMed] [Google Scholar]
- 9.Kingston, R. L., Baase, W. A. & Gay, L. S. (2004) J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 10.Longhi, S., Receveur-Brechot, V., Karlin, D., Johansson, K., Darbon, H., Bhella, D., Yeo, R., Finet, S. & Canard, B. (2003) J. Biol. Chem. 278, 18638-18648. [DOI] [PubMed] [Google Scholar]
- 11.Bourhis, J. M., Johansson, K., Receveur-Brechot, V., Oldfield, C. J., Dunker, K. A., Canard, B. & Longhi, S. (2004) Virus Res. 99, 157-167. [DOI] [PubMed] [Google Scholar]
- 12.Gill, S. C. & von Hippel, P. H. (1989) Anal. Biochem. 182, 319-326. [DOI] [PubMed] [Google Scholar]
- 13.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277-293. [DOI] [PubMed] [Google Scholar]
- 14.Farrow, N. A., Muhandiram, R., Singer, A. U., Pascal, S. M., Kay, C. M., Gish, G., Shoelson, S. E., Pawson, T., Forman-Kay, J. D. & Kay, L. E. (1994) Biochemistry 33, 5984-6003. [DOI] [PubMed] [Google Scholar]
- 15.McAlister, M. S., Mott, H. R., van der Merwe, P. A., Campbell, I. D., Davis, S. J. & Driscoll, P. C. (1996) Biochemistry 35, 5982-5991. [DOI] [PubMed] [Google Scholar]
- 16.Kay, L. E., Keifer, P. & Saarinen, T. (1992) J. Am. Chem. Soc. 114, 10663-10665. [Google Scholar]
- 17.Zhang, O., Kay, L. E., Olivier, J. P. & Forman-Kay, J. D. (1994) J. Biomol. NMR 4, 845-858. [DOI] [PubMed] [Google Scholar]
- 18.Feeney, J., Batchelor, J. G., Albrand, J. P. & Roberts, G. C. K. (1979) J. Magn. Reson. 33, 519-529. [Google Scholar]
- 19.Lian, L. Y. & Roberts, G. C. K. (1993) in NMR of Macromolecules: A Practical Approach, ed. Roberts, G. C. K. (Oxford Univ. Press, New York), pp. 153-182.
- 20.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 21.Kissinger, C. R., Gehlhaar, D. K. & Fogel, D. B. (1999) Acta Crystallogr. D 55, 484-491. [DOI] [PubMed] [Google Scholar]
- 22.McRee, D. E. (1999) J Struct. Biol. 125, 156-165. [DOI] [PubMed] [Google Scholar]
- 23.Tronrud, D. E. (1997) Methods Enzymol. 277, 306-319. [DOI] [PubMed] [Google Scholar]
- 24.Schreiber, G. (2002) Curr. Opin. Struct. Biol. 12, 41-47. [DOI] [PubMed] [Google Scholar]
- 25.Wishart, D. S. & Sykes, B. D. (1994) J Biomol. NMR 4, 171-180. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence, M. C. & Colman, P. M. (1993) J. Mol. Biol. 234, 946-950. [DOI] [PubMed] [Google Scholar]
- 27.Kamtekar, S. & Hecht, M. H. (1995) FASEB J. 9, 1013-1022. [DOI] [PubMed] [Google Scholar]
- 28.Kohn, W. D., Mant, C. T. & Hodges, R. S. (1997) J. Biol. Chem. 272, 2583-2586. [DOI] [PubMed] [Google Scholar]
- 29.Walther, D., Eisenhaber, F. & Argos, P. (1996) J. Mol. Biol. 255, 536-553. [DOI] [PubMed] [Google Scholar]
- 30.Breyer, W. A. & Matthews, B. W. (2001) Protein Sci. 10, 1699-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dyson, H. J. & Wright, P. E. (2002) Curr. Opin. Struct. Biol. 12, 54-60. [DOI] [PubMed] [Google Scholar]
- 32.Blanchard, L., Tarbouriech, N., Blackledge, M., Timmins, P., Burmeister, W. P., Ruigrok, R. W. & Marion, D. (2004) Virology 319, 201-211. [DOI] [PubMed] [Google Scholar]
- 33.Buchholz, C. J., Retzler, C., Homann, H. E. & Neubert, W. J. (1994) Virology 204, 770-776. [DOI] [PubMed] [Google Scholar]
- 34.Curran, J., Homann, H., Buchholz, C., Rochat, S., Neubert, W. & Kolakofsky, D. (1993) J. Virol. 67, 4358-4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan, K. W., Portner, A. & Murti, K. G. (1993) Virology 193, 376-384. [DOI] [PubMed] [Google Scholar]
- 36.Koradi, R., Billeter, M. & Wüthrich, K. (1996) J. Mol. Graphics 14, 29-32, 51-55. [DOI] [PubMed] [Google Scholar]
- 37.Wishart, D. S., Bigam, C. G., Holm, A., Hodges, R. S. & Sykes, B. D. (1995) J. Biomol. NMR 5, 67-81. [DOI] [PubMed] [Google Scholar]
- 38.Carson, M. (1997) Methods Enzymol. 277, 493-505. [PubMed] [Google Scholar]
- 39.Sanner, M. F., Olson, A. J. & Spehner, J. C. (1996) Biopolymers 38, 305-320. [DOI] [PubMed] [Google Scholar]
- 40.Vorobjev, Y. N. & Hermans, J. (1997) Biophys. J. 73, 722-732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diederichs, K. & Karplus, P. A. (1997) Nat. Struct. Biol. 4, 269-275. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]